
Recessive SERPING1 Variant Leads to Kinin–Kallikrein System Control Failure in a Consanguineous Brazilian Family with Hereditary Angioedema
 , , , and
, , , and
Abstract
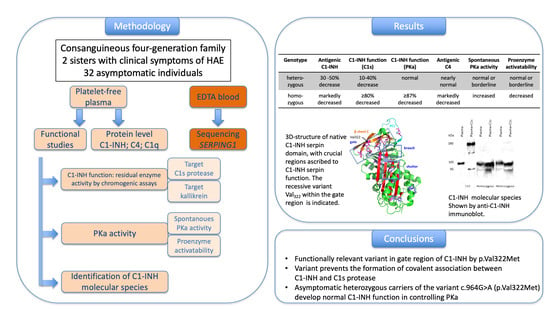
:
1. Introduction
2. Methods
2.1. Subjects
2.2. Severity Score
2.3. Levels of C1-INH, C4 and C1q
2.4. Functional Studies
2.5. Identification of C1-INH Species
2.6. Genetic Studies
2.7. Ethical Considerations
3. Results
3.1. Severity of Disease
3.2. Biochemical Studies
3.3. Studies of C1-INH Molecular Species
3.4. Genetic Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maurer, M.; Magerl, M.; Ansotegui, I.; Aygören-Pürsün, E.; Betschel, S.; Bork, K.; Bowen, T.; Boysen, H.B.; Farkas, H.; Grumach, A.S.; et al. The international WAO/EAACI guideline for the management of hereditary angioedema—The 2017 revision and update. World Allergy Organ. J. 2018, 11, 5. [Google Scholar] [CrossRef]
- Cicardi, M.; Aberer, W.; Banerji, A.; Bas, M.; Bernstein, J.A.; Bork, K.; Caballero, T.; Farkas, H.; Grumach, A.; Kaplan, A.P.; et al. Classification, diagnosis, and approach to treatment for angioedema: Consensus report from the Hereditary Angioedema International Working Group. Allergy 2014, 69, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Proper, S.P.; Lavery, W.J.; Bernstein, J.A. Definition and classification of hereditary angioedema. Allergy Asthma Proc. 2020, 41 (Suppl. 1), S3–S7. [Google Scholar] [CrossRef] [PubMed]
- Zeerleder, S. C1-inhibitor: More than a serine protease inhibitor. Semin. Thromb. Hemost. 2011, 37, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Busse, P.J.; Christiansen, S.C. Hereditary Angioedema. N. Engl. J. Med. 2020, 382, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Rosen, F.S.; Pensky, J.; Donaldson, V.; Charache, P. Hereditary angioedema: Two Genetic Variants. Science 1965, 148, 957–958. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, A.; Defendi, F.; Charignon, D.; Csopaki, F.; Favier, B.; Habib, M.; Cichon, S.; Drouet, C. Contact system activation in patients with HAE and normal C1 inhibitor function. Immunol. Allergy Clin. N. Am. 2013, 33, 513–533. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, A.; Sellier, P.; Defendi, F.; Favier, B.; Charignon, D.; Lopez-Lera, A.; Lopez-Trascasa, M.; Ponard, D.; Drouet, C. C1 inhibitor function using contact-phase proteases as target: Evaluation of an innovative assay. Allergy 2015, 70, 1103–1111. [Google Scholar] [CrossRef]
- Larrauri, B.; Hester, C.G.; Jiang, H.; Miletic, V.D.; Malbran, A.; Bork, K.; Kaplan, A.; Frank, M. Analysis of cold activation of the contact system in hereditary angioedema with normal C1 inhibitor. Mol. Immunol. 2021, 136, 150–160. [Google Scholar] [CrossRef]
- Ponard, D.; Gaboriaud, C.; Charignon, D.; Ghannam, A.; Wagenaar-Bos, I.G.A.; Roem, D.; López-Lera, A.; López-Trascasa, M.; Tosi, M.; Drouet, C. SERPING1 mutation update: Mutation spectrum and C1 inhibitor phenotypes. Hum. Mutat. 2020, 41, 38–57. [Google Scholar] [CrossRef]
