
Clinical Manifestations, Current and Future Therapy, and Long-Term Outcomes in Congenital Thrombotic Thrombocytopenic Purpura

Abstract
:1. Introduction
2. Diagnostic Flow for cTTP
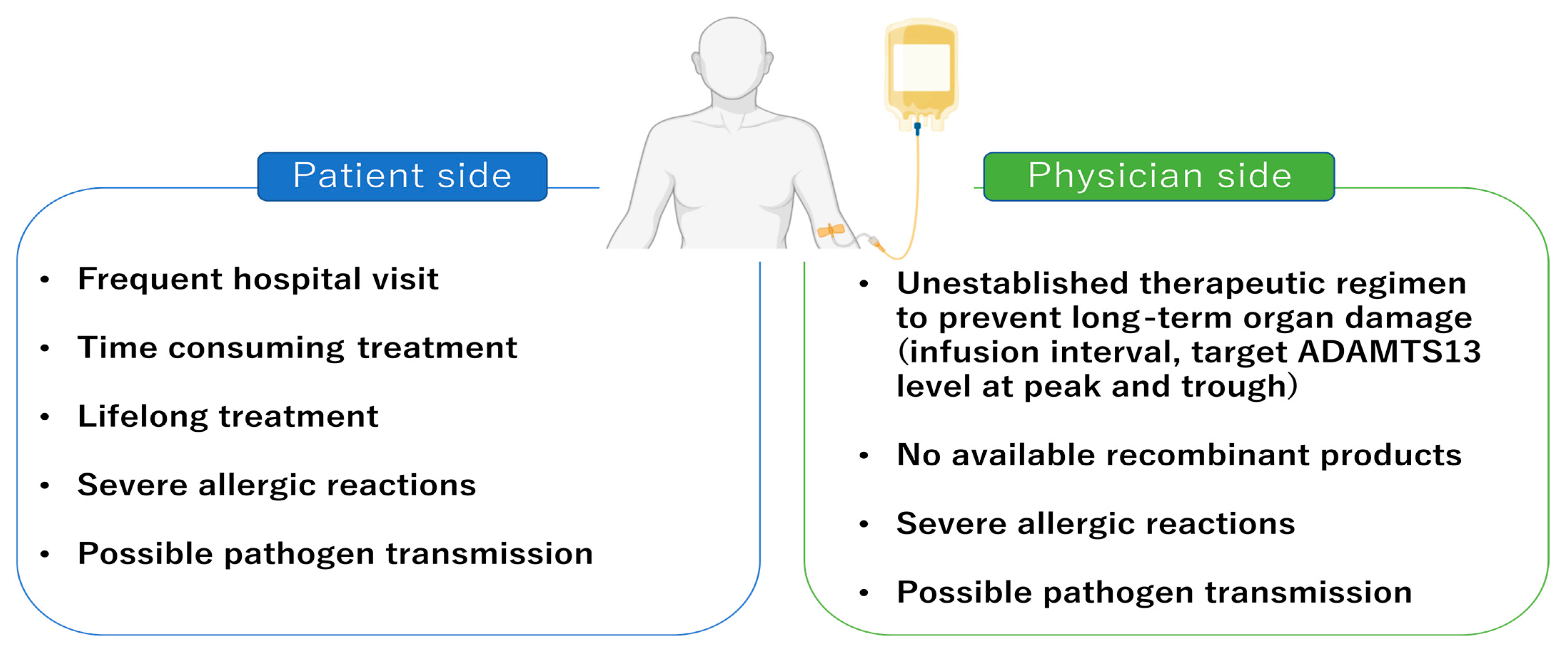
3. Current Treatments for Patients with cTTP
4. Long-Term Organ Damage and Mortality in Patients with cTTP
5. Determinants of FFP-Dependent or Asymptomatic Phenotypes in cTTP
6. Emerging Novel Therapies Will Improve the QOL and Long-Term Outcomes of Patients with cTTP
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sadler, J.E. Pathophysiology of thrombotic thrombocytopenic purpura. Blood 2017, 130, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Kremer Hovinga, J.A.; Coppo, P.; Lammle, B.; Moake, J.L.; Miyata, T.; Vanhoorelbeke, K. Thrombotic thrombocytopenic purpura. Nat. Rev. Dis. Primers 2017, 3, 17020. [Google Scholar] [CrossRef] [PubMed]
- Joly, B.S.; Coppo, P.; Veyradier, A. Thrombotic thrombocytopenic purpura. Blood 2017, 129, 2836–2846. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.L.; Vesely, S.K.; Cataland, S.R.; Coppo, P.; Geldziler, B.; Iorio, A.; Matsumoto, M.; Mustafa, R.A.; Pai, M.; Rock, G.; et al. ISTH guidelines for the diagnosis of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2020, 18, 2486–2495. [Google Scholar] [CrossRef] [PubMed]
- South, K.; Lane, D.A. ADAMTS-13 and von Willebrand factor: A dynamic duo. J. Thromb. Haemost. 2018, 16, 6–18. [Google Scholar] [CrossRef]
- Sarig, G. ADAMTS-13 in the diagnosis and management of thrombotic microangiopathies. Rambam. Maimonides. Med. J. 2014, 5, e0026. [Google Scholar] [CrossRef]
- Patschan, D.; Witzke, O.; Dührsen, U.; Erbel, R.; Philipp, T.; Herget-Rosenthal, S. Acute myocardial infarction in thrombotic microangiopathies—Clinical characteristics, risk factors and outcome. Nephrol. Dial. Transplant 2006, 21, 1549–1554. [Google Scholar] [CrossRef]
- Nichols, L.; Berg, A.; Rollins-Raval, M.A.; Raval, J.S. Cardiac injury is a common postmortem finding in thrombotic thrombocytopenic purpura patients: Is empiric cardiac monitoring and protection needed? Ther. Apher. Dial. 2015, 19, 87–92. [Google Scholar] [CrossRef]
- Furlan, M.; Robles, R.; Galbusera, M.; Remuzzi, G.; Kyrle, P.A.; Brenner, B.; Krause, M.; Scharrer, I.; Aumann, V.; Mittler, U.; et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N. Engl. J. Med. 1998, 339, 1578–1584. [Google Scholar] [CrossRef]
- Tsai, H.M.; Lian, E.C. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N. Engl. J. Med. 1998, 339, 1585–1594. [Google Scholar] [CrossRef]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001, 413, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chung, D.; Takayama, T.K.; Majerus, E.M.; Sadler, J.E.; Fujikawa, K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J. Biol. Chem. 2001, 276, 41059–41063. [Google Scholar] [CrossRef] [PubMed]
- Kremer Hovinga, J.A.; George, J.N. Hereditary thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2019, 381, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Schulman, I.; Pierce, M.; Lukens, A.; Currimbhoy, Z. Studies on thrombopoiesis. I. A factor in normal human plasma required for platelet production; chronic thrombocytopenia due to its deficiency. Blood 1960, 16, 943–957. [Google Scholar] [CrossRef]
