

Establishing a Prediction Model for the Efficacy of Platinum—Based Chemotherapy in NSCLC Based on a Two Cohorts GWAS Study
 ,
,
Abstract
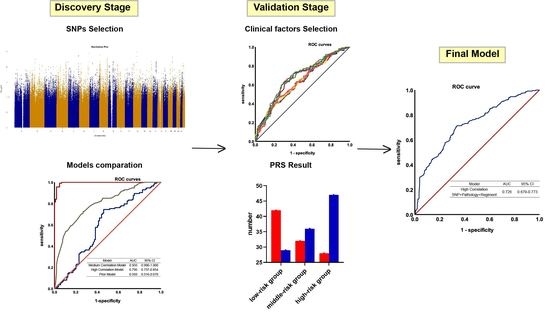
:
1. Introduction
2. Materials and Methods
2.1. Study Population and Clinical Data Collection
2.2. DNA Extraction, Genotyping, and Quality Control
2.3. Genome–Wide Association Analysis and Linkage Disequilibrium (LD) Pruning
2.4. Model Construction
3. Results
3.1. Characteristics of the Study Population
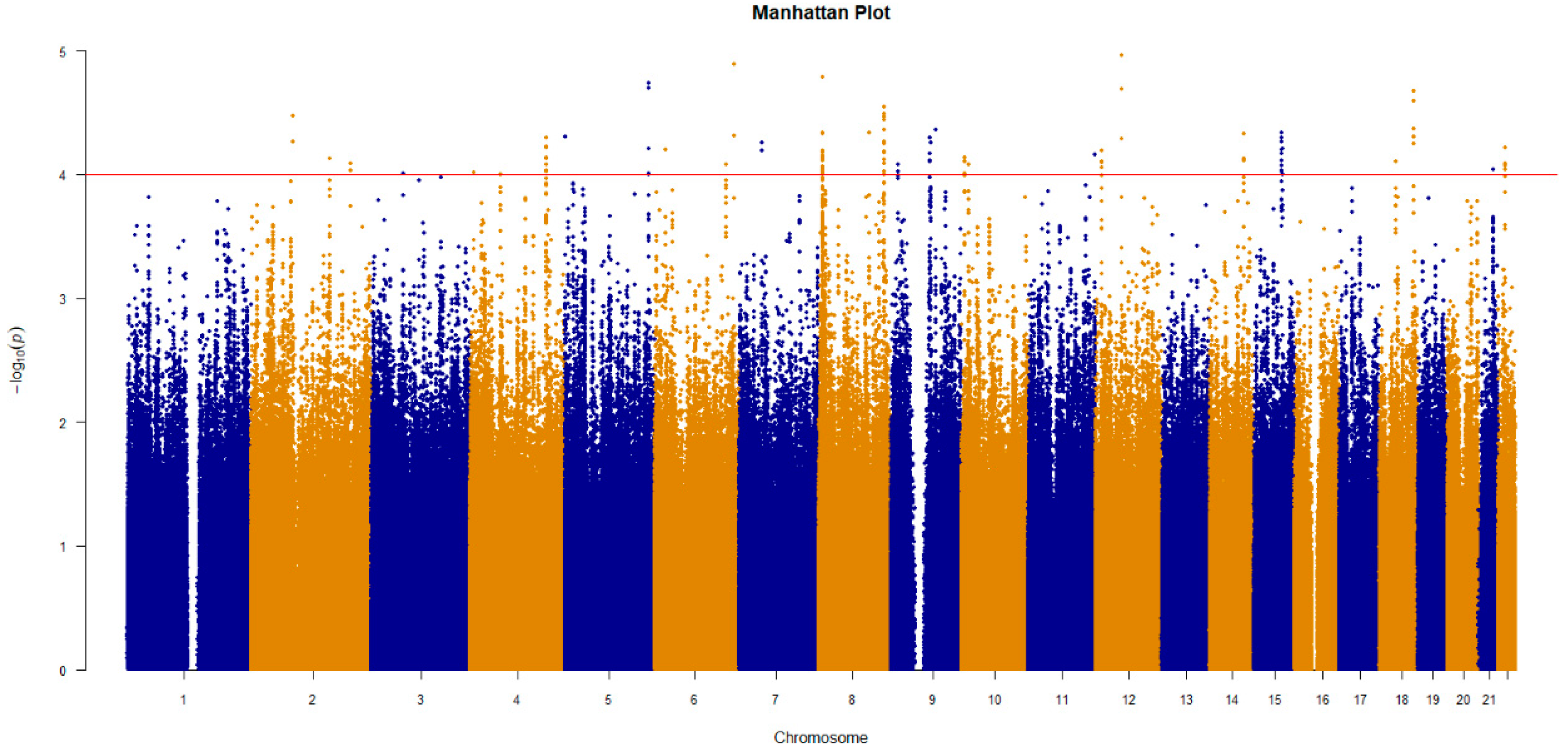
3.2. GWAS to Identify SNPs Associated with Platinum–Based Chemotherapy Response
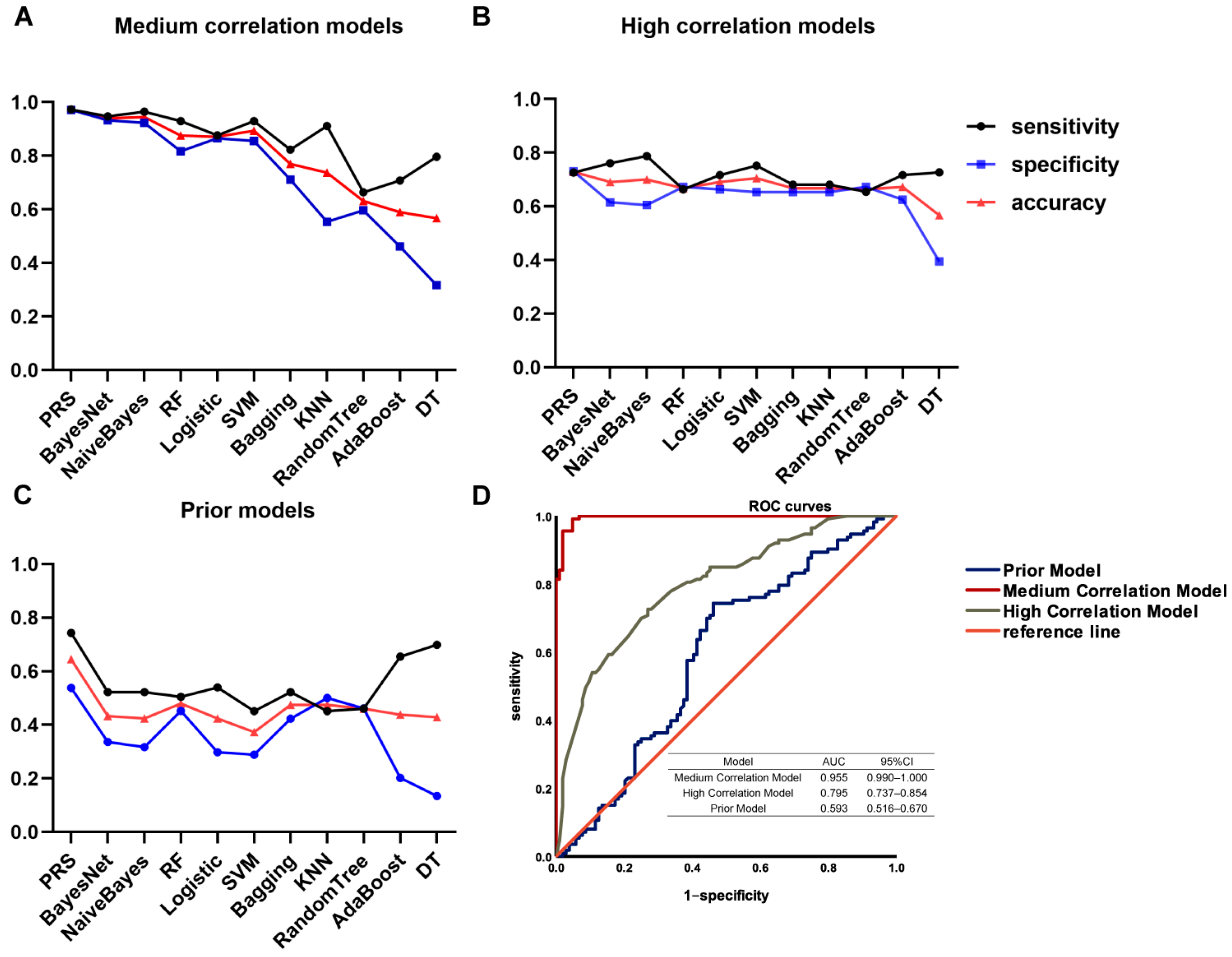
3.3. Construction of the Genetic Prediction Models in the Discovery Cohort
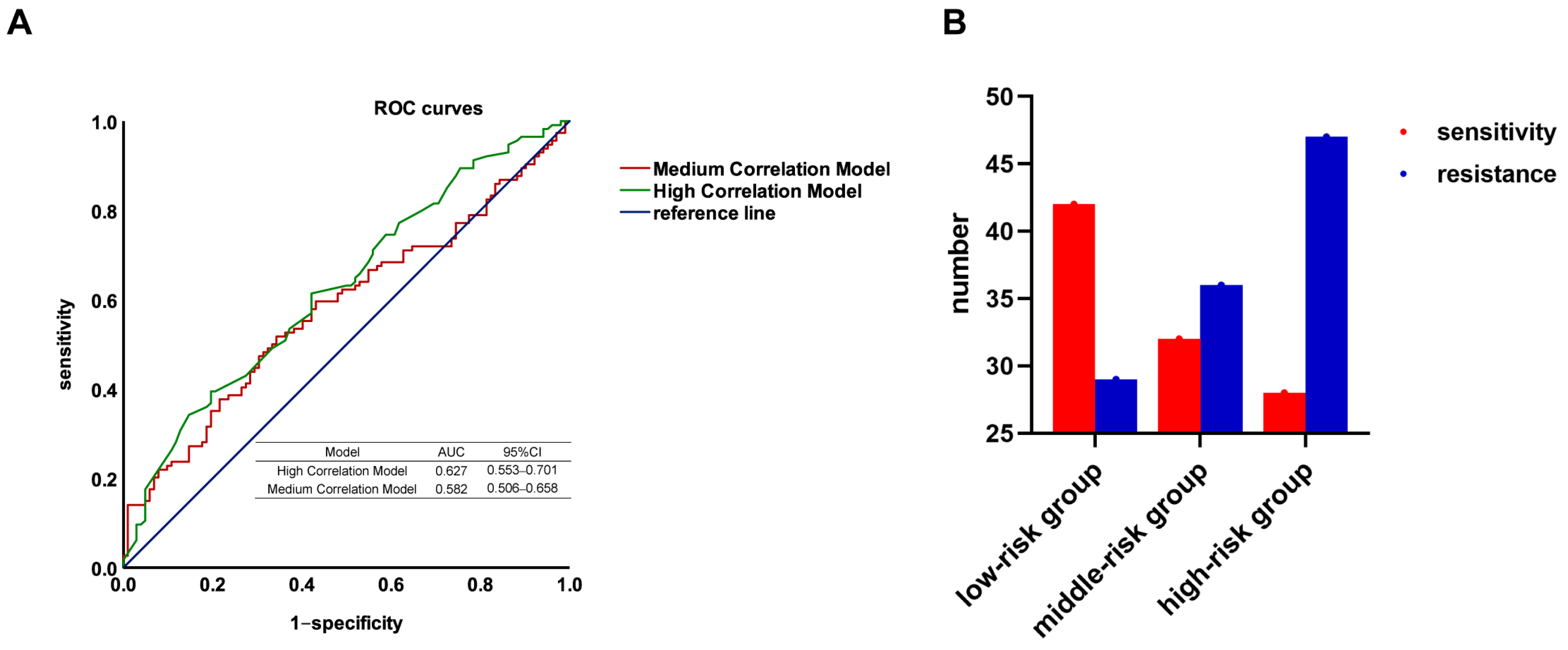
3.4. Validation of the Genetic Prediction Models
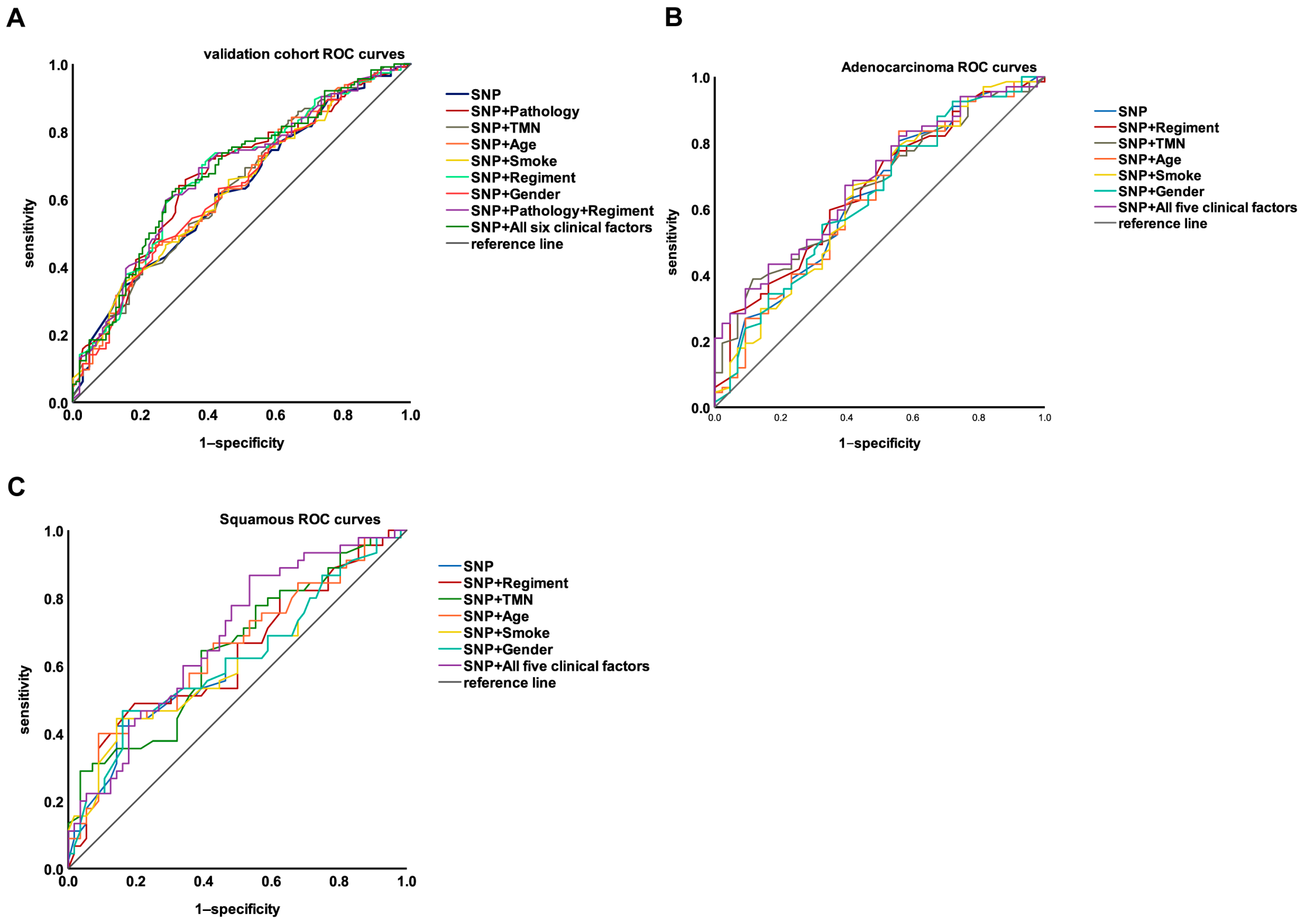
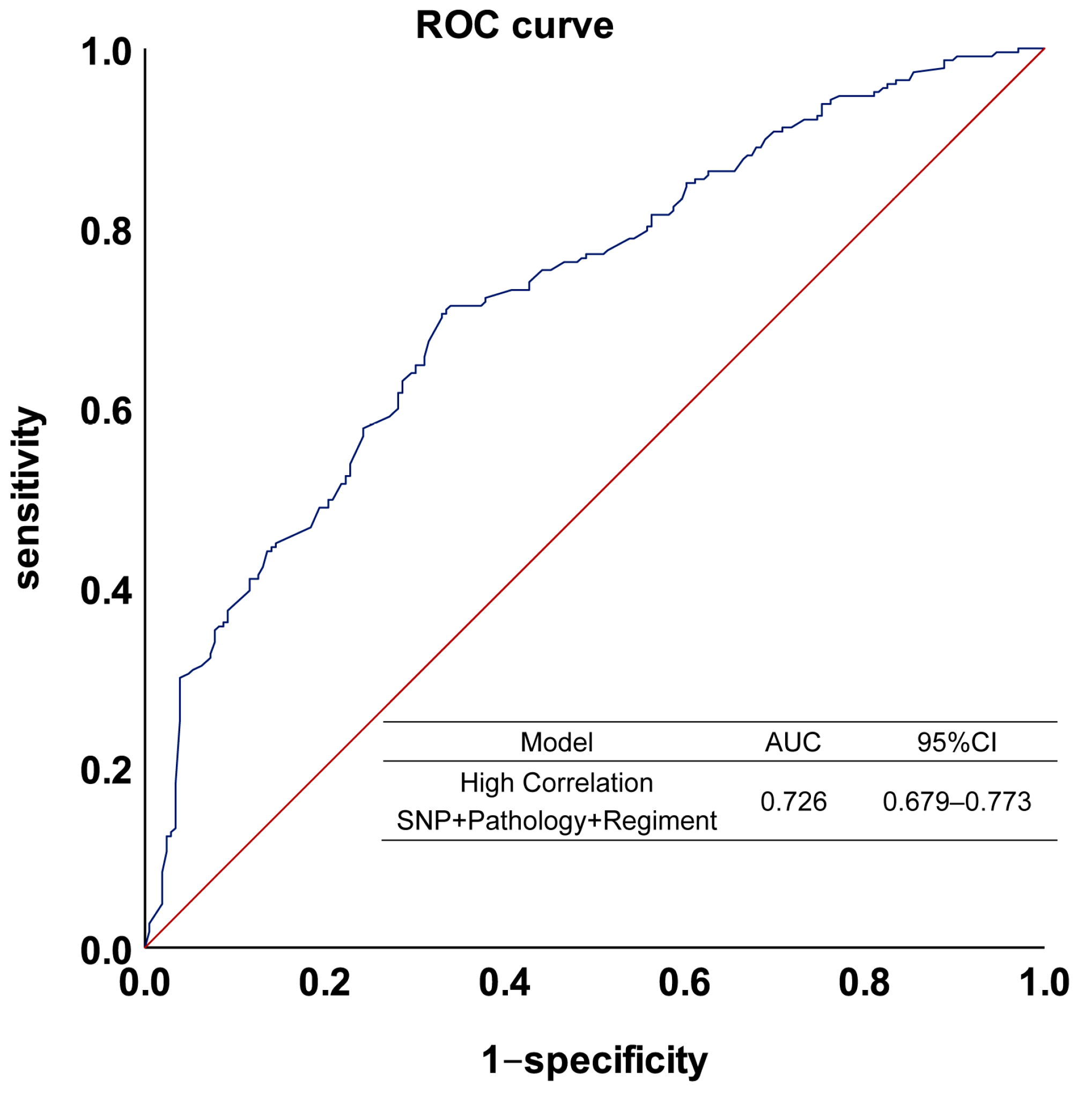
3.5. Integrating Genetic and Clinical Factors to Further Improve the Performance of the Model
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ciarimboli, G. Anticancer Platinum Drugs Update. Biomolecules 2021, 11, 1637. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Skowron, M.A.; Melnikova, M.; Van Roermund, J.G.H.; Romano, A.; Albers, P.; Thomale, J.; Schulz, W.A.; Niegisch, G.; Hoffmann, M.J. Multifaceted Mechanisms of Cisplatin Resistance in Long-Term Treated Urothelial Carcinoma Cell Lines. Int. J. Mol. Sci. 2018, 19, 590. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA Repair Pathways and Cisplatin Resistance: An Intimate Relationship. Clinics 2018, 73 (Suppl. 1), e478s. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Coinu, A.; Cabiddu, M.; Ghilardi, M.; Ardine, M.; Barni, S. Platinum rechallenge in patients with advanced NSCLC: A pooled analysis. Lung Cancer 2013, 81, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.-Y.; Li, X.; Li, X.-P.; Xiao, L.; Zheng, W.; Chen, J.; Mao, C.-X.; Fang, C.; Cui, J.-J.; Guo, C.-X.; et al. Prediction models for platinum-based chemotherapy response and toxicity in advanced NSCLC patients. Cancer Lett. 2016, 377, 65–73. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised Recist Guideline (Version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Euesden, J.; Lewis, C.M.; O’Reilly, P.F. PRSice: Polygenic Risk Score software. Bioinformatics 2014, 31, 1466–1468. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using Weka. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef]
