
The Innate Immune Microenvironment in Metastatic Breast Cancer
 , , , and
, , , and
Abstract
:1. Introduction: Immune Response and Metastatic Spread
2. The Cells of Innate Immunity and Their Role According to the Site of Metastases
2.1. Monocytes (M-DSCs)
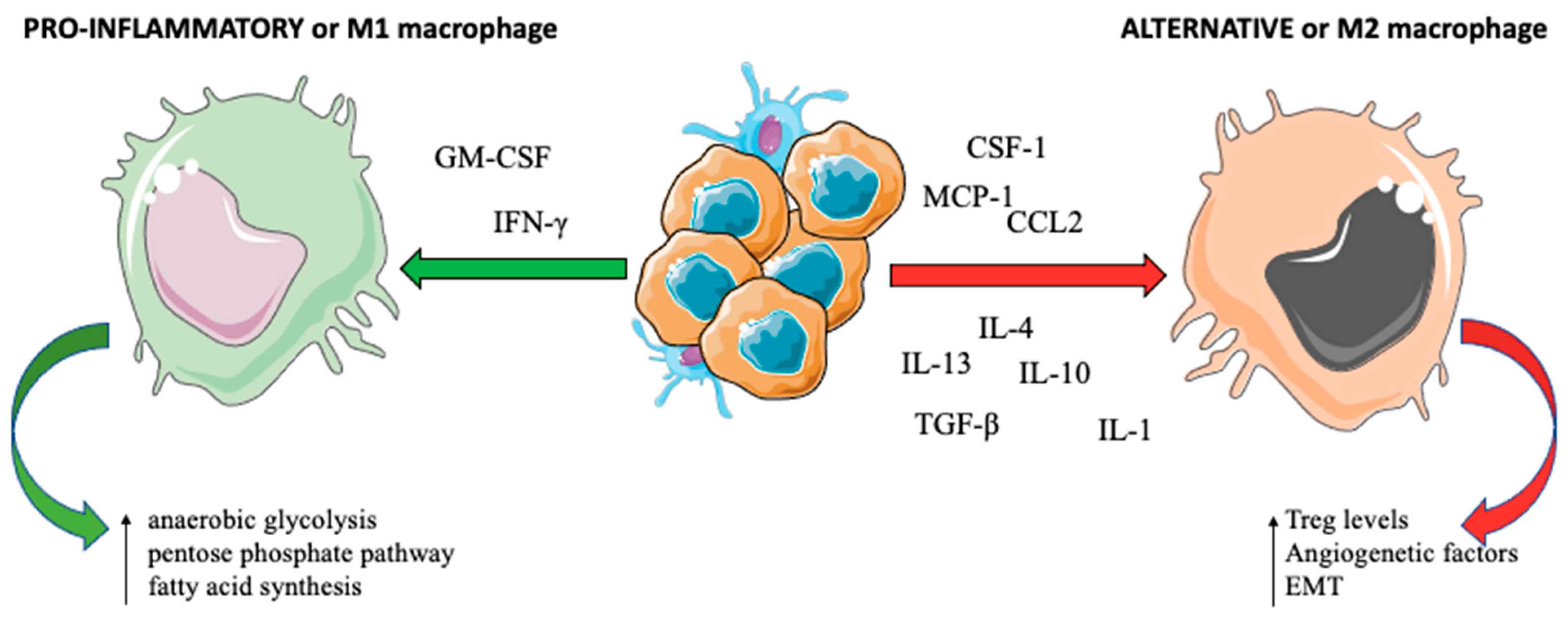
2.2. Macrophages
2.3. Dendritic Cells (DCs)
2.4. Tumor-Associated Neutrophils (TANs)
2.5. Mast Cells and Natural Killer (NK) Cells
2.6. Complement System
3. The Development of Pre-Metastatic Niches
4. Therapeutic Implications
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zardavas, D.; Baselga, J.; Piccart, M. Emerging Targeted Agents in Metastatic Breast Cancer. Nat. Rev. Clin. Oncol. 2013, 10, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.; Garrido, F.; Garcia-Lora, A.M. Metastases in Immune-Mediated Dormancy: A New Opportunity for Targeting Cancer. Cancer Res. 2014, 74, 6750–6757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Rafii, S.; Lyden, D. Preparing the “Soil”: The Premetastatic Niche. Cancer Res. 2006, 66, 11089–11093. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Psaila, B.; Lyden, D. Bone Marrow Cells in the “Pre-Metastatic Niche”: Within Bone and Beyond. Cancer Metastasis Rev. 2006, 25, 521–529. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-Positive Haematopoietic Bone Marrow Progenitors Initiate the Pre-Metastatic Niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated Regulation of Myeloid Cells by Tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P. The Interaction of Anticancer Therapies with Tumor-Associated Macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Eiró, N.; Pidal, I.; Fernandez-Garcia, B.; Junquera, S.; Lamelas, M.L.; del Casar, J.M.; González, L.O.; López-Muñiz, A.; Vizoso, F.J. Impact of CD68/(CD3+CD20) Ratio at the Invasive Front of Primary Tumors on Distant Metastasis Development in Breast Cancer. PLoS ONE 2012, 7, e52796. [Google Scholar] [CrossRef] [Green Version]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 610303. [Google Scholar] [CrossRef]
- Weiskopf, K.; Schnorr, P.J.; Pang, W.W.; Chao, M.P.; Chhabra, A.; Seita, J.; Feng, M.; Weissman, I.L. Myeloid Cell Origins, Differentiation, and Clinical Implications. Microbiol. Spectr. 2016, 4, 857–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, W.J.; Bratton, D.L.; Jakubzick, C.V.; Henson, P.M. Myeloid Cell Turnover and Clearance. Microbiol. Spectr. 2016, 4, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Coillard, A.; Segura, E. In Vivo Differentiation of Human Monocytes. Front. Immunol. 2019, 10, 1907. [Google Scholar] [CrossRef] [Green Version]
