
Fibrotic Diseases of the Human Urinary and Genital Tract: Current Understanding and Potential Strategies for Treatment
 , ,
, , (This article belongs to the Section Nephrology & Urology)
{kind=link}
Abstract
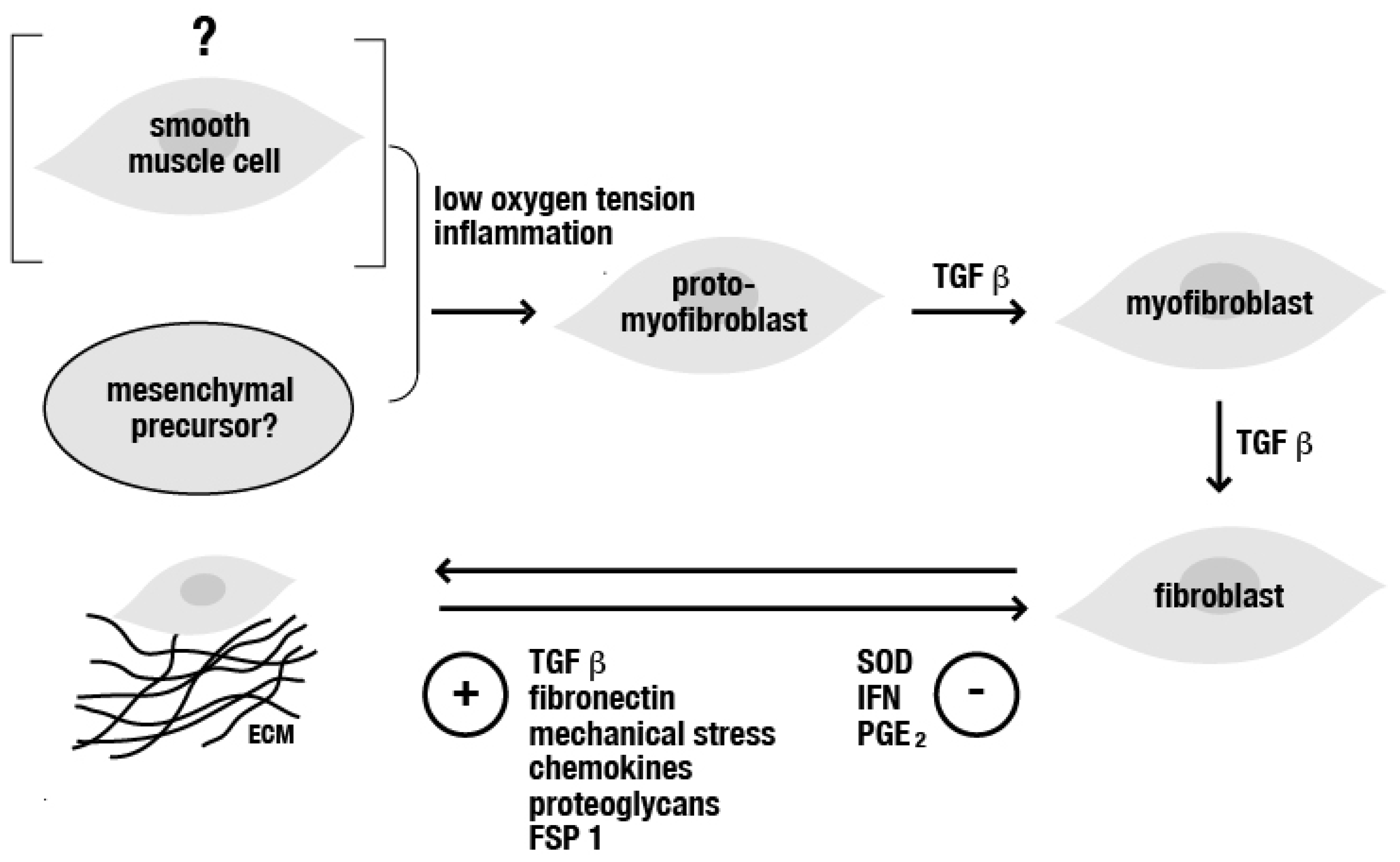
:1. General Mechanisms in the Pathophysiology of Fibrotic Diseases
2. Renal Fibrosis
3. Ureteral Fibrosis
4. Urethral Fibrosis
5. Fibrosis of the Urinary Bladder
6. Peyronie’s Disease
7. Potential Future Therapeutic Strategies to Treat Genitourinary Fibrosis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pohlers, D.; Brenmoehl, J.; Löffler, I.; Müller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF β and fibrosis in different organs—Molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Tang, P.M.K.; Li, J.; Lan, H.Y. TGF ß/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Massague, J. Integration of Smad and MAPK pathways: A link and a linker revisited. Genes Dev. 2003, 17, 2993–2997. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, T. Marfan syndrome: Demonstration of abnormal elastic fibers in skin. J. Cutan. Pathol. 1986, 13, 144–153. [Google Scholar] [CrossRef]
- Robertson, I.B.; Dias, H.F.; Osuch, I.H.; Lowe, E.D.; Jensen, S.A.; Redfield, C.; Handford, P.A. The N-terminal region of fibrillin-1 mediates a bipartite interaction with LTBP1. Structure 2017, 25, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Matt, P.; Schoenhoff, F.; Habashi, J.; Holm, T.; van Erp, C.; Loch, D.; Carlson, O.D.; Griswold, B.F.; Fu, Q.; de Backer, J.; et al. Circulating transforming growth factor-beta in Marfan syndrome. Circulation 2009, 120, 526–532. [Google Scholar] [CrossRef] [Green Version]
- Franken, R.; den Hartog, A.W.; de Waard, V.; Engele, L.; Radonic, T.; Lutter, R.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Zwinderman, A.H.; et al. Circulating transforming growth factor β as a prognostic biomarker in Marfan syndrome. Int. J. Cardiol. 2013, 168, 2441–2446. [Google Scholar] [CrossRef]
- Benke, K.; Agg, B.; Szilveszter, B.; Tarr, F.; Nagy, Z.B.; Pólos, M.; Daróczi, L.; Merkely, B.; Szabolcs, Z. The role of transforming growth factor-beta in Marfan syndrome. Cardiol. J. 2013, 20, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Siregar, S.; Farenia, R.; Sugandi, S.; Roesli, R.M. Effect of angiotensin II receptor blocker on TGF-β1, MMP-1, and collagen type I and type III concentration in New Zealand rabbit urethral stricture model. Res. Rep. Urol. 2018, 10, 127–133. [Google Scholar]
- Pyeritz, R.E. Marfan syndrome: Improved clinical history results in expanded natural history. Genet. Med. 2019, 21, 1683–1690. [Google Scholar] [CrossRef]
