
Computational and Experimental Analyses for Pathogenicity Prediction of ACVRL1 Missense Variants in Hereditary Hemorrhagic Telangiectasia
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient and Computational Pathogenicity Prediction
2.2. ACVRL1 and Endoglin Expression Plasmids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Age, Sex | Epistaxis | TE | PVM | HVM | BVM | Family History | Curaçao Criteria |
|---|---|---|---|---|---|---|---|---|
| D176Y | 67, M | + | + | − | − | − | + | Definite |
| D235Y | 67, F | + | + | − | + | − | + | Definite |
| D235Y | 61, M | + | + | − | + | − | + | Definite |
| P424T | 61, F | + | − | − | + | − | − | Definite |
| D437G | 81, F | − | − | + | + | − | + | Definite |
| D437G | 46, M | + | + | + | − | − | + | Definite |
| R479P | 28, F | + | + | − | − | − | + | Definite |
| R484L | 49, F | + | − | − | + | − | + | Definite |
2.3. Cell Culture and Plasmid Transfection
2.4. Immunocytochemistry
2.5. Luciferase Reporter Assay
2.6. Western Blot Analysis
2.7. Computational Structure Prediction
2.8. Statistical Analysis
3. Results
3.1. ACVRL1 Variants in this Study and Profiles of Unpublished Clinical Cases
3.2. Computational Pathogenicity Prediction of ACVRL1 Missense Variants
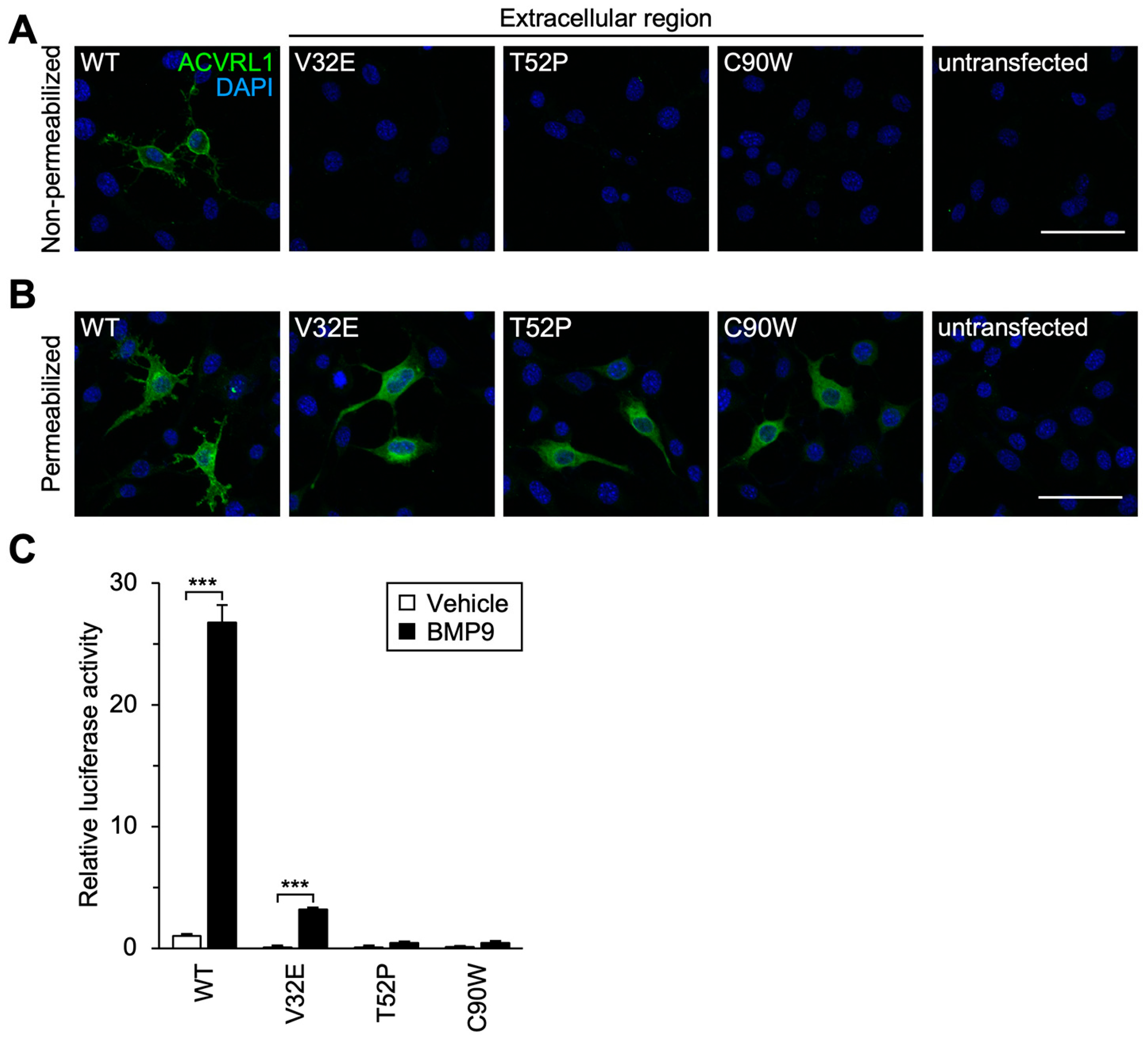
3.3. Subcellular Localization and Signal Transduction Capacity of Extracellular Residue Variants
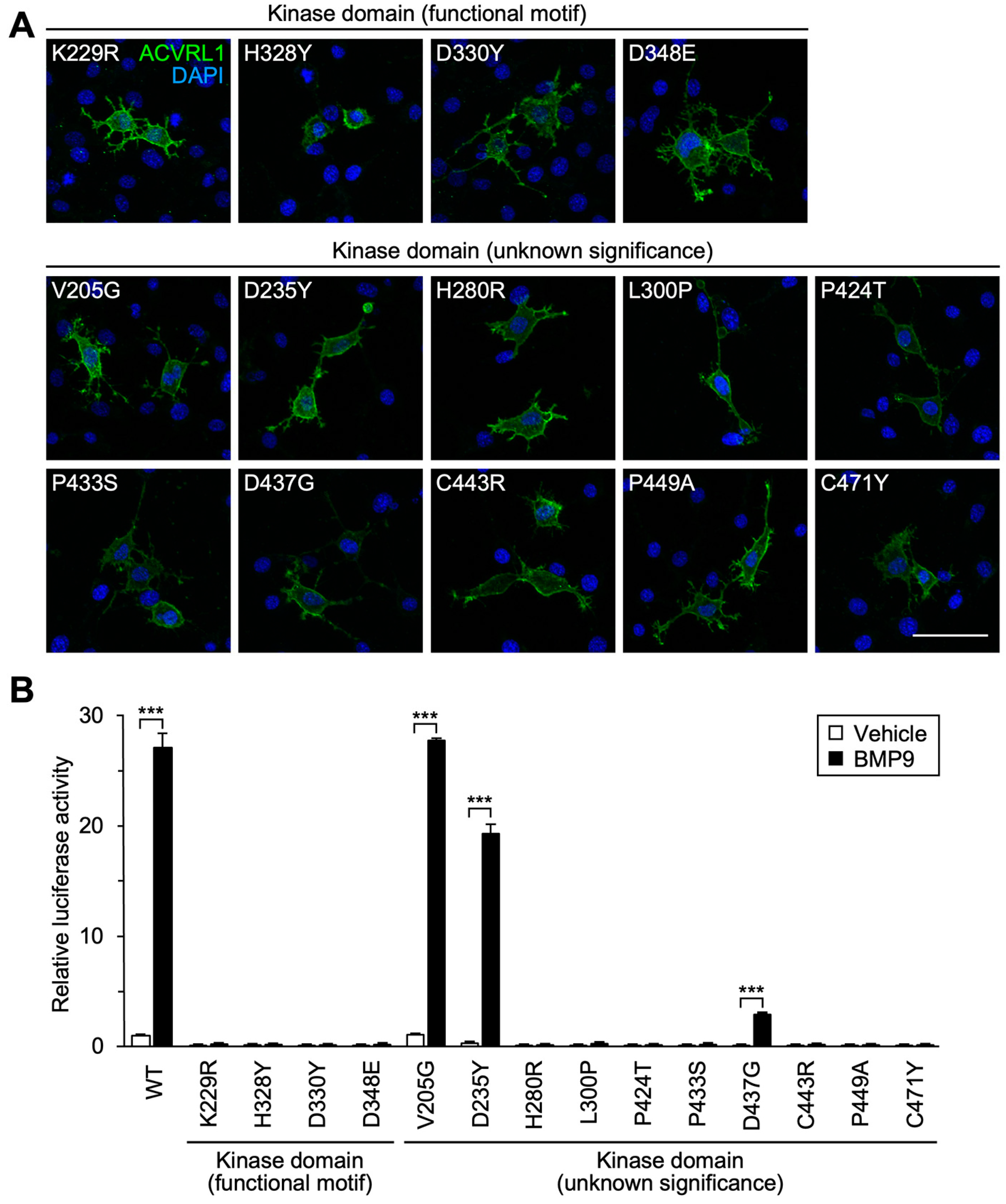
3.4. Importance of Functional Motifs and Conserved Residues in Intracellular Kinase Domain
3.5. Structural Prediction of ACVRL1-SMAD1 Interaction and Impact of Missense Variations in L45 Loop upon Signal Transduction Activity
3.6. Structural Prediction of ACVRL1-BMPR2 Interaction and Influence of Missense Variations in NANDOR and GS Domains
3.7. Detailed Characterization of Variants without Functional Defects and Responsiveness to Endoglin Co-Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Bernabeu, C.; Bayrak-Toydemir, P.; McDonald, J.; Letarte, M. Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 3571. [Google Scholar] [CrossRef] [PubMed]
- Robert, F.; Desroches-Castan, A.; Bailly, S.; Dupuis-Girod, S.; Feige, J.J. Future treatments for hereditary hemorrhagic telangiectasia. Orphanet J. Rare Dis. 2020, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; ten Dijke, P.; Kim, S.; et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Urness, L.D.; Sorensen, L.K.; Li, D.Y. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 2000, 26, 328–331. [Google Scholar] [CrossRef]
