
Impaired Terminal Erythroid Maturation in β0-Thalassemia/HbE Patients with Different Clinical Severity

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Hematologic Studies
2.3. In Vitro Culture of CD34+ Progenitor Cells
2.4. Assessment of Erythroid Maturation
2.5. Quantitative RT-PCR Analysis
2.6. Statistical Analysis
3. Results
3.1. Levels of Cellular Hb and Number of RBC in β0-Thalassemia/HbE Patients
3.2. Heterogeneity of Reticulocytes in the Circulating Blood of Anemia Patients
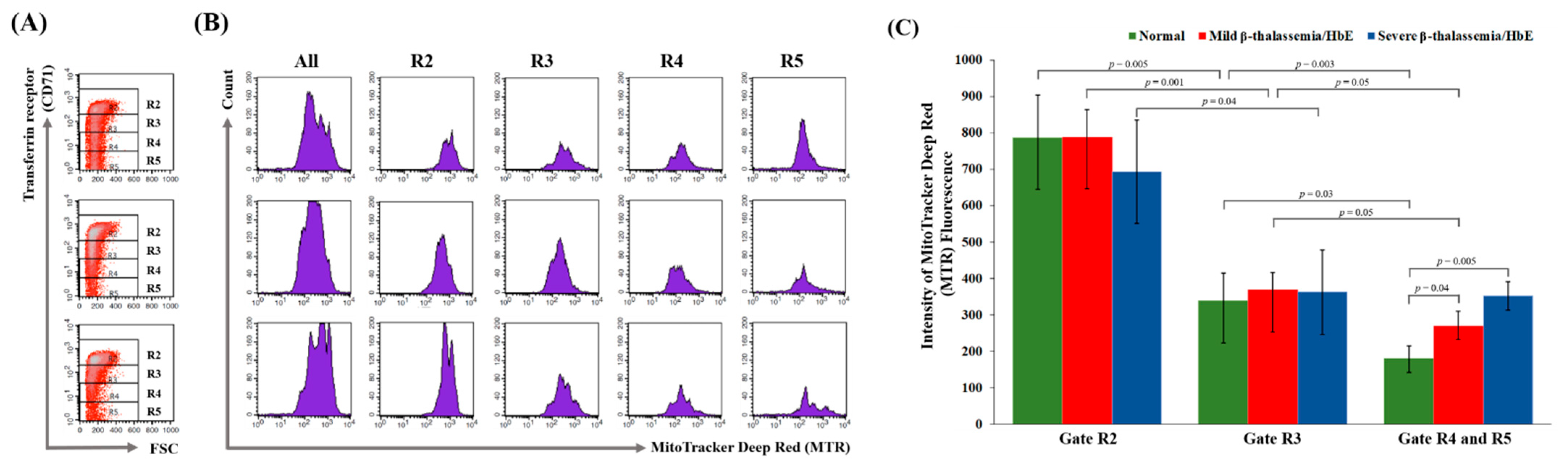
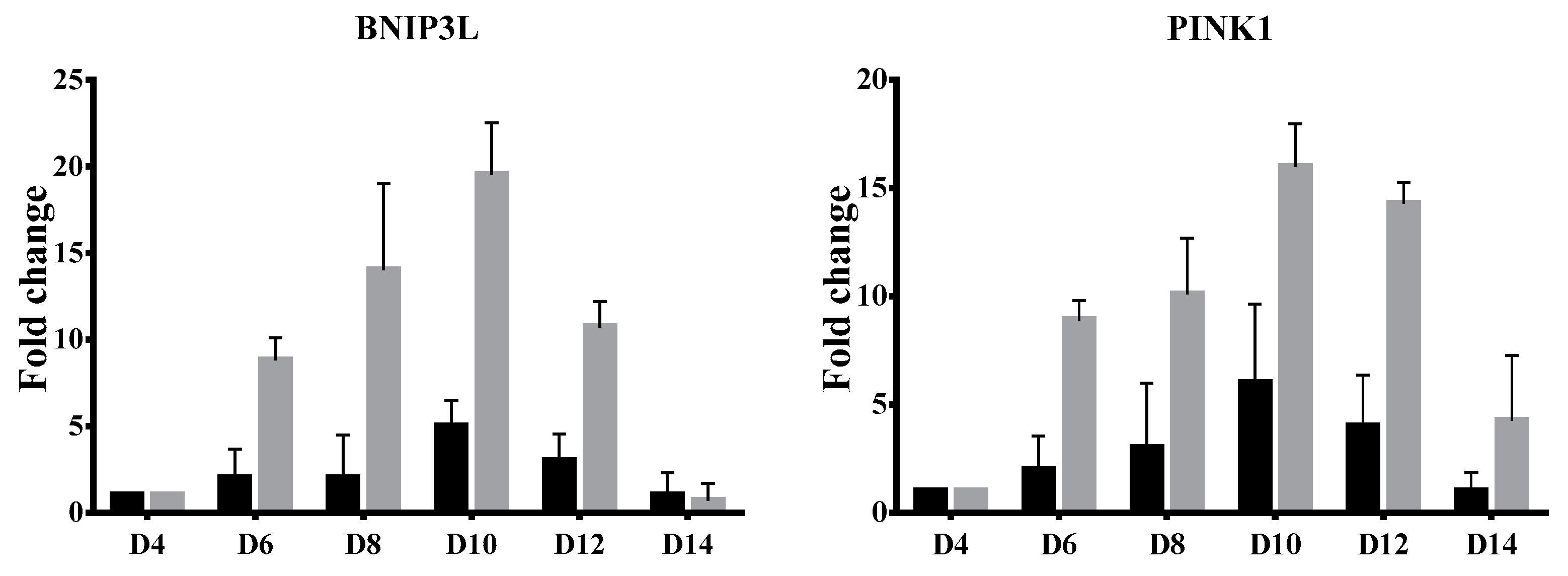
3.3. Delayed Terminal Erythroid Maturation in β-Thalassemia/HbE
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gronowicz, G.; Swift, H.; Steck, T.L. Maturation of the reticulocyte in vitro. J. Cell Sci. 1984, 71, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piva, E.; Brugnara, C.; Chiandetti, L.; Plebani, M. Automated reticulocyte counting: State of the art and clinical applications in the evaluation of erythropoiesis. Clin. Chem. Lab. Med. 2010, 48, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Torres Gomez, A.; Casaño, J.; Sánchez, J.; Madrigal, E.; Blanco, F.; Alvarez, M.A. Utility of reticulocyte maturation parameters in the differential diagnosis of macrocytic anemias. Clin. Lab. Haematol. 2003, 25, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Weatherall, D.J. The hemoglobin E thalassemias. Cold Spring Harb Perspect. Med. 2012, 2, a011734. [Google Scholar] [CrossRef] [PubMed]
- Winichagoon, P.; Thonglairoam, V.; Fucharoen, S.; Wilairat, P.; Fukumaki, Y.; Wasi, P. Severity differences in β-thalassaemia/haemoglobin E syndromes: Implication of genetic factors. Br. J. Haematol. 1993, 83, 633–639. [Google Scholar] [CrossRef]
- Sripichai, O.; Makarasara, W.; Munkongdee, T.; Kumkhaek, C.; Nuchprayoon, I.; Chuansumrit, A.; Chuncharunee, S.; Chantrakoon, N.; Boonmongkol, P.; Winichagoon, P.; et al. A scoring system for the classification of beta-thalassemia/Hb E disease severity. Am. J. Hematol. 2008, 83, 482–484. [Google Scholar] [CrossRef]
- Orkin, S.H.; Kazazian, H.H.; Antonarakis, S.E.; Ostrer, H.; Goff, S.C.; Sexton, J.P. Abnormal RNA processing due to the exon mutation of βE-globin gene. Nature 1982, 300, 768–769. [Google Scholar] [CrossRef]
- Schrier, S.L. Pathophysiology of thalassemia. Curr. Opin. Hematol. 2002, 9, 123–126. [Google Scholar] [CrossRef]