- Ferrat, L.A.; Vehik, K.; Sharp, S.A.; Lernmark, A.; Rewers, M.J.; She, J.X.; Ziegler, A.G.; Toppari, J.; Akolkar, B.; Krischer, J.P.; et al. A Combined Risk Score Enhances Prediction of Type 1 Diabetes among Susceptible Children. Nat. Med. 2020, 26, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, D.; Liu, J.; Li, M.; Zhang, B.; Liu, Y.; Wang, Z.; Wen, S.; Zhou, J. A Prediction Model of Essential Hypertension Based on Genetic and Environmental Risk Factors in Northern Han Chinese. Int. J. Med. Sci. 2019, 16, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Docherty, A.R.; Shabalin, A.A.; DiBlasi, E.; Monson, E.; Mullins, N.; Adkins, D.E.; Bacanu, S.-A.; Bakian, A.V.; Crowell, S.; Chen, D.; et al. Genome-Wide Association Study of Suicide Death and Polygenic Prediction of Clinical Antecedents. Am. J. Psychiatry 2020, 177, 917–927. [Google Scholar] [CrossRef]
- Ye, F.; Zhang, S.-F.; Xie, X.; Lu, W.-G. OPCML Gene Promoter Methylation and Gene Expression in Tumor and Stroma Cells of Invasive Cervical Carcinoma. Cancer Investig. 2008, 26, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Anglim, P.P.; Galler, J.S.; Koss, M.N.; Hagen, J.A.; Turla, S.; Campan, M.; Weisenberger, D.J.; Laird, P.W.; Siegmund, K.D.; Laird-Offringa, I.A. Identification of a panel of sensitive and specific DNA methylation markers for squamous cell lung cancer. Mol. Cancer 2008, 7, 62. [Google Scholar] [CrossRef]
- Reed, J.E.; Dunn, J.R.; Du Plessis, D.G.; Shaw, E.J.; Reeves, P.; Gee, A.L.; Warnke, P.C.; Sellar, G.C.; Moss, D.J.; Walker, C. Expression of cellular adhesion molecule ‘OPCML’ is down-regulated in gliomas and other brain tumours. Neuropathol. Appl. Neurobiol. 2007, 33, 77–85. [Google Scholar] [CrossRef]
- Mei, F.C.; Young, T.W.; Liu, J.; Cheng, X. RAS-Mediated epigenetic inactivation of OPCML in oncogenic transformation of human ovarian surface epithelial cells. FASEB J. 2005, 20, 497–499. [Google Scholar] [CrossRef]
