
Current Therapy and Liver Transplantation for Portopulmonary Hypertension in Japan

Abstract
:1. Introduction
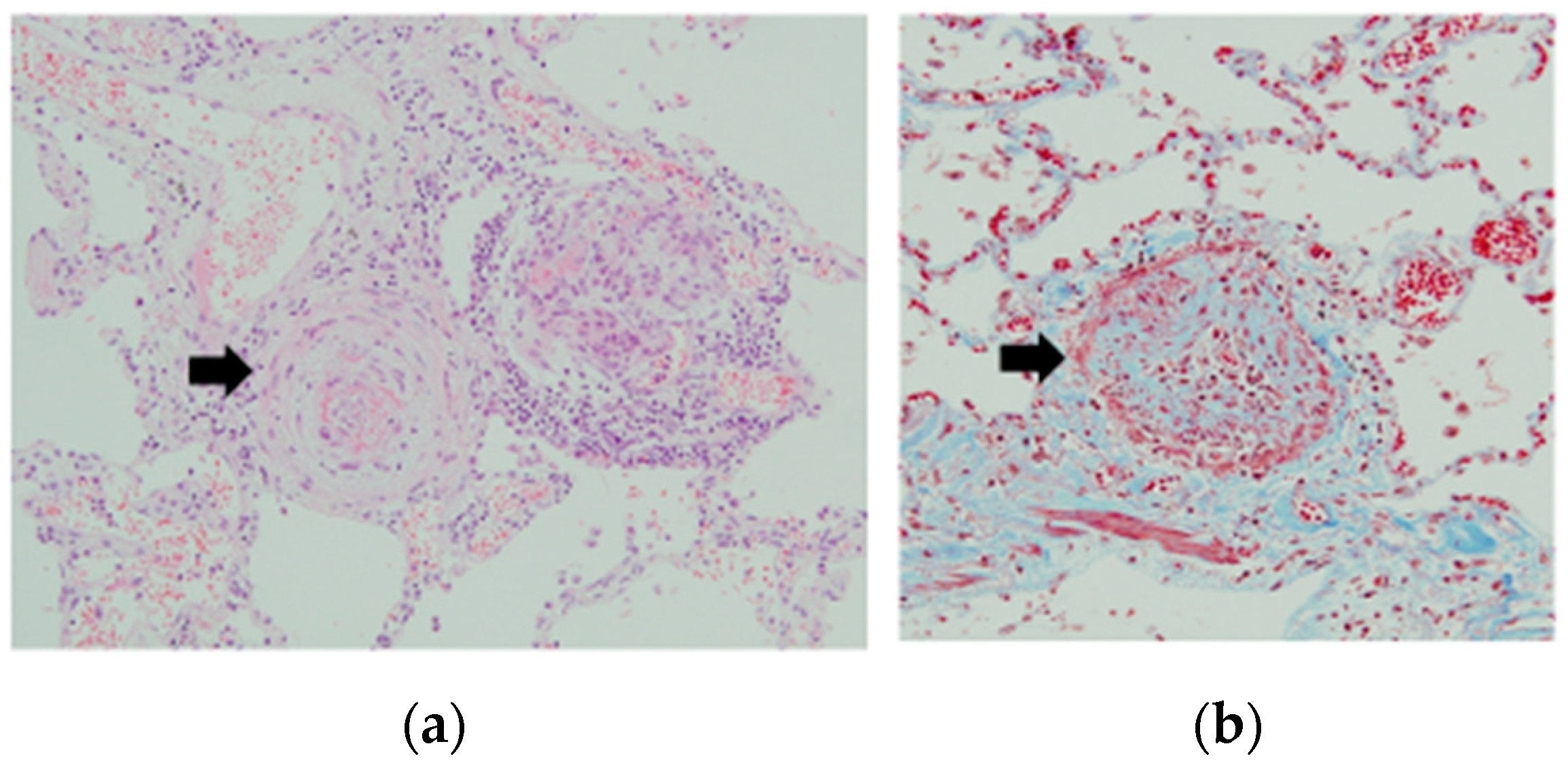
2. Clinical Features of PoPH
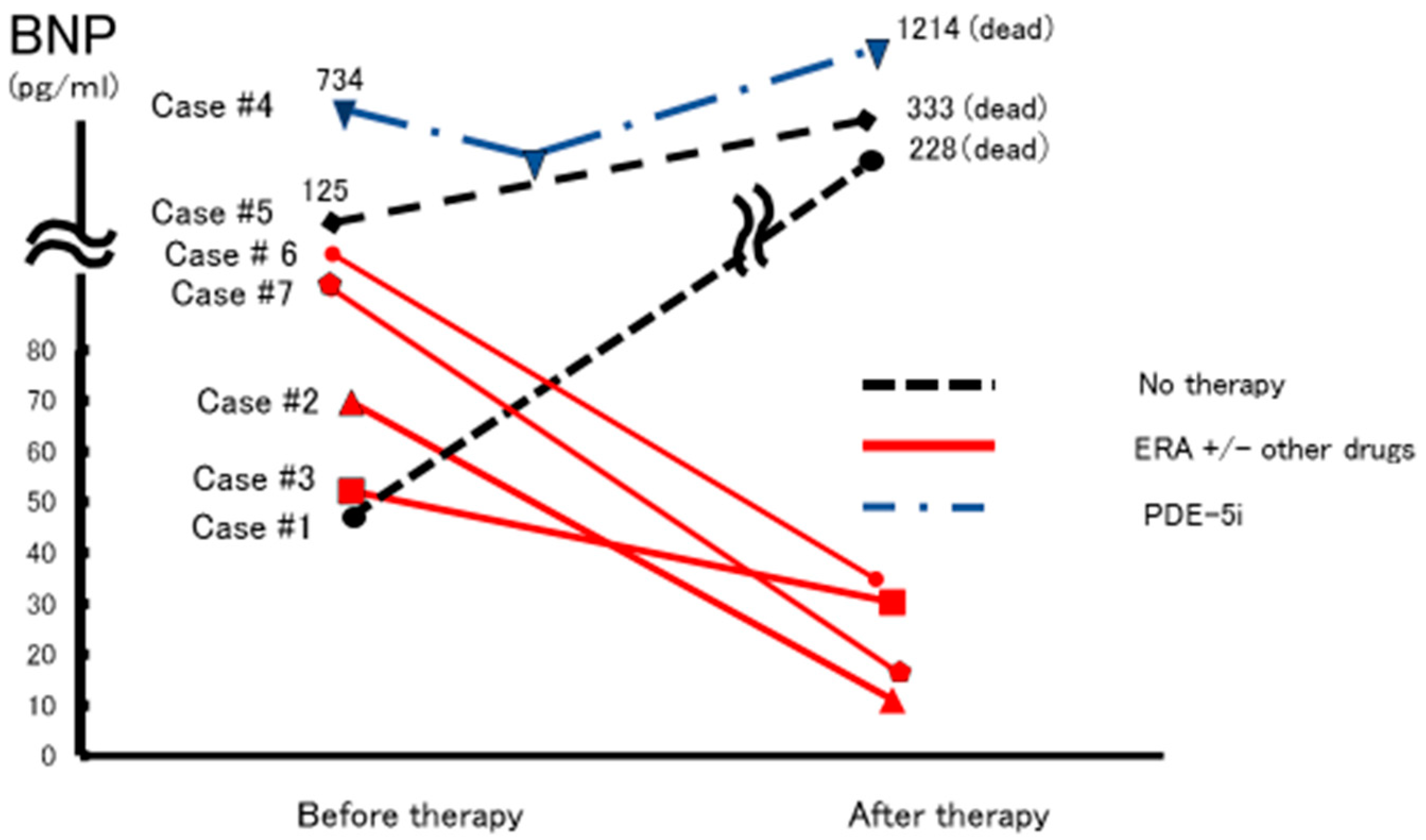
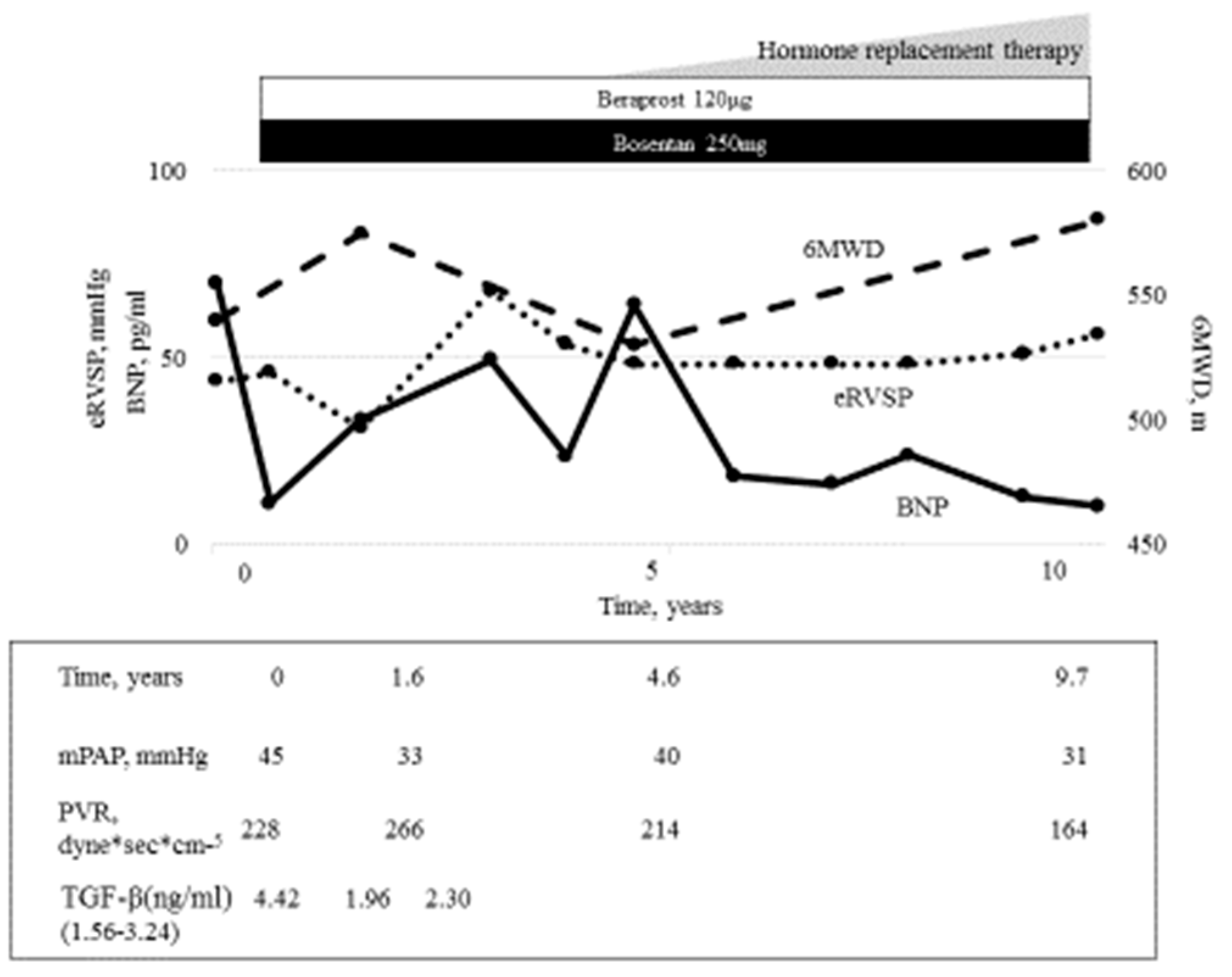
3. Drug Therapy for PoPH
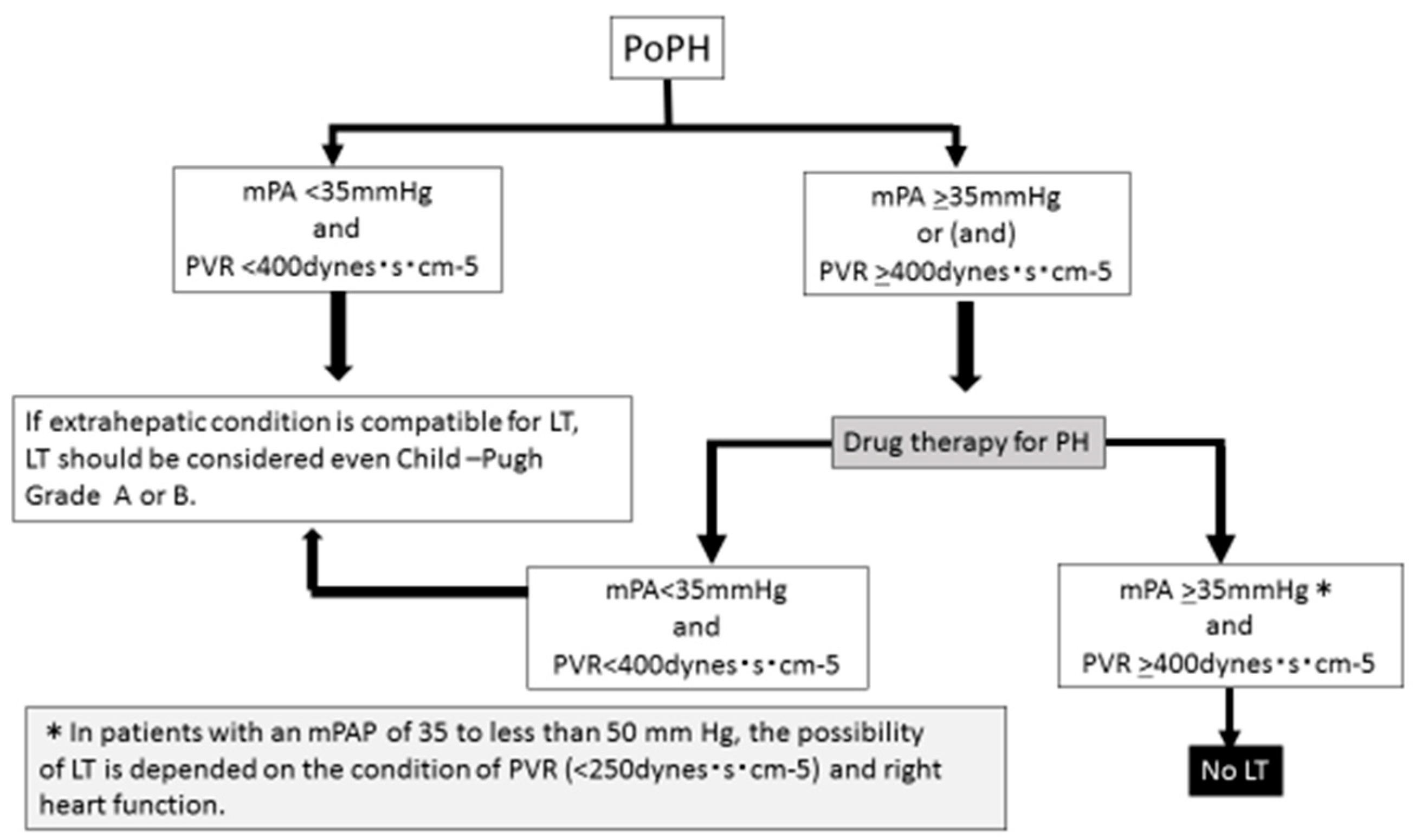
4. LT with PoPH
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Umeda, N.; Kamath, P.S. Hepatopulmonary syndrome and portopulmonary hypertension. Hepatol. Res. 2009, 39, 1020–1022. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, P.J.; Toye, P.A.; Hutchins, G.M. Primary pulmonary hypertension and cirrhosis: Are they related? Am. Rev. Respir. Dis. 1983, 127, 437–441. [Google Scholar] [CrossRef]
- Colle, I.O.; Moreau, R.; Godinho, E.; Belghiti, J.; Ettori, F.; Cohen-Solal, A.; Mal, H.; Bernuau, J.; Marty, J.; Lebrec, D.; et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: A prospective study. Hepatology 2003, 37, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Krowka, M.J.; Swanson, K.L.; Frantz, R.P.; McGoon, M.D.; Wiesner, R.H. Portopulmonary hypertension: Results from a 10-year screening algorithm. Hepatology 2006, 44, 1502–1510. [Google Scholar] [CrossRef]
- Hayashi, R.; Kogiso, T.; Kikuchi, N.; Yamamoto, K.; Nakamura, S.; Egawa, H.; Hagiwara, N.; Tokushige, K. Portopulmonary hypertension and the risk of high right ventricular systolic pressure in liver transplant candidates. PLoS ONE 2022, 17, e0267125. [Google Scholar] [CrossRef] [PubMed]