- Cicardi, M.; Zanichelli, A. Diagnosing angioedema. Immunol. Allergy Clin. N. Am. 2013, 33, 449–456. [Google Scholar] [CrossRef]
- Maurer, M.; Magerl, M.; Betschel, S.; Aberer, W.; Ansotegui, I.J.; Aygören-Pürsün, E.; Banerji, A.; Bara, N.-A.; Boccon-Gibod, I.; Bork, K.B.; et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy 2022, 77, 1961–1990. [Google Scholar] [CrossRef]
- Jindal, N.L.; Harniman, E.; Prior, N.; Perez-Fernandez, E.; Caballero, T.; Betschel, S. Hereditary angioedema: Health-related quality of life in Canadian patients as measured by the SF-36. Allergy Asthma Clin. Immunol. 2017, 13, 4. [Google Scholar] [CrossRef]
- Gobert, D.; Paule, R.; Ponard, D.; Levy, P.; Fremeaux-Bacchi, V.; Bouillet, L.; Boccon-Gibod, I.; Drouet, C.; Gayet, S.; Launay, D.; et al. A nationwide study of acquired C1-inhibitor deficiency in France: Characteristics and treatment responses in 92 patients. Medicine 2016, 95, e4363. [Google Scholar] [CrossRef]
- Agostoni, A.; Aygören-Pürsün, E.; Binkley, K.E.; Blanch, A.; Bork, K.; Bouillet, L.; Bucher, C.; Castaldo, A.J.; Cicardi, M.; Davis, A.E.; et al. Hereditary and acquired angioedema: Problems and progress: Proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J. Allergy Clin. Immunol. 2004, 114 (Suppl. 3), S51–S131. [Google Scholar] [CrossRef] [PubMed]
- Banday, A.Z.; Kaur, A.; Jindal, A.K.; Rawat, A.; Singh, S. An update on the genetics and pathogenesis of hereditary angioedema. Genes Dis. 2020, 7, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.E.; Hansen, S.; Gram, J.; Bygum, A.; Drouet, C.; Sidelmann, J.J. Presence of C1-inhibitor polymers in a subset of patients suffering from hereditary angioedema. PLoS ONE 2014, 9, e112051. [Google Scholar] [CrossRef] [PubMed]
- Potempa, J.; Korzus, E.; Travis, J. The serpin superfamily of proteinase inhibitors: Structure, function, and regulation. J. Biol. Chem. 1994, 269, 15957–15960. [Google Scholar] [CrossRef] [PubMed]
- Silverman, G.A.; Bird, P.I.; Carrell, R.W.; Church, F.C.; Coughlin, P.B.; Gettins, P.G.; Irving, J.A.; Lomas, D.A.; Luke, C.J.; Moyer, R.W.; et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 2001, 276, 33293–33296. [Google Scholar] [CrossRef] [PubMed]
- Dijk, M.; Holkers, J.; Voskamp, P.; Giannetti, B.M.; Waterreus, W.J.; van Veen, H.A.; Pannu, N.S. How Dextran Sulfate Affects C1-inhibitor Activity: A Model for Polysaccharide Potentiation. Structure 2016, 24, 2182–2189. [Google Scholar] [CrossRef]
- Stein, P.E.; Carrell, R.W. What do dysfunctional serpins tell us about molecular mobility and disease? Nat. Struct. Biol. 1995, 2, 96–113. [Google Scholar] [CrossRef] [PubMed]
- Drouet, C.; Lopez-Lera, A.; Ghannam, A.; Lopez-Trascasa, M.; Cichon, S.; Ponard, D.; Parsopoulou, F.; Grombirikova, H.; Freiberger, T.; Rijavec, M.; et al. SERPING1 variants and C1-INH biological function: A close relationship with C1-INH-HAE. Front. Allergy 2022, 3, 835503. [Google Scholar] [CrossRef] [PubMed]