- Duarte-Pereira, S.; Paiva, F.; Costa, V.L.; Ramalho-Carvalho, J.; Savva-Bordalo, J.; Rodrigues, A.; Ribeiro, F.R.; Silva, V.M.; Oliveira, J.; Henrique, R.; et al. Prognostic value of opioid binding protein/cell adhesion molecule-like promoter methylation in bladder carcinoma. Eur. J. Cancer 2011, 47, 1106–1114. [Google Scholar] [CrossRef]
- Wu, Y.; Davison, J.; Qu, X.; Morrissey, C.; Storer, B.; Brown, L.; Vessella, R.; Nelson, P.; Fang, M. Methylation profiling identified novel differentially methylated markers including OPCML and FLRT2 in prostate cancer. Epigenetics 2016, 11, 247–258. [Google Scholar] [CrossRef]
- Xing, X.; Cai, W.; Ma, S.; Wang, Y.; Shi, H.; Li, M.; Jiao, J.; Yang, Y.; Liu, L.; Zhang, X.; et al. Down-regulated expression of OPCML predicts an unfavorable prognosis and promotes disease progression in human gastric cancer. BMC Cancer 2017, 17, 268. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Ying, Y.; van Hasselt, A.; Ng, K.M.; Yu, J.; Zhang, Q.; Jin, J.; Liu, D.; Rhim, J.S.; Rha, S.Y.; et al. Opcml Is a Broad Tumor Suppressor for Multiple Carcinomas and Lymphomas with Frequently Epigenetic Inactivation. PLoS ONE 2008, 3, e2990. [Google Scholar] [CrossRef]
- Gu, F.; Ma, Y.; Zhang, J.; Qin, F.; Fu, L. Function of Slit/Robo signaling in breast cancer. Front. Med. 2015, 9, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-Cadherin Is Required for Metastasis in Multiple Models of Breast Cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Lv, J.; Zhu, M.; Wang, Y.; Qin, N.; Ma, H.; He, Y.-Q.; Zhang, R.; Tan, W.; Fan, J.; et al. Identification of risk loci and a polygenic risk score for lung cancer: A large-scale prospective cohort study in Chinese populations. Lancet Respir. Med. 2019, 7, 881–891. [Google Scholar] [CrossRef]
- Gettler, K.; Levantovsky, R.; Moscati, A.; Giri, M.; Wu, Y.; Hsu, N.Y.; Chuang, L.S.; Sazonovs, A.; Venkateswaran, S.; Korie, U.; et al. Common and Rare Variant Prediction and Penetrance of Ibd in a Large, Multi-Ethnic, Health System-Based Biobank Cohort. Gastroenterology 2021, 160, 1546–1557. [Google Scholar] [CrossRef]