- Upshaw, J.D., Jr. Congenital deficiency of a factor in normal plasma that reverses microangiopathic hemolysis and thrombocytopenia. N. Engl. J. Med. 1978, 298, 1350–1352. [Google Scholar] [CrossRef] [PubMed]
- Rock, G.A.; Shumak, K.H.; Buskard, N.A.; Blanchette, V.S.; Kelton, J.G.; Nair, R.C.; Spasoff, R.A. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N. Engl. J. Med. 1991, 325, 393–397. [Google Scholar] [CrossRef]
- Bell, W.R.; Braine, H.G.; Ness, P.M.; Kickler, T.S. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N. Engl. J. Med. 1991, 325, 398–403. [Google Scholar] [CrossRef]
- Fujimura, Y.; Matsumoto, M.; Isonishi, A.; Yagi, H.; Kokame, K.; Soejima, K.; Murata, M.; Miyata, T. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 283–301. [Google Scholar] [CrossRef]
- Van Dorland, H.A.; Taleghani, M.M.; Sakai, K.; Friedman, K.D.; George, J.N.; Hrachovinova, I.; Knobl, P.N.; von Krogh, A.S.; Schneppenheim, R.; Aebi-Huber, I.; et al. The international hereditary thrombotic thrombocytopenic purpura registry: Key findings at enrollment until 2017. Haematologica 2019, 104, 2107–2115. [Google Scholar] [CrossRef]
- Alwan, F.; Vendramin, C.; Liesner, R.; Clark, A.; Lester, W.; Dutt, T.; Thomas, W.; Gooding, R.; Biss, T.; Watson, H.G.; et al. Characterization and treatment of congenital thrombotic thrombocytopenic purpura. Blood 2019, 133, 1644–1651. [Google Scholar] [CrossRef]
- Joly, B.S.; Boisseau, P.; Roose, E.; Stepanian, A.; Biebuyck, N.; Hogan, J.; Provot, F.; Delmas, Y.; Garrec, C.; Vanhoorelbeke, K.; et al. ADAMTS13 gene mutations influence ADAMTS13 conformation and disease age-onset in the French cohort of upshaw-Schulman syndrome. Thromb. Haemost. 2018, 118, 1902–1917. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, J.J.; Khurana, M. Treatment of thrombotic thrombocytopenic purpura with plasma. N. Engl. J. Med. 1977, 297, 1386–1389. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Fujimura, Y.; Wada, H.; Kokame, K.; Miyakawa, Y.; Ueda, Y.; Higasa, S.; Moriki, T.; Yagi, H.; Miyata, T.; et al. Diagnostic and treatment guidelines for thrombotic thrombocytopenic purpura (TTP) 2017 in Japan. Int. J. Hematol. 2017, 106, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Matsumoto, M.; Matsuyama, T.; Isonishi, A.; Hiura, H.; Fujimura, Y. Novel monoclonal antibody-based enzyme immunoassay for determining plasma levels of ADAMTS13 activity. Transfusion 2006, 46, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Kokame, K.; Matsumoto, M.; Fujimura, Y.; Miyata, T. VWF73, a region from D1596 to R1668 of von Willebrand factor, provides a minimal substrate for ADAMTS-13. Blood 2004, 103, 607–612. [Google Scholar] [CrossRef]
- Kokame, K.; Nobe, Y.; Kokubo, Y.; Okayama, A.; Miyata, T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br. J. Haematol. 2005, 129, 93–100. [Google Scholar] [CrossRef]