- Roman, B.L.; Pham, V.N.; Lawson, N.D.; Kulik, M.; Childs, S.; Lekven, A.C.; Garrity, D.M.; Moon, R.T.; Fishman, M.C.; Lechleider, R.J.; et al. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development 2002, 129, 3009–3019. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Ricard, N.; Bidart, M.; Mallet, C.; Lesca, G.; Giraud, S.; Prudent, R.; Feige, J.J.; Bailly, S. Functional analysis of the BMP9 response of ALK1 mutants from HHT2 patients: A diagnostic tool for novel ACVRL1 mutations. Blood 2010, 116, 1604–1612. [Google Scholar] [CrossRef] [Green Version]
- Alaa El Din, F.; Patri, S.; Thoreau, V.; Rodriguez-Ballesteros, M.; Hamade, E.; Bailly, S.; Gilbert-Dussardier, B.; Abou Merhi, R.; Kitzis, A. Functional and splicing defect analysis of 23 ACVRL1 mutations in a cohort of patients affected by Hereditary Hemorrhagic Telangiectasia. PLoS ONE 2015, 10, e0132111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hume, A.N.; John, A.; Akawi, N.A.; Al-Awadhi, A.M.; Al-Suwaidi, S.S.; Al-Gazali, L.; Ali, B.R. Retention in the endoplasmic reticulum is the underlying mechanism of some hereditary haemorrhagic telangiectasia type 2 ALK1 missense mutations. Mol. Cell Biochem. 2013, 373, 247–257. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Thomas, P.D. PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef] [Green Version]
- Korchynskyi, O.; ten Dijke, P. Identification and Functional Characterization of Distinct Critically Important Bone Morphogenetic Protein-specific Response Elements in the Id1 Promoter. J. Biol. Chem. 2002, 277, 4883–4891. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Koinuma, D.; Tsutsumi, S.; Vasilaki, E.; Kanki, Y.; Heldin, C.H.; Aburatani, H.; Miyazono, K. ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res. 2011, 39, 8712–8727. [Google Scholar] [CrossRef]
- van Meeteren, L.A.; Thorikay, M.; Bergqvist, S.; Pardali, E.; Stampino, C.G.; Hu-Lowe, D.; Goumans, M.J.; ten Dijke, P. Anti-human activin receptor-like kinase 1 (ALK1) antibody attenuates bone morphogenetic protein 9 (BMP9)-induced ALK1 signaling and interferes with endothelial cell sprouting. J. Biol. Chem. 2012, 287, 18551–18561. [Google Scholar] [CrossRef] [Green Version]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Komiyama, M.; Ishiguro, T.; Yamada, O.; Morisaki, H.; Morisaki, T. Hereditary hemorrhagic telangiectasia in Japanese patients. J. Hum. Genet. 2014, 59, 37–41. [Google Scholar] [CrossRef]