- Safdar, Z.; Bartolome, S.; Sussman, N. Portopulmonary hypertension: An update. Liver Transpl. 2012, 18, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Sussman, N.; Kaza, V.; Barshes, N.; Stribling, R.; Goss, J.; O’Mahony, C.; Zhang, E.; Vierling, J.; Frost, A. Successful liver transplantation following medical management of portopulmonary hypertension: A single-center series. Am. J. Transplant. 2006, 6, 2177–2182. [Google Scholar] [CrossRef]
- Krowka, M.J.; Mandell, M.S.; Ramsay, M.A.; Kawut, S.M.; Fallon, M.B.; Manzarbeitia, C.; Pardo, M.; Marotta, P.; Uemoto, S.; Stoffel, M.P.; et al. Hepatopulmonary syndrome and portopulmonary hypertension: A report of the multicenter liver transplant database. Liver Transpl. 2004, 10, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhai, Z.; Huang, K.; Xie, W.; Wan, J.; Wang, C. Bosentan therapy for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: A systemic review and meta-analysis. Clin. Respir. J. 2018, 12, 2065–2074. [Google Scholar] [CrossRef]
- Feldman, J.; Habib, N.; Radosevich, J.; Dutt, M. Oral treprostinil in the treatment of pulmonary arterial hypertension. Expert Opin. Pharmacother. 2017, 18, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Ueno, Y.; Hiasa, Y.; Nishikawa, H.; Hige, S.; Takikawa, Y.; Taniai, M.; Ishikawa, T.; Yasui, K.; Takaki, A.; et al. Transition in the etiology of liver cirrhosis in Japan: A nationwide survey. J. Gastroenterol. 2020, 55, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsukawa, M.; Tsubota, A.; Hatano, M.; Kondo, C.; Shioda, K.; Ohno, H.; Kawano, T.; Hayama, K.; Arai, T.; Nakagawa-Iwashita, A.; et al. Prevalence and characteristics of portopulmonary hypertension in cirrhotic patients who underwent both hepatic