- Haslund, D.; Ryø, L.B.; Seidelin Majidi, S.; Rose, I.; Skipper, K.A.; Fryland, T.; Bohn, A.B.; Koch, C.; Thomsen, M.K.; Palarasah, Y.; et al. Dominant-negative SERPING1 variants cause intracellular retention of C1 inhibitor in hereditary angioedema. J. Clin. Investig. 2019, 129, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Blanch, A.; Roche, O.; Urrutia, I.; Gamboa, P.; Fontán, G.; López-Trascasa, M. First case of homozygous C1 inhibitor deficiency. J. Allergy Clin. Immunol. 2006, 118, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Mete Gökmen, N.; Gülbahar, O.; Onay, H.; Peker Koc, Z.; Özgül, S.; Köse, T.; Gelincik, A.; Büyüköztürk, S.; Sin, A.Z. Deletions in SERPING1 lead to lower C1 Inhibitor function: Lower C1 inhibitor function can predict disease severity. Int. Arch. Allergy Immunol. 2019, 178, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Mete Gökmen, N.; Rodriguez-Alcalde, C.; Gülbahar, O.; López-Trascasa, M.; Onay, H.; López-Lera, A. Novel homozygous variants in the SERPING1 gene in two Turkish families with hereditary angioedema of recessive inheritance. Immunol. Cell Biol. 2020, 98, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Nabilou, S.; Pak, F.; Alizadeh, Z.; Fazlollahi, M.R.; Houshmand, M.; Ayazi, M.; Mohammadzadeh, I.; Bemanian, M.H.; Fayezi, A.; Nabavi, M.; et al. Genetic study of hereditary angioedema type I and type II (first report from Iranian patients: Describing three new mutations). Immunol. Investig. 2022, 51, 170–181. [Google Scholar] [CrossRef] [PubMed]
- López-Lera, A.; Favier, B.; de la Cruz, R.M.; Garrido, S.; Drouet, C.; López-Trascasa, M. A new case of homozygous C1-inhibitor deficiency suggests a role for Arg378 in the control of kinin pathway activation. J. Allergy Clin. Immunol. 2010, 126, 1307–1310.e3. [Google Scholar] [CrossRef] [PubMed]
- Büyüköztürk, S.; Eroglu, B.K.; Gelincik, A.; Uzümcü, A.; Özseker, F.; Colakoglu, B.; Dal, M.; Uyguner, Z.O. A Turkish family with a novel mutation in the promoter region of the C1 inhibitor gene. J. Allergy Clin. Immunol. 2009, 123, 962–964. [Google Scholar] [CrossRef]
- Kesim, B.; Uyguner, Z.O.; Gelincik, A.; Mete Gökmen, N.; Sin, A.Z.; Karakaya, G.; Erdenen, F.; Ardeniz, Ö.; Özşeker, F.; Gülbahar, O.; et al. The Turkish Hereditary Angioedema Pilot Study (TURHAPS): The first Turkish series of hereditary angioedema. Int. Arch. Allergy Immunol. 2011, 156, 443–450. [Google Scholar] [CrossRef]
- Ren, Z.; Zhao, S.; Li, T.; Wedner, H.J.; Atkinson, J.P. Insights into the pathogenesis of hereditary angioedema using genetic sequencing and recombinant protein expression analyses. J. Allergy Clin. Immunol. 2023, 151, 1040–1049e5. [Google Scholar] [CrossRef] [PubMed]
- Bygum, A.; Fagerberg, C.R.; Ponard, D.; Monnier, N.; Lunardi, J.; Drouet, C. Mutational spectrum and phenotypes in Danish families with hereditary angioedema because of C1 inhibitor deficiency. Allergy 2011, 66, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.F.; Moreno, A.S.; Castelli, E.C.; Donadi, E.A.; Palma, M.S.; Arcuri, H.A.; Lange, A.P.; Bork, K.; Sarti, W.; Arruda, L.K. A single nucleotide deletion at the C1 inhibitor gene as the cause of hereditary angioedema: Insights from a Brazilian family. Allergy 2011, 66, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Defendi, F.; Charignon, D.; Ghannam, A.; Baroso, R.; Csopaki, F.; Allegret-Cadet, M.; Ponard, D.; Favier, B.; Cichon, S.; Nicolie, B.; et al. Enzymatic assays for the diagnosis of bradykinin-dependent angioedema. PLoS ONE 2013, 8, e70140. [Google Scholar] [CrossRef]