- Libani, I.V.; Guy, E.C.; Melchiori, L.; Schiro, R.; Ramos, P.; Breda, L.; Scholzen, T.; Chadburn, A.; Liu, Y.F.; Kernbach, M.; et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood 2008, 112, 875–885. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization; Centers for Disease Control and Prevention. Assessing the Iron Status of Populations: Including Literature Reviews: Report of a Joint World Health Organization/Centers for Disease Control and Prevention Technical Consultation on the Assessment of Iron Status at the Population Level, Geneva, Switzerland, 6–8 April 2004, 2nd ed; World Health Organization: Geneva, Switzerland, 2007. [Google Scholar]
- Johnson-Wimbley, T.D.; Graham, D.Y. Diagnosis and management of iron deficiency anemia in the 21st century. Ther. Adv. Gastroenterol. 2011, 4, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwart, A.; van Assendelft, O.W.; Bull, B.S.; England, J.M.; Lewis, S.M.; Zijlstra, W.G. Recommendations for reference method for haemoglobinometry in human blood (ICSH standard 1995) and specifications for international haemiglobinocyanide standard (4th edition). J. Clin. Pathol. 1996, 49, 271–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunicka, J.; Malin, M.; Zelmanovic, D.; Katzenberg, M.; Canfield, W.; Shapiro, P.; Mohandas, N. Automated quantitation of hemoglobin-based blood substitutes in whole blood samples. Am. J. Clin. Pathol. 2001, 116, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.W.; Pai, S.H. Reticulocyte subpopulations and reticulocyte maturity index (RMI) rise as body iron status falls. Am. J. Hematol. 2001, 67, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Douay, L.; Giarratana, M.C. Ex vivo generation of human red blood cells: A new advance in stem cell engineering. Methods Mol. Biol. 2009, 482, 127–140. [Google Scholar]
- Sripichai, O.; Fucharoen, S. Fetal hemoglobin regulation in β-thalassemia: Heterogeneity, modifiers and therapeutic approaches. Expert Rev. Hematol. 2016, 9, 1129–1137. [Google Scholar] [CrossRef]
- Riley, R.S.; Ben-Ezra, J.M.; Goel, R.; Tidwell, A. Reticulocytes and reticulocyte enumeration. J. Clin. Lab. Anal. 2001, 15, 267–294. [Google Scholar] [CrossRef]
- Mast, A.E.; Blinder, M.A.; Dietzen, D.J. Reticulocyte hemoglobin content. Am. J. Hematol. 2008, 83, 307–310. [Google Scholar] [CrossRef]
- Waugh, R.E.; McKenney, J.B.; Bauserman, R.G.; Brooks, D.M.; Valeri, C.R. Surface area and volume changes during maturation of reticulocytes in the circulation of the baboon. J. Lab. Clin. Med. 1997, 129, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Waugh, R.E.; Mantalaris, A.; Bauserman, R.G.; Hwang, W.C.; Wu, J.H.D. Membrane instability in late-stage erythropoiesis. Blood 2001, 97, 1869–1875. [Google Scholar] [CrossRef] [Green Version]