- Valsecchi, C.; Mirabet, M.; Mancini, I.; Biganzoli, M.; Schiavone, L.; Faraudo, S.; Mane-Padros, D.; Giles, D.; Serra-Domenech, J.; Blanch, S.; et al. Evaluation of a new, rapid, fully automated assay for the measurement of ADAMTS13 activity. Thromb. Haemost. 2019, 119, 1767–1772. [Google Scholar] [CrossRef]
- Meyer, S.C.; Sulzer, I.; Lämmle, B.; Kremer Hovinga, J.A. Hyperbilirubinemia interferes with ADAMTS-13 activity measurement by FRETS-VWF73 assay: Diagnostic relevance in patients suffering from acute thrombotic microangiopathies. J. Thromb. Haemost. 2007, 5, 866–867. [Google Scholar] [CrossRef]
- Scully, M.; Cataland, S.; Coppo, P.; de la Rubia, J.; Friedman, K.D.; Kremer Hovinga, J.; Lammle, B.; Matsumoto, M.; Pavenski, K.; Sadler, E.; et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J. Thromb. Haemost. 2017, 15, 312–322. [Google Scholar] [CrossRef]
- Lotta, L.A.; Valsecchi, C.; Pontiggia, S.; Mancini, I.; Cannavò, A.; Artoni, A.; Mikovic, D.; Meloni, G.; Peyvandi, F. Measurement and prevalence of circulating ADAMTS13-specific immune complexes in autoimmune thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2014, 12, 329–336. [Google Scholar] [CrossRef]
- Sakai, K.; Hamada, E.; Kokame, K.; Matsumoto, M. Congenital thrombotic thrombocytopenic purpura: Genetics and emerging therapies. Ann. Blood, 2022; 1–14, in press. [Google Scholar]
- Kokame, K.; Matsumoto, M.; Soejima, K.; Yagi, H.; Ishizashi, H.; Funato, M.; Tamai, H.; Konno, M.; Kamide, K.; Kawano, Y.; et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc. Natl. Acad. Sci. USA 2002, 99, 11902–11907. [Google Scholar] [CrossRef] [PubMed]
- Eura, Y.; Kokame, K.; Takafuta, T.; Tanaka, R.; Kobayashi, H.; Ishida, F.; Hisanaga, S.; Matsumoto, M.; Fujimura, Y.; Miyata, T. Candidate gene analysis using genomic quantitative PCR: Identification of ADAMTS13 large deletions in two patients with Upshaw-Schulman syndrome. Mol. Genet. Genomic. Med. 2014, 2, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, K.I.; Meyer, D.; Holcomb, D.D.; Kames, J.; Hamasaki-Katagiri, N.; Katneni, U.K.; Hunt, R.C.; Ibla, J.C.; Kimchi-Sarfaty, C. Synonymous ADAMTS13 variants impact molecular characteristics and contribute to variability in active protein abundance. Blood. Adv. 2022, 6, 5364–5378. [Google Scholar] [CrossRef]
- Zheng, X.L.; Vesely, S.K.; Cataland, S.R.; Coppo, P.; Geldziler, B.; Iorio, A.; Matsumoto, M.; Mustafa, R.A.; Pai, M.; Rock, G.; et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2020, 18, 2496–2502. [Google Scholar] [CrossRef]
- Zheng, X.L.; Vesely, S.K.; Cataland, S.R.; Coppo, P.; Geldziler, B.; Iorio, A.; Matsumoto, M.; Mustafa, R.A.; Pai, M.; Rock, G.; et al. Good practice statements (GPS) for the clinical care of patients with thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2020, 18, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Tarasco, E.; Butikofer, L.; Friedman, K.D.; George, J.N.; Hrachovinova, I.; Knobl, P.N.; Matsumoto, M.; von Krogh, A.S.; Aebi-Huber, I.; Cermakova, Z.; et al. Annual incidence and severity of acute episodes in hereditary thrombotic thrombocytopenic purpura. Blood 2021, 137, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Yoshioka, A.; Park, Y.D.; Ishizashi, H.; Konno, M.; Funato, M.; Matsui, T.; Titani, K.; Yagi, H.; Matsumoto, M.; et al. Upshaw-Schulman syndrome revisited: A concept of congenital thrombotic thrombocytopenic purpura. Int. J. Hematol. 