
Acute Encephalopathy Caused by Inherited Metabolic Diseases
Abstract
:1. Introduction
2. Methods
3. Prevalence of Inherited Metabolic Diseases
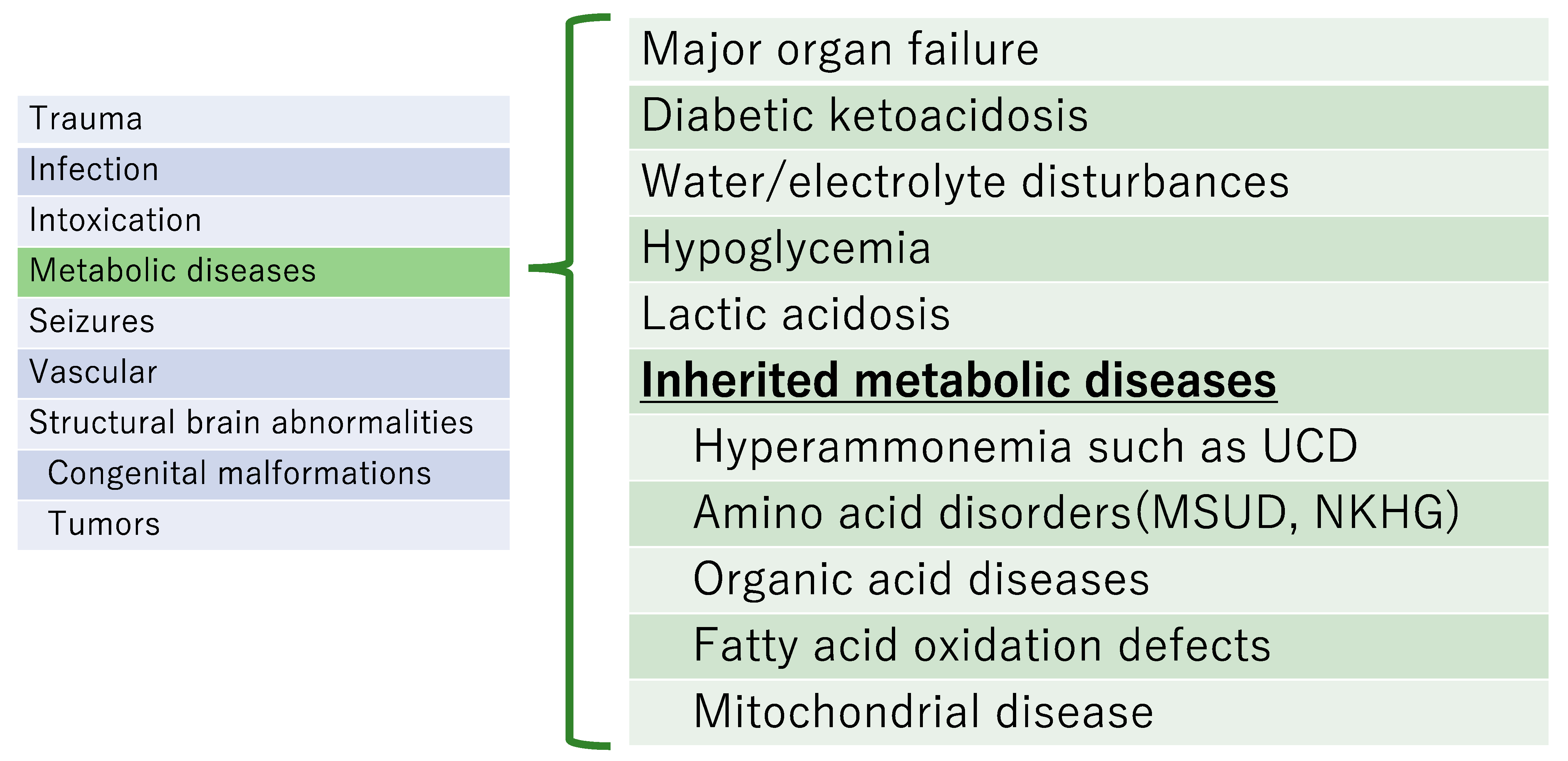
4. Diagnosis When an Inherited Metabolic Disease Is Clinically Suspected (Figure 1)
- Often occur with little warning in a previously healthy infant or child;
- Could be missed because the early signs may be mistaken as a behavior disorder;
- Often progress rapidly, and may fluctuate markedly;
- Usually show no focal neurologic deficits;
- Can result in encephalopathy, which can be triggered by fever.