- Kitayama, K.; Ishiguro, T.; Komiyama, M.; Morisaki, T.; Morisaki, H.; Minase, G.; Hamanaka, K.; Miyatake, S.; Matsumoto, N.; Kato, M.; et al. Mutational and clinical spectrum of Japanese patients with hereditary hemorrhagic telangiectasia. BMC Med. Genom. 2021, 14, 288. [Google Scholar] [CrossRef]
- Nishimoto, Y.; Morisaki, H.; Yamada, O.; Ichinose, Y.; Suzuki, N. Japanese case of hereditary hemorrhagic telangiectasia type 2 with a novel mutation, c.154A>C (p.Thr52Pro), in the ALK1/ACVRL1 gene. Neurol. Clin. Neurosci. 2014, 2, 126–128. [Google Scholar] [CrossRef]
- Iwasa, T.; Yamada, O.; Ohuchi, H.; Shiraishi, I.; Morisaki, H.; Morisaki, T.; Kurosaki, K. A Case of Spontaneously Improved Heritable Pulmonary Arterial Hypertension Diagnosed as Severe Primary Pulmonary Hypertension in Childhood. J. Pediatr. Cardiol. Card. Surg. 2020, 4, 9–13. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argyriou, L.; Twelkemeyer, S.; Panchulidze, I.; Wehner, L.E.; Teske, U.; Engel, W.; Nayernia, K. Novel mutations in the ENG and ACVRL1 genes causing hereditary hemorrhagic teleangiectasia. Int. J. Mol. Med. 2006, 17, 655–659. [Google Scholar] [CrossRef] [Green Version]
- Bossler, A.D.; Richards, J.; George, C.; Godmilow, L.; Ganguly, A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): Correlation of genotype with phenotype. Hum. Mutat. 2006, 27, 667–675. [Google Scholar] [CrossRef]
- Lesca, G.; Plauchu, H.; Coulet, F.; Lefebvre, S.; Plessis, G.; Odent, S.; Riviere, S.; Leheup, B.; Goizet, C.; Carette, M.F.; et al. Molecular screening of ALK1/ACVRL1 and ENG genes in hereditary hemorrhagic telangiectasia in France. Hum. Mutat. 2004, 23, 289–299. [Google Scholar] [CrossRef]
- Heimdal, K.; Dalhus, B.; Rødningen, O.K.; Kroken, M.; Eiklid, K.; Dheyauldeen, S.; Røysland, T.; Andersen, R.; Kulseth, M.A. Mutation analysis in Norwegian families with hereditary hemorrhagic telangiectasia: Founder mutations in ACVRL1. Clin. Genet. 2016, 89, 182–186. [Google Scholar] [CrossRef]
- Richards-Yutz, J.; Grant, K.; Chao, E.C.; Walther, S.E.; Ganguly, A. Update on molecular diagnosis of hereditary hemorrhagic telangiectasia. Hum. Genet. 2010, 128, 61–77. [Google Scholar] [CrossRef]