2001, 74, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Fujimura, Y.; Miyata, T.; Isonishi, A.; Kokame, K.; Matsumoto, M. Current prophylactic plasma infusion protocols do not adequately prevent long-term cumulative organ damage in the Japanese congenital thrombotic thrombocytopenic purpura cohort. Br. J. Haematol. 2021, 194, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Mahoney, D.H. Successful treatment of congenital TTP with a novel approach using plasma-derived factor VIII. J. Pediatr. Hematol. Oncol. 2013, 35, 551–553. [Google Scholar] [CrossRef]
- Scully, M.; Gattens, M.; Khair, K.; Liesner, R. The use of intermediate purity factor VIII concentrate BPL 8Y as prophylaxis and treatment in congenital thrombotic thrombocytopenic purpura. Br. J. Haematol. 2006, 135, 101–104. [Google Scholar] [CrossRef]
- McGonigle, A.M.; Patel, E.U.; Waters, K.M.; Moliterno, A.R.; Thoman, S.K.; Vozniak, S.O.; Ness, P.M.; King, K.E.; Tobian, A.A.R.; Lokhandwala, P.M. Solvent detergent treated pooled plasma and reduction of allergic transfusion reactions. Transfusion 2020, 60, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, D.; Snyder, E.L.; Tormey, C.A. Two approaches to the clinical dilemma of treating TTP with therapeutic plasma exchange in patients with a history of anaphylactic reactions to plasma. J. Clin. Apher. 2017, 32, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Garraud, O.; Malot, S.; Herbrecht, R.; Ojeda-Uribe, M.; Lin, J.S.; Veyradier, A.; Payrat, J.M.; Liu, K.; Corash, L.; Coppo, P. Amotosalen-inactivated fresh frozen plasma is comparable to solvent-detergent inactivated plasma to treat thrombotic thrombocytopenic purpura. Transfus. Apher. Sci. 2019, 58, 102665. [Google Scholar] [CrossRef] [PubMed]
- Boothby, A.; Mazepa, M. Caplacizumab for congenital thrombotic thrombocytopenic purpura. Am. J. Hematol. 2022, 97, e420–e421. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knobl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Borogovac, A.; Tarasco, E.; Kremer Hovinga, J.A.; Friedman, K.D.; Asch, A.S.; Vesely, S.K.; Prodan, C.; Terrell, D.R.; George, J.N. Prevalence of neuropsychiatric symptoms and stroke in patients with hereditary thrombotic thrombocytopenic purpura. Blood 2022, 140, 785–789. [Google Scholar] [CrossRef]
- Borogovac, A.; Reese, J.A.; Gupta, S.; George, J.N. Morbidities and mortality in patients with hereditary thrombotic thrombocytopenic purpura. Blood Adv. 2022, 6, 750–759. [Google Scholar] [CrossRef]
- Ocak, G.; Roest, M.; Verhaar, M.C.; Rookmaaker, M.B.; Blankestijn, P.J.; Bos, W.J.W.; Fijnheer, R.; Péquériaux, N.C.; Dekker, F.W. Von Willebrand factor, ADAMTS13 and mortality in dialysis patients. BMC Nephrol. 2021, 22, 222. [Google Scholar] [CrossRef]
