
Clinical Developmental Cardiology for Understanding Etiology of Congenital Heart Disease

Abstract
:1. Introduction
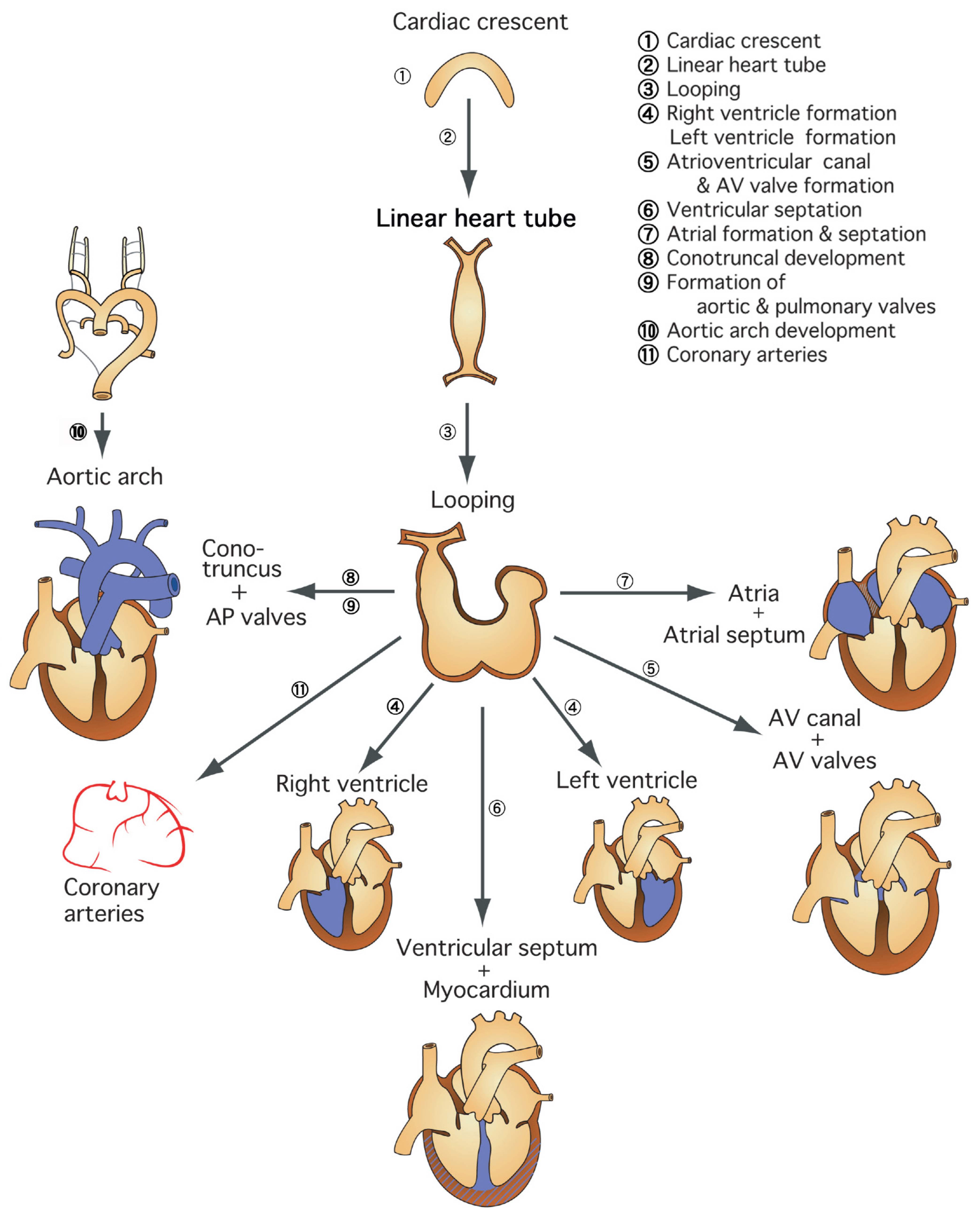
1.1. Region-Specific Step-by-Step Understanding of Cardiovascular Development for Congenital Heart Disease
1.2. Early Step of Cardiovascular Development and Heterotaxy Syndrome
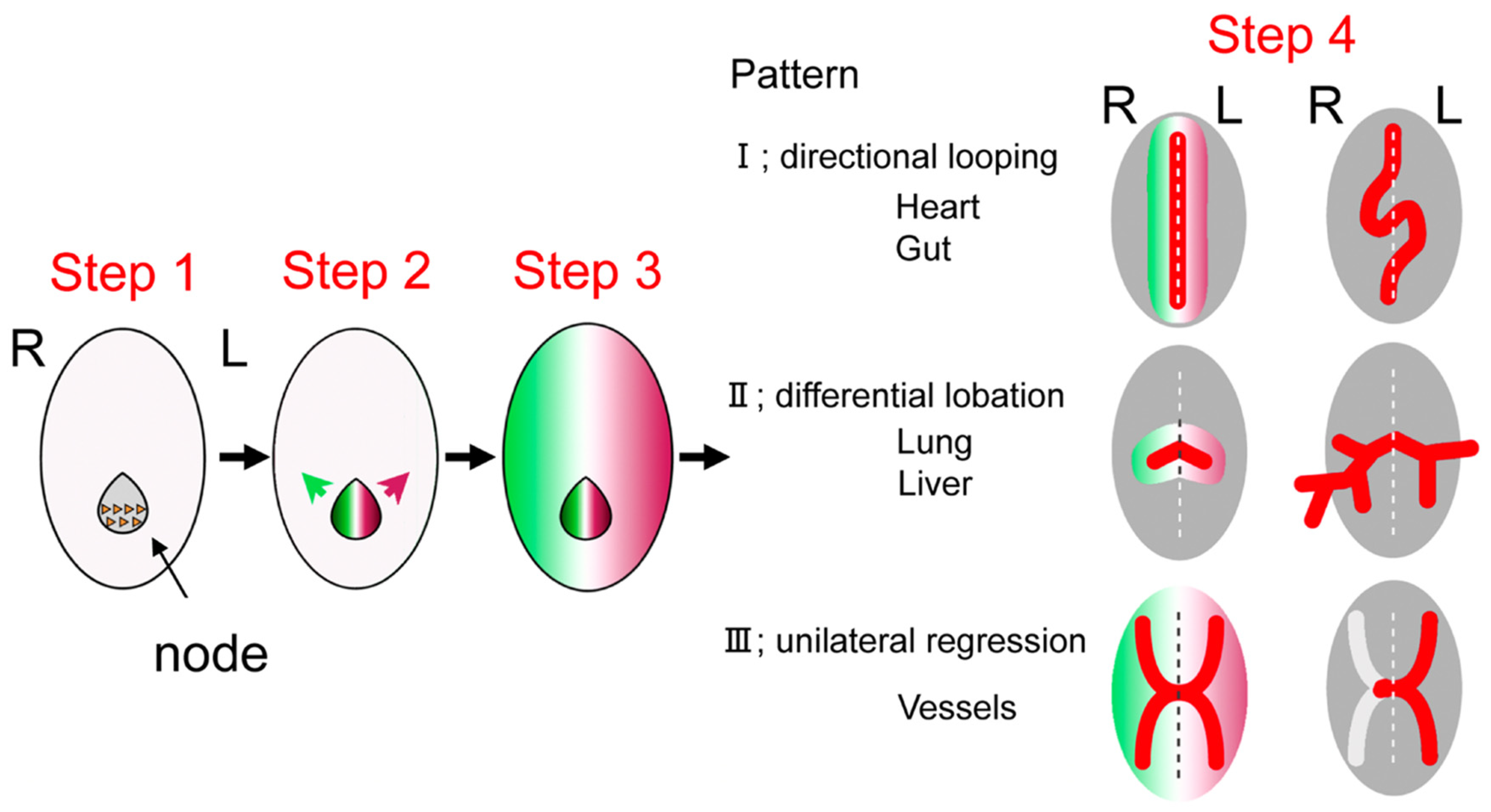
1.3. Development of Left–Right Axis
1.4. Developmental Defects of Left–Right Axis and Heterotaxy Syndrome
1.5. Morphological Characteristics of Heterotaxy Syndrome
1.6. Characteristics of Asplenia Syndrome and Necessary Medical Care
1.7. Characteristics of Polysplenia Syndrome and Necessary Medical Care
1.8. Fontan Circulation in Heterotaxy Syndrome: Recent Concepts for Pulmonary Hypertensive Vascular Disease and Protein-Loosing Enteropathy
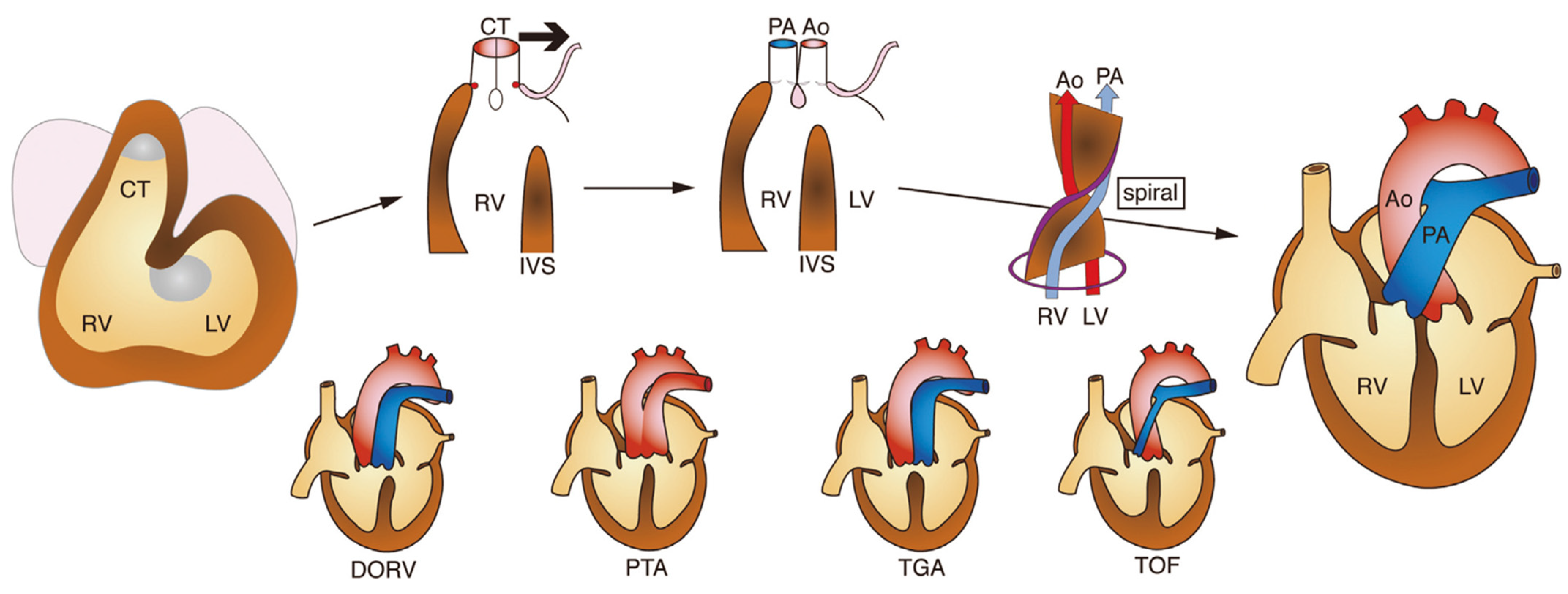
1.9. Development of the Outflow Tract Region of the Heart and Congenital Heart Disease
1.10. Congenital Outflow Tract Defects: Recent Concepts from Developmental Cardiology
2. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Morton, S.; Quiat, D.; Seidman, J.; Christine, E.; Seidman, C.E. Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 2022, 19, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.; Gelb, B.; Seidman, J.; Seidman, C.E. Genetics of congenital heart disease: The glass half empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, H.; Maeda, J.; Uchida, K.; Tsuchihashi, T.; Nakazawa, M.; Aramaki, M.; Kodo, K.; Yamagishi, C. Molecular embryology for an understanding of congenital heart diseases. Anat. Sci. Int. 2009, 84, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Meno, C.; Watanabe, D.; Saijoh, Y. Establishment of vertebrate left-right asymmetry. Nat. Rev. Genet. 