- Hentzen, C.; Turmel, N.; Chesnel, C.; le Breton, F.; Sheikh Ismael, S.; Amarenco, G. Urinary disorders and Marfan Syndrome: A series of four cases. Urol. Int. 2018, 101, 369–371. [Google Scholar] [CrossRef]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-β1/p53 signaling in renal fibrogenesis. Cell. Signal. 2018, 43, 1–10. [Google Scholar] [CrossRef]
- Luo, G.H.; Lu, Y.P.; Yang, L.; Song, J.; Shi, Y.J.; Li, Y.P. Epithelial to mesenchymal transformation in tubular epithelial cells undergoing anoxia. Transplant. Proc. 2008, 40, 2800–2803. [Google Scholar] [CrossRef]
- le Bleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 10471053. [Google Scholar]
- Wang, X.; Gao, Y.; Tian, N.; Wang, T.; Shi, Y.; Xu, J.; Wu, B. Astra-galoside IV inhibits glucose-induced epithelial-mesenchymal transition of podocytes through autophagy enhancement via the SIRT—NFκB p65 axis. Sci. Rep. 2019, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; de Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Lovisa, S.; le Bleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; de Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The epithelial-to-mesenchymal transition as a possible therapeutic target in fibrotic disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef]
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef]
- Klahr, S.; Morrissey, J. Obstructive nephropathy and renal fibrosis. Am. J. Physiol. (Ren. Physiol.) 2002, 283, 52–55. [Google Scholar] [CrossRef] [Green Version]
- Tie, Y.; Tang, F.; Peng, D.; Zhang, Y.; Shi, H. TGF-beta signal transduction: Biology, function and therapy for diseases. Mol. Biomed. 2022, 3, 45. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Larrue, R.; Fellah, S.; van der Hauwaert, C.; Hennino, M.F.; Perrais, M.; Lionet, A.; Glowacki, F.; Pottier, N.; Cauffiez, C. The versatile role of miR-21 in renal homeostasis and diseases. Cells 2022, 11, 3525. [Google Scholar] [CrossRef]
- Loboda, A.; Sobczak, M.; Jozkowicz, A.; Dulak, J. TGF-β1/Smads and miR-21 in renal fibrosis and inflammation. Mediat. Inflamm. 2016, 83, 19283. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V. Pathophysiology of AKI: Injury and Normal and Abnormal Repair. In Cardiorenal Syndromes in Critical Care; Karger: Basel, Switzerland, 2010; pp. 9–17. [Google Scholar]
- Ueshima, E.; Fujimori, M.; Kodama, H.; Felsen, D.; Chen, J.; Durack, J.C.; Solomon, S.B.; Coleman, J.A.; Srimathveeravalli, G. Macrophage-secreted TGF-β 1 contributes to fibroblast activation and ureteral stricture after ablation injury. Am. J. Physiol. (Renal Physiol.) 2019, 317, F52–F64. [Google Scholar] [CrossRef]