- Liyanarachchi, S.; Gudmundsson, J.; Ferkingstad, E.; He, H.; Jonasson, J.G.; Tragante, V.; Asselbergs, F.W.; Xu, L.; Kiemeney, L.A.; Netea-Maier, R.T.; et al. Assessing thyroid cancer risk using polygenic risk scores. Proc. Natl. Acad. Sci. USA 2020, 117, 5997–6002. [Google Scholar] [CrossRef]
- Klein, R.J.; Vertosick, E.; Sjoberg, D.; Ulmert, D.; Rönn, A.-C.; Häggström, C.; Thysell, E.; Hallmans, G.; Dahlin, A.; Stattin, P.; et al. Prostate cancer polygenic risk score and prediction of lethal prostate cancer. NPJ Precis. Oncol. 2022, 6, 1–8. [Google Scholar] [CrossRef]
- Thomas, M.; Sakoda, L.C.; Hoffmeister, M.; Rosenthal, E.A.; Lee, J.K.; van Duijnhoven, F.J.; Platz, E.A.; Wu, A.H.; Dampier, C.H.; de la Chapelle, A.; et al. Genome-wide Modeling of Polygenic Risk Score in Colorectal Cancer Risk. Am. J. Hum. Genet. 2020, 107, 432–444. [Google Scholar] [CrossRef]
- Mars, N.; Gen, F.; Koskela, J.T.; Ripatti, P.; Kiiskinen, T.T.J.; Havulinna, A.S.; Lindbohm, J.V.; Ahola-Olli, A.; Kurki, M.; Karjalainen, J.; et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat. Med. 2020, 26, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.E.; Han, X.; Qassim, A.; Hassall, M.; Bailey, J.N.C.; Kinzy, T.G.; Khawaja, A.P.; An, J.; Marshall, H.; Gharahkhani, P.; et al. Multitrait Analysis of Glaucoma Identifies New Risk Loci and Enables Polygenic Prediction of Disease Susceptibility and Progression. Nat. Genet. 2020, 52, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Chen, J.; Zou, T.; Zhou, Y.; Liu, J.; Li, X.; Li, X.; Li, M.; Pan, P.; Zhuo, W.; et al. Genome-wide analysis identify novel germline genetic variations in ADCY1 influencing platinum-based chemotherapy response in non-small cell lung cancer. Acta Pharm. Sin. B 2022, 12, 1514–1522. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Wang, C.-J.; Xiao, D.-S.; He, B.-M.; Li, M.; Yi, X.-P.; Zhang, W.; Yin, J.-Y.; Liu, Z.-Q. eIF3a R803K mutation mediates chemotherapy resistance by inducing cellular senescence in small cell lung cancer. Pharmacol. Res. 2021, 174, 105934. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Moily, N.S.; Kaur, H.; Jajodia, A.; Jain, S.; Kukreti, R. Identifying a predictive model for response to atypical antipsychotic monotherapy treatment in south Indian schizophrenia patients. Genomics 2013, 102, 131–135. [Google Scholar] [CrossRef]