- Mandruzzato, S.; Solito, S.; Falisi, E.; Francescato, S.; Chiarion-Sileni, V.; Mocellin, S.; Zanon, A.; Rossi, C.R.; Nitti, D.; Bronte, V.; et al. IL4Ralpha+ Myeloid-Derived Suppressor Cell Expansion in Cancer Patients. J. Immunol. 2009, 182, 6562–6568. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Doughty-Shenton, D.; Cassetta, L.; Fragkogianni, S.; Brownlie, D.; Kato, Y.; Carragher, N.; Pollard, J.W. Monocytes Differentiate to Immune Suppressive Precursors of Metastasis-Associated Macrophages in Mouse Models of Metastatic Breast Cancer. Front. Immunol. 2017, 8, 2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, A.-L.; Zhu, J.-K.; Sun, J.-T.; Yang, M.-X.; Neckenig, M.R.; Wang, X.-W.; Shao, Q.-Q.; Song, B.-F.; Yang, Q.-F.; Kong, B.-H.; et al. CD16+ Monocytes in Breast Cancer Patients: Expanded by Monocyte Chemoattractant Protein-1 and May Be Useful for Early Diagnosis. Clin. Exp. Immunol. 2011, 164, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Shen, C.-Y.; Lin, S.-C.; Kuo, W.-H.; Kuo, Y.-T.; Hsu, Y.-L.; Wang, W.-C.; Lin, K.-T.; Wang, L.-H. Monocytes Secrete CXCL7 to Promote Breast Cancer Progression. Cell Death Dis. 2021, 12, 1090. [Google Scholar] [CrossRef]
- Kumar, A.; Taghi Khani, A.; Sanchez Ortiz, A.; Swaminathan, S. GM-CSF: A Double-Edged Sword in Cancer Immunotherapy. Front. Immunol. 2022, 13, 901277. [Google Scholar] [CrossRef]
- Thorn, M.; Guha, P.; Cunetta, M.; Espat, N.J.; Miller, G.; Junghans, R.P.; Katz, S.C. Tumor-Associated GM-CSF Overexpression Induces Immunoinhibitory Molecules via STAT3 in Myeloid-Suppressor Cells Infiltrating Liver Metastases. Cancer Gene Ther. 2016, 23, 188–198. [Google Scholar] [CrossRef]
- Jiang, P.; Gao, W.; Ma, T.; Wang, R.; Piao, Y.; Dong, X.; Wang, P.; Zhang, X.; Liu, Y.; Su, W.; et al. CD137 Promotes Bone Metastasis of Breast Cancer by Enhancing the Migration and Osteoclast Differentiation of Monocytes/Macrophages. Theranostics 2019, 9, 2950–2966. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.-Y.; Zhang, H.; Li, X.-F.; Zhang, C.-B.; Selli, C.; Tagliavini, G.; Lam, A.D.; Prost, S.; Sims, A.H.; Hu, H.-Y.; et al. Monocyte-Derived Macrophages Promote Breast Cancer Bone Metastasis Outgrowth. J. Exp. Med. 2020, 217, e20191820. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- Standiford, T.J.; Keshamouni, V.G.; Reddy, R.C. Peroxisome Proliferator-Activated Receptor-{gamma} as a Regulator of Lung Inflammation and Repair. Proc. Am. Thorac. Soc. 2005, 2, 226–231. [Google Scholar] [CrossRef]
- Wen, S.W.; Ager, E.I.; Christophi, C. Bimodal Role of Kupffer Cells during Colorectal Cancer Liver Metastasis. Cancer Biol. Ther. 2013, 14, 606–613. [Google Scholar] [CrossRef] [Green Version]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Richardsen, E.; Uglehus, R.D.; Johnsen, S.H.; Busund, L.-T. Macrophage-Colony Stimulating Factor (CSF1) Predicts Breast Cancer Progression and Mortality. Anticancer Res. 2015, 35, 865–874. [Google Scholar] [PubMed]
- Sullivan, A.R.; Pixley, F.J. CSF-1R Signaling in Health and Disease: A Focus on the Mammary Gland. J. Mammary Gland Biol. Neoplasia 2014, 19, 149–159. [Google Scholar] [CrossRef]
- Imamura, M.; Li, T.; Li, C.; Fujisawa, M.; Mukaida, N.; Matsukawa, A.; Yoshimura, T. Crosstalk between Cancer Cells and Fibroblasts for the Production of Monocyte Chemoattractant Protein-1 in the Murine 4T1 Breast Cancer. Curr. Issues Mol. Biol. 2021, 43, 1726–1740. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Zhang, M.; Yan, L.; Kim, J.A. Modulating Mammary Tumor Growth, Metastasis and Immunosuppression by SiRNA-Induced MIF Reduction in Tumor Microenvironment. Cancer Gene Ther. 2015, 22, 463–474. [Google Scholar] [CrossRef]