2002, 3, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Miyakawa, S.; Sawa, Y. Molecular Mechanism Underlying Heterotaxy and Cardiac Isomerism. Pediatric Cardiol. Card. Surg. 2017, 33, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, H. Left-right axis determination of the body and heterotaxy Syndrome. Pediatric Cardiol. Card. Surg. 2018, 34, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, T.; Aragaki, Y.; Ohki, H.; Ichida, F.; Oana, S.; Ogawa, K.; Ono, Y.; Kamisago, M.; Koh, E.; Oyama, K.; et al. Epidemiology of severe infections in asplenia and polysplenia syndrome. Pediatric Cardiol. Card. Surg. 2009, 25, 221–223. [Google Scholar]
- Shibata, A.; Mori, H.; Kodo, K.; Nakanishi, T.; Yamagishi, H. Polysplenia syndrome as a risk factor for early progression of pulmonary hypertension. Circ. J. 2019, 83, 831–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiPaola, F.; Trout, A.; Walther, A.; Gupta, A.; Sheridan, R.; Campbell, K.M.; Tiao, G.; Bezerra, J.A.; Bove, K.E.; Patel, M.; et al. Congenital portosystemic shunts in children: Associations, complications, and outcomes. Dig. Dis. Sci. 2020, 65, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Cerro, M.; Abman, S.; Diaz, G.; Freudenthal, A.H.; Freudenthal, F.; Harikrishnan, S.; Haworth, S.G.; Ivy, D.; Lopes, A.A.; Raj, J.U.; et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: Report from the PVRI Pediatric Taskforce, Panama. Pulm. Circ. 2011, 1, 286–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itkin, M.; Piccoli, D.A.; Nadolski, G.; Rychik, J.; DeWitt, A.; Pinto, E.; Rome, J.; Dori, Y. Protein-losing enteropathy in patients with congenital heart disease. J. Am. Coll. Cardiol. 2017, 69, 2929–2937. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, H. Caridiac Neural Crest. Cold Spring Harb. Perspect. Biol. 2021, 13, a036715. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.; Waldo, K.L. Role of neural crest in congenital heart disease. Circulation 1990, 82, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, V.; Yamagishi, C.; Hu, T.; Kathiriya, I.S.; Yamagishi, H.; Srivastava, D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev. Biol. 2001, 235, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, J.; Yamagishi, H.; McAnally, J.; Yamagishi, C.; Srivastava, D. Tbx1 is regulated by forkhead proteins in the secondary heart field. Dev. Dyn. 2006, 235, 701–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siwik, E.; Patel, C.; Zahka, K.G. Tetralogy of Fallot. In Moss and Adams’ Heart Disease in Infants, Children, and Adolescents including the Fetus and Young Adult, 6th ed.; Allen, H., Gutgesell, H., Clark, E., Driscoll, D.J., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 881–900. [Google Scholar]
- Kodo, K.; Nishizawa, T.; Furutani, M.; Arai, S.; Yamamura, E.; Joo, K.; Takahashi, T.; Matsuoka, R.; Yamagishi, H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13933–13938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodo, K.; Shibata, S.; Miyagawa-Toita, S.; Ong, S.G.; Takahashi, H.; Kume, T.; Okano, H.; Matsuoka, R.; Yamagishi, H. Regulation of Sema3c and the interaction between cardiac neural crest and second heart field during outflow tract development. Sci. Rep. 2017, 7, 6771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuhara, J.; Garg, V. Genetics of congenital heart disease: A narrative review of recent advances and clinical implications. Transl. Pediatrics 2021, 10, 2366–2386. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Function | Related CHD |