- Malleret, B.; Xu, F.; Mohandas, N.; Suwanarusk, R.; Chu, C.; Leite, J.A.; Low, K.; Turner, C.; Sriprawat, K.; Zhang, R.; et al. Significant biochemical, biophysical and metabolic diversity in circulating human cord blood reticulocytes. PLoS ONE 2013, 8, e76062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chasis, J.A.; Schrier, S.L. Membrane deformability and the capacity for shape change in the erythrocyte. Blood 1989, 74, 2562–2568. [Google Scholar] [CrossRef] [Green Version]
- Al-Huniti, N.H.; Widness, J.A.; Schmidt, R.L.; Veng-Pedersen, P. Pharmacodynamic analysis of changes in reticulocyte subtype distribution in phlebotomy-induced stress erythropoiesis. J. Pharmacokinet. Pharmacodyn. 2005, 32, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.; Alfrey, C.P. The negative regulation of red cell mass by neocytolysis: Physiologic and pathophysiologic manifestations. Cell Physiol. Biochem. 2005, 15, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Sivilotti, M.L. Oxidant stress and haemolysis of the human erythrocyte. Toxicol. Rev. 2004, 23, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, and apoptosis. Am. J. Med. Genet. 2001, 106, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Ney, P.A. Normal and disordered reticulocyte maturation. Curr. Opin. Hematol. 2011, 18, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Xu, W.; Xu, L.; Kong, Q.; Fang, J. Mitophagy is increased during erythroid differentiation in β-thalassemia. Int. J. Hematol. 2017, 105, 162–173. [Google Scholar] [CrossRef]
- Lithanatudom, P.; Wannatung, T.; Leecharoenkiat, A.; Svasti, S.; Fucharoen, S.; Smith, D.R. Enhanced activation of autophagy in β-thalassemia/Hb E erythroblasts during erythropoiesis. Ann. Hematol. 2011, 90, 747–758. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Yang, C.; Hashimoto, M.; Lin, Q.X.X.; Tan, D.Q.; Suda, T. Sphingosine-1-phosphate signaling modulates terminal erythroid differentiation through the regulation of mitophagy. Exp. Hematol. 2019, 72, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Characteristics | Normal | Hb 7.5–9.0 g/dL | Hb 4.0–6.5 g/dL | ||||
|---|---|---|---|---|---|---|---|
| Iron Deficiency Anemia | Mild β-Thalassemia/HbE | p-Value * | Iron Deficiency Anemia | Severe β-Thalassemia/HbE | p-Value * | ||
| No. of cases | 90 | 12 | 60 | 9 | 30 | ||
| Red blood cell | |||||||
| Hb (g/dL) | 13.4 ± 1.1 | 8.0 ± 0.9 | 7.9 ± 0.5 | 0.75 | 5.4 ± 0.8 | 5.4 ± 0.7 b | 0.87 |