- Inazaki, K.; Kanamaru, Y.; Kojima, Y.; Sueyoshi, N.; Okumura, K.; Kaneko, K.; Yamashiro, Y.; Ogawa, H.; Nakao, A. Smad 3 deficiency attenuates renal fibrosis, inflammation, and apoptosis after unilateral ureteral obstruction. Kidney Int. 2004, 66, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Klimova, E.; Aparicio-Trejo, O.E.; Tapia, E.; Pedraza-Chaverri, J. Unilateral ureteral obstruction as a model to Investigate fibrosis-attenuating treatments. Biomolecules 2019, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Simsek, A.; Aldamanhori, R.; Chapple, C.R.; MacNeil, S. Overcoming scarring in the urethra: Challenges for tissue engineering. Asian J. Urol. 2018, 5, 69–77. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, J.; Zhang, D.; Wang, L.; Zhao, W.; Lin, D.Y.; Chen, R.; Xie, H.; Hu, X.; Fang, X.; et al. MicroRNA expression profiles of scar and normal tissue from patients with posterior urethral stricture caused by pelvic fracture urethral distraction defects. Int. J. Mol. Med. 2018, 41, 2733–2743. [Google Scholar] [CrossRef]
- Lin, H.; Guo, S.; Li, S.; Shen, J.; He, J.; Zheng, Y.; Gao, Z. Exploring relevant mRNAs and miRNAs in injured urethral tissues of rats with high-throughput sequencing. Genes 2022, 13, 824. [Google Scholar] [CrossRef]
- Rodriguez-Nieves, J.A.; Macoska, J.A. Prostatic fibrosis, lower urinary tract symptoms and BPH. Nat. Rev. Urol. 2013, 10, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Kim, J.; Na, Y.G.; Kim, K.H. Irreversible bladder remodeling induced by fibrosis. Int. Neurourol. J. 2021, 25 (Suppl. 1), S3–S7. [Google Scholar] [CrossRef]
- Qiao, L.Y.; Xia, C.; Shen, S.; Lee, S.H.; Ratz, P.H.; Fraser, M.O.; Miner, A.; Speich, J.E.; Lysiak, J.J.; Steers, W.D. Urinary bladder organ hypertrophy is partially regulated by Akt1-mediated protein synthesis pathway. Life Sci. 2018, 201, 63–71. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Anumanthan, G.; Pope, J.C. Transforming growth factor-ß signaling in bladder fibrosis. Ann. Rev. Biomed. Sci. 2008, 10, 1–5. [Google Scholar] [CrossRef]
- Chorbińska, J.; Krajewski, W.; Zdrojowy, R. Urological complications after radiation therapy—Nothing ventured, nothing gained: A narrative review. Transl. Cancer Res. 2021, 10, 1096–1118. [Google Scholar] [CrossRef]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Dunsmuir, W.D.; Kirby, R.S. François de la Peyronie (1678–1747): The man and the disease he described. Br. J. Urol. (BJU) 1996, 78, 613–622. [Google Scholar] [CrossRef]
- Hauck, E.W.; Weidner, W. François de la Peyronie and the disease named after him. Lancet 2001, 357, 2049–2051. [Google Scholar] [CrossRef]
- Smith, B.H. Peyronie’s disease. Am. J. Clin. Pathol. 1966, 45, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Pryor, J.P.; Ralph, D.J. Clinical presentations of Peyronie’s disease. Int. J. Impot. Res. (IJIR) 2002, 14, 414–417. [Google Scholar] [CrossRef] [Green Version]