|---|---|---|
| ZIC3 | Transcription factor | Heterotaxy |
| NODAL | TGF-β signal | Heterotaxy |
| CFC1 | Nodal pathway | Heterotaxy |
| ACVR2B | Nodal pathway | Heterotaxy |
| FOXH1 | Nodal pathway | Heterotaxy |
| LEFTY A | Nodal pathway | Heterotaxy |
| GDF1 | Nodal pathway | Heterotaxy |
| SESN1 | Nodal pathway | Heterotaxy |
| CRELD1 | EGF-like protein | Heterotaxy |
| DNAI1 | Dynein arm component | Heterotaxy |
| DNAH5 | Dynein arm component | Heterotaxy |
| NKX2-5 | Transcription factor | ASD, TOF, HLHS |
| NKX2-6 | Transcription factor | PTA |
| GATA4 | Transcription factor | ASD, AVSD, TOF |
| GATA6 | Transcription factor | PTA, TOF |
| TBX1 | Transcription factor | PTA, TOF, IAA |
| TBX5 | Transcription factor | AVSD, ASD, VSD |
| TBX20 | Transcription factor | ASD, VSD |
| ZFPM2/FOG2 | Transcription factor | TOF, DORV |
| NOTCH1 | Notch pathway | BAV, AS, TOF |
| VEGFA | Cell signaling | TOF, AS, IAA |
| ELN | Structural protein | SVAS |
| MYH6 | Structural protein | ASD |
| MYH7 | Structural protein | Ebstein’s anomaly |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamagishi, H. Clinical Developmental Cardiology for Understanding Etiology of Congenital Heart Disease. J. Clin. Med. 2022, 11, 2381. https://doi.org/10.3390/jcm11092381
Yamagishi H. Clinical Developmental Cardiology for Understanding Etiology of Congenital Heart Disease. Journal of Clinical Medicine. 2022; 11(9):2381. https://doi.org/10.3390/jcm11092381
Chicago/Turabian StyleYamagishi, Hiroyuki. 2022. "Clinical Developmental Cardiology for Understanding Etiology of Congenital Heart Disease" Journal of Clinical Medicine 11, no. 9: 2381. https://doi.org/10.3390/jcm11092381