- Drobin, K.; Marczyk, M.; Halle, M.; Danielsson, D.; Papiez, A.; Sangsuwan, T.; Bendes, A.; Hong, M.G.; Qundos, U.; Harms-Ringdahl, M.; et al. Molecular Profiling for Predictors of Radiosensitivity in Patients with Breast or Head-and-Neck Cancer. Cancers 2020, 12, 753. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Discovery Stage | Validation Stage | ||||
|---|---|---|---|---|---|---|
| Responders (%) (n = 104) | Non-Responders (%) (n = 113) | p Value | Responders (%) (n = 102) | Non-Responders (%) (n = 114) | p Value | |
| Histology | ||||||
| Adenocarcinoma | 51 (49.04) | 56 (49.56) | 0.74 | 43 (42.16) | 67 (58.77) | 0.05 |
| Squamous | 51 (49.04) | 54 (47.79) | 56 (54.90) | 45 (39.47) | ||
| Other | 2 (1.92) | 3 (2.65) | 3 (2.94) | 2 (1.76) | ||
| Stage | ||||||
| I | NA | 2 (1.77) | 0.72 | 1 (0.98) | NA | 0.40 |
| II | 1 (0.96) | 1 (0.88) | 3 (2.94) | 1 (0.88) | ||
| III | 32 (30.77) | 33 (29.20) | 36 (35.29) | 36 (31.58) | ||
| IV | 71 (68.27) | 77 (68.15) | 62 (60.79) | 77 (67.54) | ||
| age | ||||||
| ≤55 | 44 (42.31) | 50 (44.25) | 0.22 | 43 (42.16) | 52 (45.61) | 0.16 |
| >55 | 60 (57.69) | 63 (55.75) | 59 (57.84) | 62 (54.39) | ||
| Smoking status | ||||||
| Nonsmoker | 38 (36.54) | 36 (31.86) | 0.66 | 27 (26.47) | 40 (35.09) | 0.17 |
| Smoker | 66 (63.46) | 77 (68.14) | 75 (73.53) | 74 (64.91) | ||
| Sex | ||||||
| Male | 76 (73.08) | 94 (83.19) | 0.41 | 84 (82.35) | 89 (78.07) | 0.43 |
| Female | 28 (26.92) | 19 (16.81) | 18 (17.65) | 25 (21.93) | ||
| Chemotherapeutic regimens | ||||||
| platinum/pemetrexed | 42 (40.39) | 50 (44.25) | 0.84 | 38 (37.25) | 61 (53.51) | 0.11 |
| platinum/gemcitabine | 40 (38.46) | 44 (38.94) | 38 (37.25) | 34 (29.82) | ||
| platinum/paclitaxel | 15 (14.42) | 12 (10.62) | 18 (17.65) | 14 (12.28) | ||
| platinum/docetaxel | 7 (6.73) | 7 (6.19) | 8 (7.85) | 5 (4.39) | ||
| SNP | CHR | OR | p | Gene | Mutation Type |
|---|---|---|---|---|---|
| rs7463048 | 8 | 2.45 | 4.60 × 10−5 | LOC105375676 | Intron |
| rs17176196 | 15 | 0.34 | 6.16 × 10−5 | ANKRD34C-AS1 | Intron |
| rs527646 | 11 | 0.34 | 6.96 × 10−5 | OPCML | Intron |
| rs11134542 | 5 | 2.44 | 9.89 × 10−5 | SLIT3, LOC105377713 | Intron, Non-Coding Transcription |