- Gedge, F.; McDonald, J.; Phansalkar, A.; Chou, L.S.; Calderon, F.; Mao, R.; Lyon, E.; Bayrak-Toydemir, P. Clinical and analytical sensitivities in hereditary hemorrhagic telangiectasia testing and a report of de novo mutations. J. Mol. Diagn. 2007, 9, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Faughnan, M.E.; Krings, T.; Chakinala, M.; Gossage, J.R.; Young, W.L.; Kim, H.; Pourmohamad, T.; Henderson, K.J.; Schrum, S.D.; et al. Brain arteriovenous malformations associated with hereditary hemorrhagic telangiectasia: Gene-phenotype correlations. Am. J. Med. Genet. A 2012, 158a, 2829–2834. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, C.; Pagella, F.; Semino, L.; Lanzarini, L.; Valacca, C.; Pilotto, A.; Corno, S.; Scappaticci, S.; Manfredi, G.; Buscarini, E.; et al. Analysis of ENG and ACVRL1 genes in 137 HHT Italian families identifies 76 different mutations (24 novel). Comparison with other European studies. J. Hum. Genet. 2007, 52, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Fontalba, A.; Fernandez-L, A.; García-Alegria, E.; Albiñana, V.; Garrido-Martin, E.M.; Blanco, F.J.; Zarrabeitia, R.; Perez-Molino, A.; Bernabeu-Herrero, M.E.; Ojeda, M.-L.; et al. Mutation study of Spanish patients with Hereditary Hemorrhagic Telangiectasia. BMC Med. Genet. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argyriou, L.; Pfitzmann, R.; Wehner, L.E.; Twelkemeyer, S.; Neuhaus, P.; Nayernia, K.; Engel, W. ALK-1 mutations in liver transplanted patients with hereditary hemorrhagic telangiectasia. Liver Transpl. 2005, 11, 1132–1135. [Google Scholar] [CrossRef]
- Letteboer, T.G.; Zewald, R.A.; Kamping, E.J.; de Haas, G.; Mager, J.J.; Snijder, R.J.; Lindhout, D.; Hennekam, F.A.; Westermann, C.J.; Ploos van Amstel, J.K. Hereditary hemorrhagic telangiectasia: ENG and ALK-1 mutations in Dutch patients. Hum. Genet. 2005, 116, 8–16. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.; Damjanovich, K.; Millson, A.; Wooderchak, W.; Chibuk, J.M.; Stevenson, D.A.; Gedge, F.; Bayrak-Toydemir, P. Molecular diagnosis in hereditary hemorrhagic telangiectasia: Findings in a series tested simultaneously by sequencing and deletion/duplication analysis. Clin. Genet. 2011, 79, 335–344. [Google Scholar] [CrossRef]
- Brakensiek, K.; Frye-Boukhriss, H.; Malzer, M.; Abramowicz, M.; Bahr, M.J.; von Beckerath, N.; Bergmann, C.; Caselitz, M.; Holinski-Feder, E.; Muschke, P.; et al. Detection of a significant association between mutations in the ACVRL1 gene and hepatic involvement in German patients with hereditary haemorrhagic telangiectasia. Clin. Genet. 2008, 74, 171–177. [Google Scholar] [CrossRef]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, J.N.; Gallione, C.J.; Stenzel, T.T.; Johnson, D.W.; Allen, W.P.; Schwartz, C.E.; Jackson, C.E.; Porteous, M.E.M.; Marchuk, D.A. The Activin Receptor-Like Kinase 1 Gene: Genomic Structure and Mutations in Hereditary Hemorrhagic Telangiectasia Type 2. Am. J. Hum. Genet. 