- Green, D.; Roberts, P.R.; New, D.I.; Kalra, P.A. Sudden cardiac death in hemodialysis patients: An in-depth review. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2011, 57, 921–929. [Google Scholar] [CrossRef]
- Makar, M.S.; Pun, P.H. Sudden cardiac death among hemodialysis patients. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2017, 69, 684–695. [Google Scholar] [CrossRef]
- Itami, H.; Hara, S.; Matsumoto, M.; Imamura, S.; Kanai, R.; Nishiyama, K.; Ishimura, M.; Ohga, S.; Yoshida, M.; Tanaka, R.; et al. Complement activation associated with ADAMTS13 deficiency may contribute to the characteristic glomerular manifestations in Upshaw-Schulman syndrome. Thromb. Res. 2018, 170, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Fujimura, Y.; Nagata, Y.; Higasa, S.; Moriyama, M.; Isonishi, A.; Konno, M.; Kajiwara, M.; Ogawa, Y.; Kaburagi, S.; et al. Success and limitations of plasma treatment in pregnant women with congenital thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2020, 18, 2929–2941. [Google Scholar] [CrossRef] [PubMed]
- Von Krogh, A.S.; Kremer Hovinga, J.A.; Tjonnfjord, G.E.; Ringen, I.M.; Lammle, B.; Waage, A.; Quist-Paulsen, P. The impact of congenital thrombotic thrombocytopenic purpura on pregnancy complications. Thromb. Haemost. 2014, 111, 1180–1183. [Google Scholar] [CrossRef]
- Schiviz, A.; Wuersch, K.; Piskernik, C.; Dietrich, B.; Hoellriegl, W.; Rottensteiner, H.; Scheiflinger, F.; Schwarz, H.P.; Muchitsch, E.M. A new mouse model mimicking thrombotic thrombocytopenic purpura: Correction of symptoms by recombinant human ADAMTS13. Blood 2012, 119, 6128–6135. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Knobl, P.; Kentouche, K.; Rice, L.; Windyga, J.; Schneppenheim, R.; Kremer Hovinga, J.A.; Kajiwara, M.; Fujimura, Y.; Maggiore, C.; et al. Recombinant ADAMTS-13: First-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood 2017, 130, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- Sarode, R. Recombinant ADAMTS-13: Goodbye, allergic reactions! Blood 2017, 130, 2045–2046. [Google Scholar] [CrossRef] [PubMed]
- Asmis, L.M.; Serra, A.; Krafft, A.; Licht, A.; Leisinger, E.; Henschkowski-Serra, J.; Ganter, M.T.; Hauptmann, S.; Tinguely, M.; Kremer Hovinga, J.A. Recombinant ADAMTS13 for hereditary thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2022, 387, 2356–2361. [Google Scholar] [CrossRef]
- Stubbs, M.J.; Kendall, G.; Scully, M. Recombinant ADAMTS13 in severe neonatal thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2022, 387, 2391–2392. [Google Scholar] [CrossRef]
- Chauhan, A.K.; Motto, D.G.; Lamb, C.B.; Bergmeier, W.; Dockal, M.; Plaimauer, B.; Scheiflinger, F.; Ginsburg, D.; Wagner, D.D. Systemic antithrombotic effects of ADAMTS13. J. Exp. Med. 2006, 203, 767–776. [Google Scholar] [CrossRef]
- Xu, H.; Cao, Y.; Yang, X.; Cai, P.; Kang, L.; Zhu, X.; Luo, H.; Lu, L.; Wei, L.; Bai, X.; et al. ADAMTS13 controls vascular remodeling by modifying VWF reactivity during stroke recovery. Blood 2017, 130, 11–22. [Google Scholar] [CrossRef]
- South, K.; Saleh, O.; Lemarchand, E.; Coutts, G.; Smith, C.J.; Schiessl, I.; Allan, S.M. Robust thrombolytic and anti-inflammatory action of a constitutively active ADAMTS13 variant in murine stroke models. Blood 2022, 139, 1575–1587. [Google Scholar] [CrossRef]