vein and pulmonary artery catheterization. Hepatol. Res. 2020, 50, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Kawut, S.M.; Krowka, M.J.; Trotter, J.F.; Roberts, K.E.; Benza, R.L.; Badesch, D.B.; Taichman, D.B.; Horn, E.M.; Zacks, S.; Kaplowitz, N.; et al. Clinical risk factors for portopulmonary hypertension. Hepatology 2008, 48, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Kodama, K.; Serizawa, N.; Hashimoto, E. Current status of portopulmonary hypertension and hepatopulmonary syndrome. EC Gastroenterol. Dig. Syst. 2020, 7, 63–69. [Google Scholar]
- Suzuki, K.; Kanamoto, M.; Hinohara, H.; Saito, S. A case of hypopituitarism complicated by non-alcoholic steatohepatitis and severe pulmonary hypertension. Am. J. Case Rep. 2021, 22, e928004. [Google Scholar] [CrossRef]
- Justino, H.; Sanders, K.; McLin, V.A. Rapid progression from hepatopulmonary syndrome to portopulmonary hypertension in an adolescent female with hypopituitarism. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 334–336. [Google Scholar] [CrossRef]
- Torii, N.; Ichihara, A.; Mizuguchi, Y.; Seki, Y.; Hashimoto, E.; Tokushige, K. Hormone-replacement therapy for hepatopulmonary syndrome and NASH associated with hypopituitarism. Intern. Med. 2018, 57, 1741–1745. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Sun, M.; Ramchandran, R.; Raj, J.U. IGF-1 signaling in neonatal hypoxia-induced pulmonary hypertension: Role of epigenetic regulation. Vasc. Pharmacol. 2015, 73, 20–31. [Google Scholar] [CrossRef] [Green Version]