1997, 61, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Tadaka, S.; Hishinuma, E.; Komaki, S.; Motoike, I.N.; Kawashima, J.; Saigusa, D.; Inoue, J.; Takayama, J.; Okamura, Y.; Aoki, Y.; et al. jMorp updates in 2020: Large enhancement of multi-omics data resources on the general Japanese population. Nucleic Acids Res. 2020, 49, D536–D544. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallet, C.; Lamribet, K.; Giraud, S.; Dupuis-Girod, S.; Feige, J.J.; Bailly, S.; Tillet, E. Functional analysis of endoglin mutations from hereditary hemorrhagic telangiectasia type 1 patients reveals different mechanisms for endoglin loss of function. Hum. Mol. Genet. 2015, 24, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- González-Núñez, M.; Muñoz-Félix, J.M.; López-Novoa, J.M. The ALK-1/Smad1 pathway in cardiovascular physiopathology. A new target for therapy? Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1492–1510. [Google Scholar] [CrossRef] [Green Version]
- Chaikuad, A.; Alfano, I.; Kerr, G.; Sanvitale, C.E.; Boergermann, J.H.; Triffitt, J.T.; von Delft, F.; Knapp, S.; Knaus, P.; Bullock, A.N. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem. 2012, 287, 36990–36998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Hackert, E.; Sundan, A.; Holien, T. Receptor binding competition: A paradigm for regulating TGF-β family action. Cytokine Growth Factor Rev. 2021, 57, 39–54. [Google Scholar] [CrossRef]
- Wu, G.; Chen, Y.G.; Ozdamar, B.; Gyuricza, C.A.; Chong, P.A.; Wrana, J.L.; Massagué, J.; Shi, Y. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science 2000, 287, 92–97. [Google Scholar] [CrossRef]
- Wu, J.-W.; Hu, M.; Chai, J.; Seoane, J.; Huse, M.; Li, C.; Rigotti, D.J.; Kyin, S.; Muir, T.W.; Fairman, R.; et al. Crystal Structure of a Phosphorylated Smad2: Recognition of Phosphoserine by the MH2 Domain and Insights on Smad Function in TGF-β Signaling. Mol. Cell 2001, 8, 1277–1289. [Google Scholar] [CrossRef]
- Agnew, C.; Ayaz, P.; Kashima, R.; Loving, H.S.; Ghatpande, P.; Kung, J.E.; Underbakke, E.S.; Shan, Y.; Shaw, D.E.; Hata, A.; et al. Structural basis for ALK2/BMPR2 receptor complex signaling through kinase domain oligomerization. Nat. Commun. 2021, 12, 4950. [Google Scholar] [CrossRef]