- Cakan, M.; Akman, T.; Oktar, T.; Gurkan, L.; Celtik, M.; Kadioglu, A. The clinical characteristics of Peyronie’s patients with notching deformity. J. Sex. Med. 2007, 4, 1174–1178. [Google Scholar] [CrossRef]
- el-Sakka, A.I.; Hassoba, H.M.; Pillarisetty, R.J.; Nunes, L.; Dahiya, R.; Lue, T.F. Peyronie’s disease is associated with an increase in transforming growth factor beta protein expression. J. Urol. 1997, 158, 1391–1394. [Google Scholar] [CrossRef]
- Gonzalez-Cadavid, N.; Rajfer, J. Mechanisms of disease: New insights into the cellular and molecular pathology of Peyronie´s disease. Clin. Pract. Urol. 2005, 2, 291–297. [Google Scholar] [CrossRef]
- Weidner, W.; Hauck, E.W.; Schnitker, J.; for the Peyronie’s Disease Study Group of the Andrological Group of German Urologists. Potassium para-aminobenzoate (POTABA) in the treatment of Peyronie’s disease: A prospective, placebo-controlled, randomized study. Eur. Urol. 2005, 47, 530–535; Discussion 535–536. [Google Scholar] [CrossRef]
- Egui-Rojo, M.A.; Moncada-Iribarren, I.; Carballido-Rodriguez, J.; Martinez-Salamanca, J.I. Experience in the use of collagenase clostridium histolyticum in the management of Peyronie’s disease: Current data and future prospects. Ther. Adv. Urol. 2014, 6, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Vernet, D.; Ferrini, M.G.; Valente, E.G.; Magee, T.R.; Bou-Gharios, G.; Rajfer, J.; Gonzalez-Cadavid, N.F. Effect of nitric oxide on the differentiation of fibroblasts into myofibroblasts in the Peyronie’s fibrotic plaque and in its rat model. Nitric Oxide 2002, 7, 262–276. [Google Scholar] [CrossRef]
- Gonzalez-Cadavid, N.F.; Rajfer, J. Treatment of Peyronie’s disease with PDE5 inhibitors: An antifibrotic strategy. Nat. Rev. Urol. 2010, 7, 215–221. [Google Scholar] [CrossRef]
- Ilg, M.M.; Mateus, M.; Stebbeds, W.J.; Milenkovic, U.; Christopher, N.; Muneer, A.; Albersen, M.; Ralph, D.J.; Cellek, S. Antifibrotic synergy between phosphodiesterase type 5 inhibitors and selective oestrogen receptor modulators in Peyronie’s Disease models. Eur. Urol. 2019, 75, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Ilg, M.M.; Stafford, S.J.; Mateus, M.; Bustin, S.A.; Carpenter, M.J.; Muneer, A.; Bivalacqua, T.J.; Ralph, D.J.; Cellek, S. Phosphodiesterase type 5 inhibitors and selective estrogen receptor modulators can prevent but not reverse myofibroblast transformation in Peyronie’s Disease. J. Sex. Med. 2020, 17, 1848–1864. [Google Scholar] [CrossRef]
- Mateus, M.; Ilg, M.M.; Stebbeds, W.J.; Christopher, N.; Muneer, A.; Ralph, D.J.; Cellek, S. Understanding the role of adenosine receptors in the myofibroblast transformation in Peyronie’s Disease. J. Sex. Med. 2018, 15, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Qin, F.; Jiuhong, J. Molecular mechanisms and current pharmacotherapy of Peyronie’s Disease: A review. Front. Pharmacol. 2021, 12, 643641. [Google Scholar] [CrossRef]