- Connolly, M.; Garfield, B.E.; Crosby, A.; Morrell, N.W.; Wort, S.J.; Kemp, P.R. MiR-322-5p targets IGF-1 and is suppressed in the heart of rats with pulmonary hypertension. FEBS Open Bio. 2018, 8, 339–348. [Google Scholar] [CrossRef]
- Kudo, M. Recent advances in systemic therapy for hepatocellular carcinoma in an aging society: 2020 update. Liver Cancer 2020, 9, 640–662. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Current status of molecularly targeted therapy for hepatocellular carcinoma: Clinical practice. Int. J. Clin. Oncol. 2010, 15, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Sasaki, R.; Matsuda, T.; Saeki, I.; Takami, T.; Wada, Y.; Yano, M.; Sakaida, I. Successful management with dual therapy of lenvatinib and macitentan for HCC With portopulmonary hypertension. Hepatology 2021, 74, 2300–2303. [Google Scholar] [CrossRef]
- Yoshimaru, K.; Matsuura, T.; Takahashi, Y.; Yanagi, Y.; Nagata, H.; Ohga, S.; Taguchi, T. The efficacy of serum brain natriuretic peptide for the early detection of portopulmonary hypertension in biliary atresia patients before liver transplantation. Pediatr. Transplant. 2018, 22, e13203. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Elliott, C.G.; Levine, D.J.; Bossone, E.; Duvall, L.; Fagan, K.; Frantsve-Hawley, J.; Kawut, S.M.; Ryan, J.J.; Rosenzweig, E.B.; et al. Therapy for pulmonary arterial hypertension in adults: Update of the CHEST guideline and expert panel report. Chest 2019, 155, 565–586. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Seyfarth, H.J.; Hoeffken, G.; Wirtz, H.; Spiekerkoetter, E.; Pletz, M.W.; Welte, T.; Halank, M. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur. Respir. J. 2007, 30, 1096–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitbon, O.; Bosch, J.; Cottreel, E.; Csonka, D.; de Groote, P.; Hoeper, M.M.; Kim, N.H.; Martin, N.; Savale, L.; Krowka, M. Macitentan for the treatment of portopulmonary hypertension (PORTICO): A multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir. Med. 2019, 7, 594–604. [Google Scholar] [CrossRef]
- Savale, L.; Guimas, M.; Ebstein, N.; Fertin, M.; Jevnikar, M.; Renard, S.; Horeau-Langlard, D.; Tromeur, C.; Chabanne, C.; Prevot, G.; et al. Portopulmonary hypertension in the current era of pulmonary hypertension management. J. Hepatol. 2020, 73, 130–139. [Google Scholar] [CrossRef]
- Takahashi, Y.; Yamamoto, K.; Sakao, S.; Takeuchi, T.; Suda, R.; Tanabe, N.; Tatsumi, K. The clinical characteristics, treatment, and survival of portopulmonary hypertension in Japan. BMC Pulm. Med. 2021, 21, 89. [Google Scholar] [CrossRef] [PubMed]
- Martinet, J.P.; Legault, L.; Cernacek, P.; Roy, L.; Dufresne, M.P.; Spahr, L.; Fenyves, D.; Pomier-Layrargues, G. Changes in plasma endothelin-1 and big endothelin-1 induced by transjugular intrahepatic portosystemic shunts in patients with cirrhosis and refractory ascites. J. Hepatol. 1996, 25, 700–706. [Google Scholar] [CrossRef]
- Feng, H.Q.; Weymouth, N.D.; Rockey, D.C. Endothelin antagonism in portal hypertensive mice: Implications for endothelin receptor-specific signaling in liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G27–G33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sogni, P.; Moreau, R.; Gomola, A.; Gadano, A.; Cailmail, S.; Calmus, Y.; Clozel, M.; Lebrec, D. Beneficial hemodynamic effects of bosentan, a mixed ET(A) and ET(B) receptor antagonist, in portal hypertensive rats. Hepatology 1998, 28, 655–659. [Google Scholar] [CrossRef]