- Garamszegi, N.; Doré, J.J.E.; Penheiter, S.G.; Edens, M.; Yao, D.; Leof, E.B. Transforming Growth Factor β Receptor Signaling and Endocytosis Are Linked through a COOH Terminal Activation Motif in the Type I Receptor. Mol. Biol. Cell 2001, 12, 2881–2893. [Google Scholar] [CrossRef]
- Doré, J.J., Jr.; Yao, D.; Edens, M.; Garamszegi, N.; Sholl, E.L.; Leof, E.B. Mechanisms of transforming growth factor-beta receptor endocytosis and intracellular sorting differ between fibroblasts and epithelial cells. Mol. Biol. Cell 2001, 12, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Liu, Y.; Cao, D.; Chen, J.; Liu, Z.; Ji, H.; Chen, Y.; Zhang, W.; Zhu, P.; Xiao, D.; et al. BMP10 preserves cardiac function through its dual activation of SMAD-mediated and STAT3-mediated pathways. J. Biol. Chem. 2019, 294, 19877–19888. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.H.; Greenblatt, M.B.; Xie, M.; Schneider, M.D.; Zou, W.; Zhai, B.; Gygi, S.; Glimcher, L.H. TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 2009, 28, 2028–2041. [Google Scholar] [CrossRef] [Green Version]
- Soemedi, R.; Cygan, K.J.; Rhine, C.L.; Wang, J.; Bulacan, C.; Yang, J.; Bayrak-Toydemir, P.; McDonald, J.; Fairbrother, W.G. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 2017, 49, 848–855. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Abdalla, S.A.; Cymerman, U.; Johnson, R.M.; Deber, C.M.; Letarte, M. Disease-associated mutations in conserved residues of ALK-1 kinase domain. Eur. J. Hum. Genet. 2003, 11, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Kerr, G.; Sheldon, H.; Chaikuad, A.; Alfano, I.; von Delft, F.; Bullock, A.N.; Harris, A.L. A small molecule targeting ALK1 prevents Notch cooperativity and inhibits functional angiogenesis. Angiogenesis 2015, 18, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.G.; Massagué, J. Smad1 recognition and activation by the ALK1 group of transforming growth factor-beta family receptors. J. Biol. Chem. 1999, 274, 3672–3677. [Google Scholar] [CrossRef] [PubMed] [Green Version]







| Variant | Homology | Computational Pathogenicity Classifier | |||
|---|---|---|---|---|---|
| MCLXZ | SIFT | PolyPhen-2 | PROVEAN | PANTHER | |
| V32E | VLVWL | Tolerated | Benign | Neutral | Poss Dmgg |
| T52P | TFFYF | Tolerated | Prob Dmgg | Neutral | Poss Dmgg |
| C90W | CCCCC | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| D176Y | DDNED | Deleterious | Prob Dmgg | Deleterious | Poss Dmgg |
| L193P | LLLLL | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| T197I | TTTTT | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| R200G | RRRRR | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| V205G | VVVVV | Deleterious | Prob Dmgg | Deleterious | Poss Dmgg |