| rs10078147 | 5 | 2.15 | 1.32 × 10−4 | SLC1A3 | Intron |
| rs9378820 | 6 | 2.32 | 1.39 × 10−4 | NA | NA |
| rs7113019 | 11 | 2.40 | 1.52 × 10−4 | JHY | Intron |
| rs7005628 | 8 | 0.31 | 1.76 × 10−4 | NA | NA |
| rs11617804 | 13 | 3.68 | 1.77 × 10−4 | FAM155A | Intron |
| rs7005216 | 8 | 0.32 | 1.79 × 10−4 | NA | NA |
| rs944927 | 20 | 0.42 | 1.83 × 10−4 | LOC105372656 | Intron |
| rs11808688 | 1 | 0.44 | 1.91 × 10−4 | ATP2B4 | Intron |
| rs17121520 | 14 | 0.12 | 2.02 × 10−4 | NA | NA |
| rs1620779 | 11 | 2.23 | 2.33 × 10−4 | LINC02698 | Intron |
| rs60976228 | 6 | 0.24 | 2.37 × 10−4 | FANCE | Intron |
| rs9496862 | 6 | 0.32 | 2.67 × 10−4 | LOC105378036 | 2 KB Upstream |
| rs7225086 | 17 | 2.21 | 2.89 × 10−4 | RTN4RL1 | Intron |
| rs10859720 | 12 | 2.63 | 3.97 × 10−4 | LOC102724960 | Intron |
| rs949561 | 3 | 2.37 | 3.99 × 10−4 | LOC107986169 | Intron |
| rs12545542 | 8 | 0.47 | 4.78 × 10−4 | LOC100128993 | Intron |
| rs150131032 | 1 | 0.45 | 4.88 × 10−4 | LINC01344 | Intron |
| rs4146476 | 4 | 0.49 | 5.31 × 10−4 | NA | NA |
| rs76581411 | 14 | 0.22 | 5.69 × 10−4 | NA | NA |
| rs7313678 | 12 | 2.12 | 5.79 × 10−4 | NA | NA |
| rs2060515 | 4 | 2.26 | 6.31 × 10−4 | NA | NA |
| rs7134969 | 12 | 2.16 | 6.31 × 10−4 | LOC105370003 | Intron |
| rs1601345 | 14 | 0.48 | 6.54 × 10−4 | NA | NA |
| rs3762678 | 3 | 0.22 | 6.56 × 10−4 | ACAD11, NPHP3-ACAD11 | Intron |
| rs6873965 | 5 | 2.15 | 6.76 × 10−4 | NA | NA |
| rs9832471 | 3 | 0.27 | 6.78 × 10−4 | NA | NA |
| rs1080178 | 2 | 0.47 | 6.94 × 10−4 | UPP2 | Intron |
| rs4244459 | 8 | 1.92 | 7.06 × 10−4 | LOC105379311 | Intron |
| rs77176301 | 3 | 0.48 | 7.07 × 10−4 | NA | NA |
| rs12962513 | 18 | 2.35 | 7.18 × 10−4 | NA | NA |
| exm2267842 | 15 | 0.46 | 7.46 × 10−4 | ANP32A, ANP32A-IT1 | Intron, 2KB Upstream |
| rs4520608 | 11 | 2.14 | 7.51 × 10−4 | OPCML | Intron |
| rs77859697 | 12 | 0.30 | 8.01 × 10−4 | LINC02388, LOC100506869 | Intron |
| rs1523483 | 3 | 0.49 | 8.02 × 10−4 | NA | NA |
| rs145303018 | 9 | 0.35 | 8.10 × 10−4 | NA | NA |
| rs149864625 | 10 | 0.21 | 8.12 × 10−4 | LRMDA | Intron |
| rs1425351 | 4 | 2.91 | 8.21 × 10−4 | COL25A1 | Intron |
| rs74531987 | 22 | 2.51 | 8.26 × 10−4 | NA | NA |
| rs201395 | 12 | 0.47 | 8.45 × 10−4 | NA | NA |