- a.
- Sudden deterioration of general condition after infection or fasting;
- b.
- Specific facial and skin findings, body odor, and urine odor;
- c.
- Hypercapnia and/or respiratory disturbances associated with metabolic acidosis;
- d.
- Growth retardation and/or mental retardation;
- e.
- Cardiomyopathy;
- f.
- Hepatosplenomegaly (hepatomegaly without splenomegaly, splenomegaly without findings of portal hypertension);
- g.
- Presence of poorly related multisystemic symptoms;
- h.
- Specific imaging findings;
- i.
- Family history of inherited metabolic diseases.

5. Typical Inherited Metabolic Diseases That Cause Acute Encephalopathy (Figure 2)

5.1. Urea Cycle Disorders
5.2. Amino Acid Metabolism Disorders
5.3. Fatty Acid Metabolism Disorders
5.4. Mitochondrial Disease [8,9,10]
6. Examinations to Perform When Inherited Metabolic Diseases Are Suspected
6.1. Laboratory Tests Specific for Each Disease
6.1.1. Urea Cycle Disorders
- (1)
- Amino acid analysis in blood and urine
- (2)
- Urine organic acid analysis
- (3)
- Enzyme diagnosis or genetic analysis
- (4)
- Tandem mass test
6.1.2. Amino Acid Metabolism Disorders
MSUD (Leucine Encephalopathy)
6.1.3. Fatty Acid Metabolism Disorders
- (1)
- Acylcarnitine analysis
- (2)
- Urine organic acid analysis
- (3)
- Enzymatic diagnosis
- (4)
- In vitro probe assay (evaluation of β-oxidation capacity)
- (5)
- Immunoblotting
- (6)
- Genetic analysis
6.1.4. Mitochondrial Disease [15,16,17,18]
- (1)
- Clinical findings
- (2)
- Imaging findings
- (3)
- Hyperlactatemia
- (4)
- Enzyme analysis
- (5)
- Pathological examination
- (6)
- Genetic analysis
7. Treatment for Inherited Metabolic Diseases
7.1. When Urea Cycle Disorders Must Be Suspected
- (1)
- Check ammonia level
- (2)
- Blood glucose control
- (3)
- Protection of central nervous system
- (4)
- Administration of carglumic acid (Carbaglu®)
- (5)
- Administration of sodium phenylbutyrate (Buphenyl®), sodium benzoate, and L-arginine
- (6)
- Blood purification therapy
- (7)
- Nutritional management
- (8)
- Administration of vitamins
7.2. When Organic Acid Metabolic Diseases or Fatty Acid Metabolic Diseases Are Suspected [12]
- (1)
- First, stabilization of the patient’s ABC will be needed;
- (2)
- Blood glucose control
- (3)
- Administration of alkalizing agents
- (4)
- Hemodialysis (continuous hemodialysis [CHD] or continuous hemodiafiltration dialysis [CHDF])
- (5)
- Nutritional management
- (6)
- Administration of vitamins
7.3. When Mitochondrial Disease Is Suspected [4]
- (1)
- First, stabilization of the patient’s ABC;
- (2)
- Blood glucose control
- (3)
- Administration of alkalizing agents
- (4)
- Blood purification therapy
- (5)
- Vitamin cocktail
- (6)
- Nutritional management
- (1)
- Taurine treatment for m.3243A>G mutation (tRNA modification disorder) [27]
- (2)
- Arginine treatment for stroke prevention in MELAS [28]
8. Inherited Metabolic Diseases Causing Acute Encephalopathy and Other Related Disorders: Recent Findings
8.1. MSUD, Liver Transplantation [31]
8.2. Homocystinuria Type III (5,10-Methylenetetrahydrofolate Reductase Deficiency) [34] (Figure 7), Bethain, Newborn Screening
8.3. ECHS-1, Valine-Restricted Diet
8.4. Thiamine Metabolism Dysfunction Syndrome 2 (Biotin-Thiamine-Responsive Basal Ganglia Disease), Thiamine Treatment
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Imataka, G.; Kuwashima, S.; Yoshihara, S. A comprehensive review of pediatric acute encephalopathy. J. Clin. Med. 2022, 11, 5921. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.T.R. (Ed.) A Clinical Guide to Inherited Metabolic Diseases, 3rd ed.; Cambridge University Press: Cambridge, UK, 2006; pp. 53–89. [Google Scholar]