- Maia, L.S.M.; Moreno, A.S.; Ferriani, M.P.L.; Nunes, F.L.; Ferraro, M.F.; Dias, M.M.; Roxo-Junior, P.; Dias, F.C.; Valle, S.O.R.; Levy, S.; et al. Genotype-phenotype correlations in Brazilian patients with hereditary angioedema due to C1 inhibitor deficiency. Allergy 2019, 74, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Caccia, S.; Suffritti, C.; Carzaniga, T.; Berardelli, R.; Berra, S.; Martorana, V.; Fra, A.; Drouet, C.; Cicardi, M. Intermittent C1-Inhibitor Deficiency Associated with Recessive Inheritance: Functional and Structural Insight. Sci. Rep. 2018, 8, 977. [Google Scholar] [CrossRef]
- Gosswein, T.; Kocot, A.; Emmert, G.; Kreuz, W.; Martinez-Saguer, I.; Aygoren-Pursun, E.; Rusicke, E.; Bork, K.; Oldenburg, J.; Müller, C. Mutational spectrum of the C1INH (SERPING1) gene in patients with hereditary angioedema. Cytogenet. Genome Res. 2008, 121, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, E.; Caccia, S.; Suffritti, C.; Tordai, A.; Zingale, L.C.; Cicardi, M. Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: Functional and structural correlates. Mol Immunol. 2008, 45, 3536–3544. [Google Scholar] [CrossRef]
- Hashimura, C.; Kiyohara, C.; Fukushi, J.I.; Hirose, T.; Ohsawa, I.; Tahira, T.; Horiuchi, T. Clinical and genetic features of hereditary angioedema with and without C1-inhibitor (C1-INH) deficiency in Japan. Allergy 2021, 76, 3529–3534. [Google Scholar] [CrossRef]
- Wagenaar-Bos, I.G.; Drouet, C.; Aygoren-Pursun, E.; Bork, K.; Bucher, C.; Bygum, A.; Farkas, H.; Fust, G.; Gregorek, H.; Hack, C.E.; et al. Functional C1-inhibitor diagnostics in hereditary angioedema: Assay evaluation and recommendations. J. Immunol. Methods 2008, 338, 14–20. [Google Scholar] [CrossRef]
- Charignon, D.; Ghannam, A.; Ponard, D.; Drouet, C. Hereditary C1 inhibitor deficiency is associated with high spontaneous amidase activity. Mol. Immunol. 2017, 85, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Germenis, A.E.; Margaglione, M.; Pesquero, J.B.; Farkas, H.; Cichon, S.; Csuka, D.; Lera, A.L.; Rijavec, M.; Jolles, S.; Szilagyi, A.; et al. International consensus on the use of genetics in the management of hereditary angioedema. J. Allergy Clin. Immunol. Pract. 2020, 8, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Drouet, C.; Desormeaux, A.; Robillard, J.; Ponard, D.; Bouillet, L.; Martin, L.; Kanny, G.; Moneret-Vautrin, D.-A.; Bosson, J.-L.; Quesada, J.-L.; et al. Metallopeptidase activities in hereditary angioedema: Effect of androgen prophylaxis on plasma aminopeptidase P. J. Allergy Clin. Immunol. 