- Krowka, M.J.; Plevak, D.J.; Findlay, J.Y.; Rosen, C.B.; Wiesner, R.H.; Krom, R.A. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000, 6, 443–450. [Google Scholar] [CrossRef]
- Ramsay, M.A.; Simpson, B.R.; Nguyen, A.T.; Ramsay, K.J.; East, C.; Klintmalm, G.B. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl. Surg. 1997, 3, 494–500. [Google Scholar] [CrossRef]
- Machicao, V.I.; Balakrishnan, M.; Fallon, M.B. Pulmonary complications in chronic liver disease. Hepatology 2014, 59, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Starkel, P.; Vera, A.; Gunson, B.; Mutimer, D. Outcome of liver transplantation for patients with pulmonary hypertension. Liver Transpl. 2002, 8, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Selection Criteria of Recipients Who Expect Deceased-Donor Liver Ransplantation in Japan. Available online: https://www.mhlw.go.jp/content/10900000/000696415.p (accessed on 9 September 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Age Gender | Based Liver Disease | Shunt or Varices | BNP (pg/mL) | eRVSP (mmHg) mPAP (mmHg) | Therapy | Effect of Therapy | Prognosis |
|---|---|---|---|---|---|---|---|---|
| 1 | 61 F | PBC | E, G-varices (E-transection) | 45 | 144.6 - | No therapy | Dead | |
| 2 | 39 F | NASH-LC hypopituitarism | E-varices (EVL) | 69.7 | 61 45 | ERA, PRA | (+) | Survival |
| 3 | 29 F | Portal vein obstruction (portsystemic shunt) | Huge shunt | 52.2 | 59 34 | ERA | (+) | Survival |
| 4 | 60 F | PBC | E–varisce (EVL, EIS) | 734 | 106 53 | PDE-5i | (−) | Dead |