- Wespes, E.; Goes, P.M.; Schiffmann, S.; Deprierreux, M.; Vanderhaeghen, J.; Schulman, C.C. Computerized analysis of smooth muscle fibers in potent and impotent patients. J. Urol. 1991, 146, 1015–1017. [Google Scholar] [CrossRef]
- Ryu, J.K.; Song, S.U.; Choi, H.K.; Seong, D.H.; Yoon, S.M.; Kim, S.J.; Suh, J.K. Plasma transforming growth factor ß1 levels in patients with erectile dysfunction. Asian J. Androl. 2004, 6, 349–353. [Google Scholar]
- Moreland, R.B. Is there a role of hypoxemia in penile fibrosis: A viewpoint presented to the Society for the Study of Impotence. Int. J. Impot. Res. (IJIR) 1998, 10, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Montorsi, F.; Brock, G.; Stolzenburg, J.U.; Mulhall, J.; Moncada, I.; Patel, H.R.; Chevallier, D.; Krajka, K.; Henneges, C.; Dickson, R.; et al. Effects of tadalafil treatment on erectile function recovery following bilateral nerve-sparing radical prostatectomy: A randomised placebo-controlled study (REACTT). Eur. Urol. 2014, 65, 587–596. [Google Scholar] [CrossRef]
- Osmonov, D.K.; Jünemann, K.P.; Bannowsky, A. The Kiel Concept of long-term administration of daily low-dose sildenafil initiated in the immediate post-prostatectomy period: Evaluation and comparison with the international literature on penile rehabilitation. Sex. Med. Rev. 2017, 5, 387–392. [Google Scholar] [CrossRef]
- Bannowsky, A.; Osmonov, D.K.; Jünemann, K.P.; Ückert, S. Penile rehabilitation in the long term follow up—How Important is the preservation of nocturnal penile tumescence with daily low-dose sildenafil 6 weeks after nerve sparing radical prostatectomy? J. Sex. Med. 2018, 16 (Suppl. 1), S80. [Google Scholar] [CrossRef]
- Bannowsky, A.; Märker, V.; Osmonov, D.; Ückert, S. Vardenafil on a daily basis for rehabilitation of erectile function after nerve-sparing radical prostatectomy—Is it a matter of the dose in the longer follow-up of 2 years? J. Sex. Med. 2020, 17 (Suppl. 2), S179–S180. [Google Scholar] [CrossRef]
- McGaraughty, S.; Davis-Taber, R.A.; Zhu, C.Z.; Cole, T.B.; Nikkel, A.L.; Chhaya, M.; Doyle, K.J.; Olson, L.M.; Preston, G.M.; Grinnell, C.M.; et al. Targeting anti-TGF-beta therapy to fibrotic kidneys with a dual specificity antibody approach. J. Am. Soc. Nephrol. 2017, 28, 3616–3626. [Google Scholar] [CrossRef] [Green Version]
- Miyajima, A.; Chen, J.; Lawrence, C.; Ledbetter, S.; Soslow, R.A.; Stern, J.; Jha, S.; Pigato, J.; Lemer, M.L.; Poppas, D.P.; et al. Antibody to transforming growth factor β ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int. 2000, 58, 2301–2313. [Google Scholar] [CrossRef] [Green Version]