2008, 121, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Frazer, J.; Notin, P.; Dias, M.; Gomez, A.; Min, J.K.; Brock, K.; Gal, Y.; Marks, D.S. Disease variant prediction with deep generative models of evolutionary data. Nature 2021, 599, 91–95. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pedigree Position | Sex | Age | Genetics | Antigenic C1-INH (mg/L) * | C1-INH Function—C1s * | Antigenic C4 (mg/L) * | Antigenic C1q (mg/L) * | Kallikrein (PKa) Activity | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Spontaneous PKa Activity (nmol/mL/min) * | Proenzyme Activatability (nmol/mL/min) * | C1-INH Function—PKa (IU/mL) * | C1-INH Function—PKa (%) | ||||||||
| II.6 | F | 50 | Homozygous | 40↓ | 17%↓ | 64.4↓ | 191 | 124↑ | 1601↓ | <0.11↓ | <12.9↓ |
| II.5 | F | 52 | Homozygous | 95↓ | 16%↓ | 64.4↓ | 230 | 193.6↑ | 1769↓ | <0.11↓ | <12.9↓ |
| III.6 | M | 26 | Heterozygous | 181↓ | 32%↓ | 161↓ | Not assessed | 56.2↑ | 2203 | 0.36↓ | 42 |
| I.3 | F | 75 | Heterozygous | 169↓ | 85% | 493 | Not assessed | 42↑ | 1997 | 0.76 | 102 |
| III.7 | F | 26 | Heterozygous | 164↓ | 45%↓ | 171 | Not assessed | 20.5↑ | 1842↓ | 0.45 | 61 |
| III.8 | F | 21 | Heterozygous | 164↓ | 23%↓ | 64↓ | Not assessed | 12.6↑ | 1648↓ | 0.29↓ | 39 |
| II.4 | F | 54 | Heterozygous | 128↓ | 41%↓ | 279 | Not assessed | 12.6↑ | 1830↓ | 0.41↓ | 55 |
| III.11 | M | 28 | Heterozygous | 189↓ | 29%↓ | 242 | Not assessed | 12.2↑ | 1857 | 0.59↓ | 69 |
| III.10 | F | 31 | Heterozygous | 128↓ | 10%↓ | 161↓ | Not assessed | 10.6 | 2101 | 0.39↓ | 52 |
| II.1 | F | 42 | Heterozygous | 186↓ | 29%↓ | 346 | Not assessed | 8.5 | 1889 | 0.61 | 109 |
| II.8 | F | 46 | Wild type | 265 | Not assessed | 632 | Not assessed | 7.2 | 1936 | 1.36 | 183 |
| III.5 | M | 25 | Wild type | 164 | Not assessed | 306 | Not assessed | 6.6 | 1872 | 1.49 | 175 |
| II.7 | M | 48 | Wild type | 450 | Not assessed | 217 | Not assessed | 8.5 | 1899 | 1.27 | 149 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maia, L.S.M.; Burger, B.; Ghannam, A.; Nunes, F.L.; Ferriani, M.P.L.; Dias, M.M.; Arruda, L.K.; Drouet, C.; Cichon, S. Recessive SERPING1 Variant Leads to Kinin–Kallikrein System Control Failure in a Consanguineous Brazilian Family with Hereditary Angioedema. J. Clin. Med. 2023, 12, 7299. https://doi.org/10.3390/jcm12237299
Maia LSM, Burger B, Ghannam A, Nunes FL, Ferriani MPL, Dias MM, Arruda LK, Drouet C, Cichon S. Recessive SERPING1 Variant Leads to Kinin–Kallikrein System Control Failure in a Consanguineous Brazilian Family with Hereditary Angioedema. Journal of Clinical Medicine. 2023; 12(23):7299. https://doi.org/10.3390/jcm12237299
Chicago/Turabian StyleMaia, Luana Sella Motta, Bettina Burger, Arije Ghannam, Fernanda Leonel Nunes, Mariana Paes Leme Ferriani, Marina Mendonça Dias, Luisa Karla Arruda, Christian Drouet, and Sven Cichon. 2023. "Recessive SERPING1 Variant Leads to Kinin–Kallikrein System Control Failure in a Consanguineous Brazilian Family with Hereditary Angioedema" Journal of Clinical Medicine 12, no. 23: 7299. https://doi.org/10.3390/jcm12237299