| 5 | 82 F | LC-C | E-varices (E-transection) | 124.9 | 55 - | No therapy | Dead | |
| 6 | 63 F | PBC HCV(+) | Huge shunt | 69.7 | 122 45 | ERA, PRA PDE-5i | (+) | Survival |
| 7 | 20 F | Portal vein obstruction (portsystemic shunt) | Huge shunt E-varices | 90.8 | 77 53 | ERA, PDE-5i | (+) | Survival |
| PoPH | HPS | |
|---|---|---|
| Age (mean + S.E.) | 50.6 ± 8.3 | 49.0 ± 5.9 |
| Gender F/M (female %) | 7/0 (100%) ** | 2/5 (29%) |
| Background liver disease | PBC 3 (43%) + LC due to HCV 1 (14%) + Portal-systemic shunt 2 (29%) NASH (14%) | LC due to HCV 4 (57%) NASH 2 (29%) Alcoholic LC 1(14%) |
| Shunt or varices | Huge portosytemic shunt 3 E, G varices 4 (surgical therapy 2, endoscopic therapy 2) | E-varices 3 Portosytemic shunt 1 |
| Symptoms | Hearing failure 3 (43%) Dyspnea 3 (43%) No symptoms 1 (14%) <diagnosis during examination of liver tumor> | Dyspnea 7 (100%) |
| Prognosis and therapy |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokushige, K.; Kogiso, T.; Egawa, H. Current Therapy and Liver Transplantation for Portopulmonary Hypertension in Japan. J. Clin. Med. 2023, 12, 562. https://doi.org/10.3390/jcm12020562
Tokushige K, Kogiso T, Egawa H. Current Therapy and Liver Transplantation for Portopulmonary Hypertension in Japan. Journal of Clinical Medicine. 2023; 12(2):562. https://doi.org/10.3390/jcm12020562
Chicago/Turabian StyleTokushige, Katsutoshi, Tomomi Kogiso, and Hiroto Egawa. 2023. "Current Therapy and Liver Transplantation for Portopulmonary Hypertension in Japan" Journal of Clinical Medicine 12, no. 2: 562. https://doi.org/10.3390/jcm12020562