- Applegarth, D.A.; Toone, J.R.; Lowry, R.B. Incidence of inborn errors of metabolism in British Columbia, 1969–1996. Pediatrics 2000, 105, e10. [Google Scholar] [CrossRef] [PubMed]
- The Japanese Society of Child Neurology. Guideline on the Diagnosis and Treatment of Acute Encephalopathy in Childhood; Shindan to Chiryo Sha: Tokyo, Japan, 2016; Volume 2016. (In Japanese) [Google Scholar]
- Hoffmann, G.F.; Nyhan, W.L.; Zschocke, J.; Kahler, S.G.; Mayatepek, E. (Eds.) Inherited Metabolic Diseases, 1st ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2002; p. 34. [Google Scholar]
- Brusilow, S.W.; Horwich, A.L. Urea cycle enzymes. In The Metabolic and Molecular Basis of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 1909–1963. [Google Scholar]
- Chuang, D.T.; Shih, V.E. Maple syrup urine disease. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 1971–2005. [Google Scholar]
- Skladal, D.; Halliday, J.; Thorburn, D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Blok, R.B.; Dahl, H.H.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef]
- Pavlakis, S.G.; Phillips, P.C.; Dimauro, S.; De Vivo, D.C.; Rowland, L.P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol. 1984, 16, 481–488. [Google Scholar] [CrossRef]
- Ogawa, E.; Fushimi, T.; Ogawa-Tominaga, M.; Shimura, M.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; et al. Mortality of Japanese patients with Leigh syndrome: Effects of age at onset and genetic diagnosis. J. Inherit. Metab. Dis. 2020, 43, 819–826. [Google Scholar] [CrossRef]
- Zschochke, J.; Hoffmann, G.F. (Eds.) Essential laboratory tests. In Vade Mecum Metabolicum: Diagnosis and Treatment of Inborn Errors of Metabolism, Feuerbach; Thieme Publishing Group: New York, NY, USA, 2021. [Google Scholar]
- Mitsubuchi, H.; Owada, M.; Endo, F. Markers associated with inborn errors of branched-chain amino acids and their relevance to upper levels of intake in healthy people: An implication from clinical and molecular investigations on maple syrup urine disease. J. Nutr. 2005, 135, 1565S–1570S. [Google Scholar] [CrossRef]
- Andrew, A.M.M.; Ute, S. Disorders of mitochondrial fatty acid oxidation and related metabolic pathways. In Inborn Metabolic Disease, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 201–216. [Google Scholar]
- Bernier, F.P.; Boneh, A.; Dennett, X.; Chow, C.W.; Cleary, M.A.; Thorburn, D.R. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002, 59, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; Crawford, M.; Cleary, M.A.; Dahl, H.H.; Dennett, X.; Thorburn, D.R. Respiratory chain complex I deficiency: An underdiagnosed energy generation disorder. Neurology 1999, 52, 1255–1264. [Google Scholar] [CrossRef]
- Thorburn, D.R.; Chow, C.W.; Kirby, D.M. Respiratory chain enzyme analysis in muscle and liver. Mitochondrion 2004, 4, 363–375. [Google Scholar] [CrossRef]
- Ohtake, A.; Murayama, K.; Mori, M.; Harashima, H.; Yamazaki, T.; Tamaru, S.; Yamashita, Y.; Kishita, Y.; Nakachi, Y.; Kohda, M.; et al. Diagnosis and molecular basis of mitochondrial respiratory chain disorders: Exome sequencing for disease gene identification. Biochim. Biophys. Acta 2014, 1840, 1355–1359. [Google Scholar] [CrossRef]
- Häberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef]
- Ah Mew, N.; McCarter, R.; Daikhin, Y.; Lichter-Konecki, U.; Nissim, I.; Yudkoff, M.; Tuchman, M. Augmenting ureagenesis in patients with partial carbamyl phosphate synthetase 1 deficiency with N-carbamyl-L-glutamate. J. Pediatr. 2014, 165, 401–403.e3. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, Y.; Shimura, M.; Ogawa-Tominaga, M.; Ebihara, T.; Kinouchi, Y.; Isozaki, K.; Matsuhashi, T.; Tajika, M.; Fushimi, T.; Ichimoto, K.; et al. Therapeutic effect of N-carbamylglutamate in CPS1 deficiency. Mol. Genet. Metab. Rep. 2020, 24, 100622. [Google Scholar] [CrossRef] [PubMed]