- Teicher, B.A. TGF β-directed therapeutics: 2020. Pharmacol. Ther. 2021, 217, 107666. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGF β signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lu, H.; Xie, S.; Wu, C.; Guo, Y.; Xiao, Y.; Zheng, S.; Zhu, H.; Zhang, Y.; Ba, Y. Resveratrol suppresses the myofibroblastic phenotype and fibrosis formation in kidneys via proliferation-related signaling pathways. Br. J. Pharmacol. 2019, 176, 4745–4759. [Google Scholar] [CrossRef]
- Zhao, J.L.; Zhang, T.; Shao, X.; Zhu, J.J.; Guo, M.Z. Curcumin ameliorates peritoneal fibrosis via inhibition of transforming growth factor-activated kinase1 (TAK1) pathway in a rat model of peritoneal dialysis. BMC Complement. Altern. Med. 2019, 19, 280. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ming, Y.; Jia, H.; Wang, G. Poricoic acid A suppresses TGF β1-induced renal fibrosis and proliferation via the PDGF-C, Smad 3 and MAPK pathways. Exp. Ther. Med. 2021, 21, 289. [Google Scholar] [CrossRef]
- Wang, M.; Chen, D.Q.; Chen, L.; Cao, G.; Zhao, H.; Liu, D.; Vaziri, N.D.; Guo, Y.; Zhao, Y.Y. Novel inhibitors of the cellular renin-angiotensin system components, poricoic acids, target Smad 3 phosphorylation and Wnt/β-catenin pathway against renal fibrosis. Br. J. Pharmacol. 2018, 175, 2689–2708. [Google Scholar] [CrossRef]
- Zhiqiang, W.; Juan, C.; Xu, Z.; Di, Y.; Deyu, X.; Guoyuan, L. EPA attenuates epithelial-mesenchymal transition and fibrosis through the TGF β1/Smad 3/ILK pathway in renal tubular epithelial HK-2 cells by up-regulating miR-541. Int. J. Clin. Exp. Pathol. 2019, 12, 2516–2525. [Google Scholar]
- Park, J.H.; Park, B.; Park, K.K. Suppression of hepatic epithelial-to-mesenchymal transition by melittin via blocking of TGF β/Smad and MAPK-JNK signaling pathways. Toxins 2017, 9, 138. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahardjo, H.E.; Märker, V.; Tsikas, D.; Kuczyk, M.A.; Ückert, S.; Bannowsky, A. Fibrotic Diseases of the Human Urinary and Genital Tract: Current Understanding and Potential Strategies for Treatment. J. Clin. Med. 2023, 12, 4770. https://doi.org/10.3390/jcm12144770
Rahardjo HE, Märker V, Tsikas D, Kuczyk MA, Ückert S, Bannowsky A. Fibrotic Diseases of the Human Urinary and Genital Tract: Current Understanding and Potential Strategies for Treatment. Journal of Clinical Medicine. 2023; 12(14):4770. https://doi.org/10.3390/jcm12144770
Chicago/Turabian StyleRahardjo, Harrina E., Viktoria Märker, Dimitrios Tsikas, Markus A. Kuczyk, Stefan Ückert, and Andreas Bannowsky. 2023. "Fibrotic Diseases of the Human Urinary and Genital Tract: Current Understanding and Potential Strategies for Treatment" Journal of Clinical Medicine 12, no. 14: 4770. https://doi.org/10.3390/jcm12144770