| rs4518067 | 3 | 0.46 | 8.47 × 10−4 | NA | NA |
| rs1472094 | 4 | 0.18 | 8.56 × 10−4 | LOC105374528 | Intron |
| rs56845228 | 7 | 0.27 | 8.70 × 10−4 | LOC105375523 | Intron |
| rs11957972 | 5 | 0.15 | 8.99 × 10−4 | NA | NA |
| rs7019568 | 9 | 0.31 | 9.07 × 10−4 | NA | NA |
| rs7864626 | 9 | 0.48 | 9.08 × 10−4 | GNA14, GNA14-AS1 | Intron |
| rs244046 | 4 | 2.15 | 9.15 × 10−4 | NA | NA |
| rs139740488 | 5 | 2.08 | 9.18 × 10−4 | NA | NA |
| rs76443044 | 20 | 0.22 | 9.37 × 10−4 | CDH4 | Intron |
| rs2050346 | 10 | 0.35 | 9.48 × 10−4 | LOC105376391 | Intron |
| rs1860139 | 14 | 0.40 | 9.48 × 10−4 | LINC02274 | Non-Coding Transcription |
| rs2324596 | 13 | 0.49 | 9.71 × 10−4 | NA | NA |
| rs9285510 | 6 | 3.03 | 9.93 × 10−4 | STXBP5-AS1 | Intron Variant |
| rs28517685 | 19 | 3.01 | 9.97 × 10−4 | LOC105372349, LOC100420587 | Non-Coding Transcription, Intron |
| (A) | ||||
| Methods | Sensitivity | Specificity | Accuracy | AUC |
| PRS | 0.97 | 0.97 | 0.97 | 0.995 |
| BayesNet | 0.95 | 0.93 | 0.94 | 0.988 |
| NaiveBayes | 0.96 | 0.92 | 0.94 | 0.988 |
| RF | 0.93 | 0.82 | 0.88 | 0.956 |
| Logistic | 0.88 | 0.87 | 0.87 | 0.946 |
| SVM | 0.93 | 0.86 | 0.89 | 0.893 |
| KNN (IBK) | 0.91 | 0.55 | 0.74 | 0.750 |
| Bagging | 0.82 | 0.71 | 0.77 | 0.859 |
| Random Tree | 0.66 | 0.60 | 0.63 | 0.612 |
| DT | 0.80 | 0.32 | 0.57 | 0.551 |
| AdaBoost | 0.71 | 0.46 | 0.59 | 0.673 |
| (B) | ||||
| Methods | Sensitivity | Specificity | Accuracy | AUC |
| PRS | 0.73 | 0.73 | 0.73 | 0.795 |
| BayesNet | 0.76 | 0.62 | 0.69 | 0.773 |
| NaiveBayes | 0.79 | 0.61 | 0.70 | 0.776 |
| RF | 0.66 | 0.67 | 0.69 | 0.712 |
| Logistic | 0.72 | 0.66 | 0.69 | 0.761 |
| SVM | 0.75 | 0.65 | 0.71 | 0.702 |
| KNN (IBK) | 0.68 | 0.65 | 0.67 | 0.715 |
| Bagging | 0.68 | 0.65 | 0.67 | 0.724 |
| Random Tree | 0.65 | 0.67 | 0.66 | 0.682 |
| DT | 0.73 | 0.39 | 0.57 | 0.525 |
| AB | 0.72 | 0.63 | 0.67 | 0.756 |
| (C) | ||||
| Methods | Sensitivity | Specificity | Accuracy | AUC |
| PRS | 0.74 | 0.54 | 0.64 | 0.593 |
| BayesNet | 0.52 | 0.34 | 0.43 | 0.406 |
| NaiveBayes | 0.52 | 0.32 | 0.42 | 0.402 |
| RF | 0.50 | 0.45 | 0.48 | 0.490 |
| Logistic | 0.54 | 0.30 | 0.42 | 0.397 |
| SVM | 0.45 | 0.29 | 0.37 | 0.370 |
| KNN (IBK) | 0.45 | 0.50 | 0.47 | 0.485 |