- Kido, J.; Nakamura, K.; Mitsubuchi, H.; Ohura, T.; Takayanagi, M.; Matsuo, M.; Yoshino, M.; Shigematsu, Y.; Yorifuji, T.; Kasahara, M.; et al. Long-term outcome and intervention of urea cycle disorders in Japan. J. Inherit. Metab. Dis. 2012, 35, 777–785. [Google Scholar] [CrossRef]
- Murayama, K.; Ohtake, A. Children’s toxicology from bench to bed—Liver Injury (4): Mitochondrial Respiratory Chain Disorder and Liver Disease in Children. J. Toxicol. Sci. 2009, 34 (Suppl. S2), SP237–SP243. [Google Scholar] [CrossRef]
- Omata, T.; Fujii, K.; Takanashi, J.; Murayama, K.; Takayanagi, M.; Muta, K.; Kodama, K.; Iida, Y.; Watanabe, Y.; Shimojo, N. Drugs indicated for mitochondrial dysfunction as treatments for acute encephalopathy with onset of febrile convulsive status epileptics. J. Neurol. Sci. 2016, 360, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, M.R.; Hörster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Sechi, G.P.; Bardanzellu, F.; Pintus, M.C.; Sechi, M.M.; Marcialis, M.A.; Fanos, V. Thiamine as a possible neuroprotective strategy in neonatal hypoxic-ischemic encephalopathy. Antioxidants 2022, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, Y.; Hagiwara, H.; Nishimatsu, S.I.; Hirakawa, A.; Kamimura, N.; Ohtsubo, H.; Fukai, Y.; Murakami, T.; Koga, Y.; Goto, Y.I.; et al. Taurine supplementation for prevention of stroke-like episodes in MELAS: A multicentre, open-label, 52-week phase III trial. J. Neurol. Neurosurg. Psychiatry 2019, 90, 529–536. [Google Scholar] [CrossRef]
- Koga, Y.; Povalko, N.; Inoue, E.; Nakamura, H.; Ishii, A.; Suzuki, Y.; Yoneda, M.; Kanda, F.; Kubota, M.; Okada, H.; et al. Therapeutic regimen of L-arginine for patients with MELAS: 9-year, prospective, multicentre, clinical research integrating the data from two 2-year clinical trials with 7-year follow-up. Neurology 2018, 90 (Suppl. S15), S54.008. [Google Scholar]
- Murayama, K.; Shimura, M.; Liu, Z.; Okazaki, Y.; Ohtake, A. Recent topics: The diagnosis, molecular genesis, and treatment of mitochondrial diseases. J. Hum. Genet. 2019, 64, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Distelmaier, F.; Haack, T.B.; Wortmann, S.B.; Mayr, J.A.; Prokisch, H. Treatable mitochondrial diseases: Cofactor metabolism and beyond. Brain 2017, 140, e11. [Google Scholar] [CrossRef] [PubMed]
- Suryawan, A.; Hawes, J.W.; Harris, R.A.; Shimomura, Y.; Jenkins, A.E.; Hutson, S.M. A molecular model of human branched-chain amino acid metabolism. Am. J. Clin. Nutr. 1998, 68, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Frazier, D.M.; Allgeier, C.; Homer, C.; Marriage, B.J.; Ogata, B.; Rohr, F.; Splett, P.L.; Stembridge, A.; Singh, R.H. Nutrition management guideline for maple syrup urine disease: An evidence- and consensus-based approach. Mol. Genet. Metab. 2014, 112, 210–217. [Google Scholar] [CrossRef]
- Mazariegos, G.V.; Morton, D.H.; Sindhi, R.; Soltys, K.; Nayyar, N.; Bond, G.; Shellmer, D.; Shneider, B.; Vockley, J.; Strauss, K.A. Liver transplantation for classical maple syrup urine disease: Long-term follow-up in 37 patients and comparative United Network for Organ Sharing experience. J. Pediatr. 2012, 160, 116–121.e1. [Google Scholar] [CrossRef]
- Huemer, M.; Kožich, V.; Rinaldo, P.; Baumgartner, M.R.; Merinero, B.; Pasquini, E.; Ribes, A.; Blom, H.J. Newborn screening for homocystinurias and methylation disorders: Systematic review and proposed guidelines. J. Inherit. Metab. Dis. 2015, 38, 1007–1019. [Google Scholar] [CrossRef]