- Zhou, S.; Guo, J.; Liao, X.; Zhou, Q.; Qiu, X.; Jiang, S.; Xu, N.; Wang, X.; Zhao, L.; Hu, W.; et al. rhADAMTS13 reduces oxidative stress by cleaving VWF in ischaemia/reperfusion-induced acute kidney injury. Acta Physiol. 2022, 234, e13778. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, M.A.; de Maat, M.P.; Portegies, M.L.; Kavousi, M.; Hofman, A.; Turecek, P.L.; Rottensteiner, H.; Scheiflinger, F.; Koudstaal, P.J.; Ikram, M.A.; et al. Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood 2015, 126, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Witsch, T.; Martinod, K.; Sorvillo, N.; Portier, I.; De Meyer, S.F.; Wagner, D.D. Recombinant human ADAMTS13 treatment improves myocardial remodeling and functionality after pressure overload injury in mice. J. Am. Heart Assoc. 2018, 7, e007004. [Google Scholar] [CrossRef] [PubMed]
- Newnham, M.; South, K.; Bleda, M.; Auger, W.R.; Barberà, J.A.; Bogaard, H.; Bunclark, K.; Cannon, J.E.; Delcroix, M.; Hadinnapola, C.; et al. The ADAMTS13-VWF axis is dysregulated in chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801805. [Google Scholar] [CrossRef] [PubMed]
- Zitomersky, N.L.; Demers, M.; Martinod, K.; Gallant, M.; Cifuni, S.M.; Biswas, A.; Snapper, S.; Wagner, D.D. ADAMTS13 deficiency worsens colitis and exogenous ADAMTS13 administration decreases colitis severity in mice. TH Open 2017, 1, e11–e23. [Google Scholar] [CrossRef] [PubMed]
- Pipe, S.W.; Leebeek, F.W.G.; Recht, M.; Key, N.S.; Castaman, G.; Miesbach, W.; Lattimore, S.; Peerlinck, K.; Van der Valk, P.; Coppens, M.; et al. Gene therapy with etranacogene dezaparvovec for hemophilia B. N. Engl. J. Med. 2023, 388, 706–718. [Google Scholar] [CrossRef]
- Cook, K.; Forbes, S.P.; Adamski, K.; Ma, J.J.; Chawla, A.; Garrison, L.P., Jr. Assessing the potential cost-effectiveness of a gene therapy for the treatment of hemophilia A. J. Med. Econ. 2020, 23, 501–512. [Google Scholar] [CrossRef]
- Dekimpe, C.; Roose, E.; Sakai, K.; Tersteeg, C.; De Meyer, S.F.; Vanhoorelbeke, K. Toward gene therapy for congenital thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2022, 21, 1090–1099. [Google Scholar] [CrossRef]
- Laje, P.; Shang, D.; Cao, W.; Niiya, M.; Endo, M.; Radu, A.; DeRogatis, N.; Scheiflinger, F.; Zoltick, P.W.; Flake, A.W.; et al. Correction of murine ADAMTS13 deficiency by hematopoietic progenitor cell-mediated gene therapy. Blood 2009, 113, 2172–2180. [Google Scholar] [CrossRef]
- Niiya, M.; Endo, M.; Shang, D.; Zoltick, P.W.; Muvarak, N.E.; Cao, W.; Jin, S.Y.; Skipwith, C.G.; Motto, D.G.; Flake, A.W.; et al. Correction of ADAMTS13 deficiency by in utero gene transfer of lentiviral vector encoding ADAMTS13 genes. Mol. Ther. 2009, 17, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Trionfini, P.; Tomasoni, S.; Galbusera, M.; Motto, D.; Longaretti, L.; Corna, D.; Remuzzi, G.; Benigni, A. Adenoviral-mediated gene transfer restores plasma ADAMTS13 antigen and activity in ADAMTS13 knockout mice. Gene Ther. 2009, 16, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.Y.; Xiao, J.; Bao, J.; Zhou, S.; Wright, J.F.; Zheng, X.L. AAV-mediated expression of an ADAMTS13 variant prevents shigatoxin-induced thrombotic thrombocytopenic purpura. Blood 2013, 121, 3825–3829. [Google Scholar] [CrossRef] [PubMed]