| Bagging | 0.52 | 0.42 | 0.47 | 0.472 |
| Random Tree | 0.46 | 0.46 | 0.46 | 0.492 |
| DT | 0.70 | 0.13 | 0.43 | 0.380 |
| AB | 0.65 | 0.20 | 0.44 | 0.419 |
| (A) | ||
| Model | AUC | 95% CI |
| SNP | 0.627 | 0.553–0.701 |
| SNP + Regimen | 0.673 | 0.602–0.745 |
| SNP + Pathology | 0.670 | 0.597–0.742 |
| SNP + TMN | 0.635 | 0.562–0.709 |
| SNP + Age | 0.637 | 0.563–0.710 |
| SNP + Smoke | 0.633 | 0.559–0.706 |
| SNP + Gender | 0.633 | 0.559–0.707 |
| SNP + Pathology + Regimen | 0.675 | 0.603–0.746 |
| SNP + Regimen + Pathology + TMN + Age + Smoke + Gender | 0.679 | 0.608–0.750 |
| (B) | ||
| Model | AUC | 95% CI |
| SNP | 0.638 | 0.531–0.746 |
| SNP + Regimen | 0.661 | 0.558–0.764 |
| SNP + TMN | 0.659 | 0.557–0.761 |
| SNP + Age | 0.634 | 0.526–0.741 |
| SNP + Smoke | 0.64 | 0.532–0.747 |
| SNP + Gender | 0.635 | 0.527–0.742 |
| SNP + Regimen + TMN + Age + Smoke + Gender | 0.685 | 0.585–0.784 |
| (C) | ||
| Model | AUC | 95% CI |
| SNP | 0.612 | 0.499–0.724 |
| SNP + Regimen | 0.632 | 0.521–0.743 |
| SNP + TMN | 0.646 | 0.537–0.754 |
| SNP + Age | 0.648 | 0.538–0.757 |
| SNP + Smoke | 0.614 | 0.501–0.727 |
| SNP + Gender | 0.617 | 0.505–0.729 |
| SNP + Regimen + TMN + Age + Smoke + Gender | 0.681 | 0.577–0.785 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Q.; Mao, C.; Gao, Y.; Huang, H.; Yu, B.; Yu, L.; Li, X.; Mao, X.; Zhang, W.; Yin, J.; et al. Establishing a Prediction Model for the Efficacy of Platinum—Based Chemotherapy in NSCLC Based on a Two Cohorts GWAS Study. J. Clin. Med. 2023, 12, 1318. https://doi.org/10.3390/jcm12041318
Xiao Q, Mao C, Gao Y, Huang H, Yu B, Yu L, Li X, Mao X, Zhang W, Yin J, et al. Establishing a Prediction Model for the Efficacy of Platinum—Based Chemotherapy in NSCLC Based on a Two Cohorts GWAS Study. Journal of Clinical Medicine. 2023; 12(4):1318. https://doi.org/10.3390/jcm12041318
Chicago/Turabian StyleXiao, Qi, Chenxue Mao, Ying Gao, Hanxue Huang, Bing Yu, Lulu Yu, Xi Li, Xiaoyuan Mao, Wei Zhang, Jiye Yin, and et al. 2023. "Establishing a Prediction Model for the Efficacy of Platinum—Based Chemotherapy in NSCLC Based on a Two Cohorts GWAS Study" Journal of Clinical Medicine 12, no. 4: 1318. https://doi.org/10.3390/jcm12041318