| RBC (×106/uL) | 4.6 ± 0.4 | 4.0 ± 0.5 | 4.2 ± 0.3 | 0.11 | 3.5 ± 0.6 | 3.1 ± 0.4 b | 0.03 |
| MCV (fL) | 89.5 ± 2.7 | 70.5 ± 7.2 | 60.3 ± 4.2 | 4.3 × 10−9 | 60.0 ± 8.9 | 60.2 ± 4.7 | 0.94 |
| MCH (pg) | 29.2 ± 1.4 | 19.9 ± 2.4 | 18.9 ± 1.7 | 0.09 | 15.7 ± 2.6 | 17.6 ± 1.6 a | 0.01 |
| MCHC (g/dL) | 32.7 ± 1.3 | 28.2 ± 1.6 | 31.3 ± 1.4 | 5.6 × 10−9 | 26.3 ± 3.5 | 29.2 ± 1.1 b | 3.5 × 10−4 |
| Reticulocyte | |||||||
| Retic (×103/uL) | 62 ± 21.1 | 118 ± 45.8 | 187 ± 41.0 | 1.8 × 10−6 | 101 ± 78.0 | 180 ± 39.3 | 1.4 × 10−5 |
| retHb (g/dL) | 0.6 ± 0.2 | 0.3 ± 0.1 | 0.1 ± 0.0 | 1.1 × 10−10 | 0.3 ± 0.2 | 0.1 ± 0.0 | 7.3 × 10−6 |
| rbcHb/retHb ratio | 25.2 ± 9.7 | 34.9 ± 13.0 | 70.9 ± 18.1 | 8.2 × 10−9 | 29.2 ± 27.4 | 48.2 ± 14.8 b | 9.4 × 10−3 |
| Retic (%) | 1.4 ± 0.4 | 3.0 ± 1.2 | 4.4 ± 1.0 | 1.6 × 10−5 | 2.9 ± 2.3 | 5.9 ± 1.3 b | 1.4 × 10−5 |
| L-Retic (%) | 93.8 ± 2.8 | 85.3 ± 4.6 | 75.8 ± 5.9 | 1.5 × 10−6 | 84.1 ± 6.8 | 64.0 ± 8.8 b | 2.2 × 10−7 |
| M-Retic (%) | 5.6 ± 2.7 | 12.9 ± 3.6 | 16.1 ± 3.0 | 1.4 × 10−3 | 12.3 ± 4.7 | 18.5 ± 3.4 b | 9.2 × 10−5 |
| H-Retic (%) | 0.6 ± 0.4 | 1.8 ± 1.3 | 8.1 ± 3.5 | 7.3 × 10−8 | 3.5 ± 2.5 | 17.5 ± 6.0 a | 6.1 × 10−8 |
| IRF (%) | 6.2 ± 2.8 | 14.7 ± 4.6 | 24.2 ± 5.9 | 1.5 × 10−6 | 15.9 ± 6.8 | 36.0 ± 8.7 b | 2.2 × 10−7 |
| M/H ratio | 11.6 ± 8.9 | 14.0 ± 15.0 | 2.3 ± 0.8 | 4.4 × 10−8 | 4.4 ± 1.9 | 1.1 ± 0.3 b | 3.5 × 10−11 |
| MCVr (fL) | 102.4 ± 3.5 | 97.7 ± 10.4 | 77.8 ± 5.0 | 1.8 × 10−15 | 79.4 ± 12.0 | 78.9 ± 4.9 | 0.83 |
| CHr (pg) | 32.4 ± 1.4 | 25.9 ± 3.9 | 20.8 ± 1.4 | 1.3 × 10−11 | 19.3 ± 3.2 | 20.0 ± 1.3 | 0.34 |
| CHCMr (g/dL) | 31.8 ± 1.3 | 26.5 ± 1.5 | 27.0 ± 1.1 | 0.18 | 24.5 ± 2.1 | 25.7 ± 0.7 b | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suriyun, T.; Winichagoon, P.; Fucharoen, S.; Sripichai, O. Impaired Terminal Erythroid Maturation in β0-Thalassemia/HbE Patients with Different Clinical Severity. J. Clin. Med. 2022, 11, 1755. https://doi.org/10.3390/jcm11071755
Suriyun T, Winichagoon P, Fucharoen S, Sripichai O. Impaired Terminal Erythroid Maturation in β0-Thalassemia/HbE Patients with Different Clinical Severity. Journal of Clinical Medicine. 2022; 11(7):1755. https://doi.org/10.3390/jcm11071755
Chicago/Turabian StyleSuriyun, Thunwarat, Pranee Winichagoon, Suthat Fucharoen, and Orapan Sripichai. 2022. "Impaired Terminal Erythroid Maturation in β0-Thalassemia/HbE Patients with Different Clinical Severity" Journal of Clinical Medicine 11, no. 7: 1755. https://doi.org/10.3390/jcm11071755