- Verhenne, S.; Vandeputte, N.; Pareyn, I.; Izsvák, Z.; Rottensteiner, H.; Deckmyn, H.; De Meyer, S.F.; Vanhoorelbeke, K. Long-Term Prevention of Congenital Thrombotic Thrombocytopenic Purpura in ADAMTS13 Knockout Mice by Sleeping Beauty Transposon-Mediated Gene Therapy. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 836–844. [Google Scholar] [CrossRef]


{kind=link}
| Summary | Reference |
|---|---|
| The prophylactic FFP group received a lower FFP dose than suggested in the ISTH guidelines (13.2 mL/kg/month vs. 20–30 mL/kg/month). These patients showed mild thrombocytopenia immediately before FFP infusions (median 138 × 109/L). Chronic kidney disease was the most prevalent organ damage among these patients (32%), followed by cerebral infarction (15%) and cardiac hypofunction (2%) during follow-up. | Japanese registry [39] |
| Sixty-eight percent of patients developed strokes as they aged and 44% had stroke-related sequelae. | Oklahoma registry [47] |
| Among patients over 40 years of age, 51% had a major comorbidity, and 28% of patients experienced a major morbidity recurrence after initiating prophylactic FFP. | Literature review [48] |
| The FFP on-demand group maintained platelet counts within a normal range without regular treatment and did not develop long-term organ damage during follow-up. | Japanese registry [39] |
| Code | Age at Death (Years) | Sex | Follow-Up (Years) | Cause of Death | Renal Impairment | Complications/ Backgrounds | Prophylactic FFP Infusion |
|---|---|---|---|---|---|---|---|
| C3 | 38 | M | 30 | Unknown * | ESRD (HD) | Yes | |
| H3 | 52 | M | 1 | Uremia | ESRD (HD) | GIH | No |
| R5 | 37 | F | 14 | Suicide | Yes | ||
| X5 | 44 | F | 4 | Unknown * | SLE | No | |
| 2G2 | 79 | M | 3 | Cerebral infarction | Yes | ||
| 2N4 | 23 | F | 0 | Unknown * | Pregnancy | No | |
| 2O | 41 | M | 2 | Unknown * | ESRD (HD) | No | |
| 2P4 | 44 | F | 17 | Status epilepticus, NOMI | Paralysis after stroke | Yes | |
| 2R | 48 | M | 14 | Unknown * | ESRD (HD) | Yes | |
| 2T | 66 | M | 1 | Sepsis | ESRD (HD) | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakai, K.; Matsumoto, M. Clinical Manifestations, Current and Future Therapy, and Long-Term Outcomes in Congenital Thrombotic Thrombocytopenic Purpura. J. Clin. Med. 2023, 12, 3365. https://doi.org/10.3390/jcm12103365
Sakai K, Matsumoto M. Clinical Manifestations, Current and Future Therapy, and Long-Term Outcomes in Congenital Thrombotic Thrombocytopenic Purpura. Journal of Clinical Medicine. 2023; 12(10):3365. https://doi.org/10.3390/jcm12103365
Chicago/Turabian StyleSakai, Kazuya, and Masanori Matsumoto. 2023. "Clinical Manifestations, Current and Future Therapy, and Long-Term Outcomes in Congenital Thrombotic Thrombocytopenic Purpura" Journal of Clinical Medicine 12, no. 10: 3365. https://doi.org/10.3390/jcm12103365