| K229R | KKKKK | Deleterious | Prob Dmgg | Deleterious | Poss Dmgg |
| D235Y | DDDDD | Deleterious | Prob Dmgg | Deleterious | Poss Dmgg |
| D263G | DDDDD | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| T265P | TTTTT | Tolerated | Prob Dmgg | Deleterious | Prob Dmgg |
| H280R | HHHHH | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| L300P | LLLLL | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| H328Y | HHHHH | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| D330Y | DDDDD | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| D348E | DDDDD | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| P424T | PPPPP | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| P433S | PPPPP | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| D437G | DDDDE | Deleterious | Prob Dmgg | Deleterious | Poss Dmgg |
| C443R | CCCCC | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| P449A | PPPPP | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| C471Y | CCCCC | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| R479P | RRRRR | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
| R484L | RRRRR | Deleterious | Prob Dmgg | Deleterious | Prob Dmgg |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwasa, T.; Urasaki, A.; Kakihana, Y.; Nagata-Akaho, N.; Harada, Y.; Takeda, S.; Kawamura, T.; Shiraishi, I.; Kurosaki, K.; Morisaki, H.; et al. Computational and Experimental Analyses for Pathogenicity Prediction of ACVRL1 Missense Variants in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2023, 12, 5002. https://doi.org/10.3390/jcm12155002
Iwasa T, Urasaki A, Kakihana Y, Nagata-Akaho N, Harada Y, Takeda S, Kawamura T, Shiraishi I, Kurosaki K, Morisaki H, et al. Computational and Experimental Analyses for Pathogenicity Prediction of ACVRL1 Missense Variants in Hereditary Hemorrhagic Telangiectasia. Journal of Clinical Medicine. 2023; 12(15):5002. https://doi.org/10.3390/jcm12155002
Chicago/Turabian StyleIwasa, Toru, Akihiro Urasaki, Yuki Kakihana, Nami Nagata-Akaho, Yukihiro Harada, Soichi Takeda, Teruhisa Kawamura, Isao Shiraishi, Kenichi Kurosaki, Hiroko Morisaki, and et al. 2023. "Computational and Experimental Analyses for Pathogenicity Prediction of ACVRL1 Missense Variants in Hereditary Hemorrhagic Telangiectasia" Journal of Clinical Medicine 12, no. 15: 5002. https://doi.org/10.3390/jcm12155002