- Diekman, E.F.; de Koning, T.J.; Verhoeven-Duif, N.M.; Rovers, M.M.; van Hasselt, P.M. Survival and psychomotor development with early betaine treatment in patients with severe methylenetetrahydrofolate reductase deficiency. JAMA Neurol. 2014, 71, 188–194. [Google Scholar] [CrossRef]
- Huemer, M.; Diodato, D.; Schwahn, B.; Schiff, M.; Bandeira, A.; Benoist, J.F.; Burlina, A.; Cerone, R.; Couce, M.L.; Garcia-Cazorla, A.; et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J. Inherit. Metab. Dis. 2017, 40, 21–48. [Google Scholar] [CrossRef]
- Haack, T.B.; Jackson, C.B.; Murayama, K.; Kremer, L.S.; Schaller, A.; Kotzaeridou, U.; de Vries, M.C.; Schottmann, G.; Santra, S.; Büchner, B.; et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann. Clin. Transl. Neurol. 2015, 2, 492–509. [Google Scholar] [CrossRef]
- Peters, H.; Buck, N.; Wanders, R.; Ruiter, J.; Waterham, H.; Koster, J.; Yaplito-Lee, J.; Ferdinandusse, S.; Pitt, J. ECHS1 mutations in Leigh disease: A new inborn error of metabolism affecting valine metabolism. Brain 2014, 137, 2903–2908. [Google Scholar] [CrossRef]
- Peters, H.; Ferdinandusse, S.; Ruiter, J.P.; Wanders, R.J.; Boneh, A.; Pitt, J. Metabolite studies in HIBCH and ECHS1 defects: Implications for screening. Mol. Genet. Metab. 2015, 115, 168–173. [Google Scholar] [CrossRef]
- Shayota, B.J.; Soler-Alfonso, C.; Bekheirnia, M.R.; Mizerik, E.; Boyer, S.W.; Xiao, R.; Yang, Y.; Elsea, S.H.; Scaglia, F. Case report and novel treatment of an autosomal recessive Leigh syndrome caused by short-chain enoyl-CoA hydratase deficiency. Am. J. Med. Genet. Part A 2019, 179, 803–807. [Google Scholar] [CrossRef]
- Kono, S.; Miyajima, H.; Yoshida, K.; Togawa, A.; Shirakawa, K.; Suzuki, H. Mutations in a thiamine-transporter gene and Wernicke’s-like encephalopathy. N. Engl. J. Med. 2009, 360, 1792–1794. [Google Scholar] [CrossRef]
- Alfadhel, M.; Almuntashri, M.; Jadah, R.H.; Bashiri, F.A.; Al Rifai, M.T.; Al Shalaan, H.; Al Balwi, M.; Al Rumayan, A.; Eyaid, W.; Al-Twaijri, W. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: A retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J. Rare Dis. 2013, 8, 83. [Google Scholar] [CrossRef]
- Sechi, E.; Addis, A.; Fadda, G.; Minafra, L.; Bravatà, V.; Sechi, G. Teaching NeuroImages: Subacute encephalopathy in a young woman with THTR2 gene mutation. Neurology 2015, 85, e108–e109. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UCD | MSUD | OAD | FAOD | Mitochondria Disease | |
|---|---|---|---|---|---|
| Metabolic acidosis | ± | ± | +++ | ± | ++ |
| Blood glucose | → | → to ↓ | ↓↓ | ↓↓↓ | → to ↓ |
| Ketone | → | ↑↑ | ↑↑ | ↓ | → |
| Ammonia | ↑↑↑ | → | ↑↑ | ↑ | → |
| Lactate | → | → | ↑ | ± | ↑↑↑ |
| Liver dysfunction | → | → | → | ↑↑ | → |
| Free carnitine | → | → | ↓↓↓ | ↓↓ | → |
| Amino acid analysis | Specific finding | ↑ BCAA | Specific finding | No finding | ↑ Alanine |
| Urine organic acid analysis | Specific finding | Metabolites of BCAA | Specific finding | Specific finding | Specific finding |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugiyama, Y.; Murayama, K. Acute Encephalopathy Caused by Inherited Metabolic Diseases. J. Clin. Med. 2023, 12, 3797. https://doi.org/10.3390/jcm12113797
Sugiyama Y, Murayama K. Acute Encephalopathy Caused by Inherited Metabolic Diseases. Journal of Clinical Medicine. 2023; 12(11):3797. https://doi.org/10.3390/jcm12113797
Chicago/Turabian StyleSugiyama, Yohei, and Kei Murayama. 2023. "Acute Encephalopathy Caused by Inherited Metabolic Diseases" Journal of Clinical Medicine 12, no. 11: 3797. https://doi.org/10.3390/jcm12113797