- Frankenberger, C.; Rabe, D.; Bainer, R.; Sankarasharma, D.; Chada, K.; Krausz, T.; Gilad, Y.; Becker, L.; Rosner, M.R. Metastasis Suppressors Regulate the Tumor Microenvironment by Blocking Recruitment of Prometastatic Tumor-Associated Macrophages. Cancer Res. 2015, 75, 4063–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A Positive Feedback Loop between Mesenchymal-like Cancer Cells and Macrophages Is Essential to Breast Cancer Metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Hollmén, M.; Karaman, S.; Schwager, S.; Lisibach, A.; Christiansen, A.J.; Maksimow, M.; Varga, Z.; Jalkanen, S.; Detmar, M. G-CSF Regulates Macrophage Phenotype and Associates with Poor Overall Survival in Human Triple-Negative Breast Cancer. Oncoimmunology 2016, 5, e1115177. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, Q.; Zhang, Z.; Bai, N.; Liu, Z.; Xiong, M.; Wei, Y.; Xiang, R.; Tan, X. VDR Status Arbitrates the Prometastatic Effects of Tumor-Associated Macrophages. Mol. Cancer Res. 2014, 12, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Barsoum, I.B.; Hamilton, T.K.; Li, X.; Cotechini, T.; Miles, E.A.; Siemens, D.R.; Graham, C.H. Hypoxia Induces Escape from Innate Immunity in Cancer Cells via Increased Expression of ADAM10: Role of Nitric Oxide. Cancer Res. 2011, 71, 7433–7441. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, A.S.; Heikkila, P.; von Smitten, K.; Vakkila, J.; Leidenius, M. The Presence of Sinusoidal CD163(+) Macrophages in Lymph Nodes Is Associated with Favorable Nodal Status in Patients with Breast Cancer. Virchows Arch. 2012, 461, 639–646. [Google Scholar] [CrossRef]
- Mitrofanova, I.; Zavyalova, M.; Telegina, N.; Buldakov, M.; Riabov, V.; Cherdyntseva, N.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast Cancer Parenchyma Negatively Correlate with Lymphatic Metastasis after Neoadjuvant Chemotherapy. Immunobiology 2017, 222, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lin, Y.; Zhu, H.; Zhou, Y.; Mao, F.; Huang, X.; Sun, Q.; Li, C. The Prognostic and Clinical Value of Tumor-Associated Macrophages in Patients With Breast Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 2022, 12, 905846. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, W.; Bado, I.; Zhang, X.H.-F. Bone Tropism in Cancer Metastases. Cold Spring Harb. Perspect. Med. 2020, 10, a036848. [Google Scholar] [CrossRef]
- Lacey, D.L.; Boyle, W.J.; Simonet, W.S.; Kostenuik, P.J.; Dougall, W.C.; Sullivan, J.K.; San Martin, J.; Dansey, R. Bench to Bedside: Elucidation of the OPG-RANK-RANKL Pathway and the Development of Denosumab. Nat. Rev. Drug Discov. 2012, 11, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Capietto, A.-H.; Faccio, R. Immune Regulation of Bone Metastasis. Bonekey Rep. 2014, 3, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, A.F.; Appelman-Dijkstra, N.M.; van der Burg, S.H.; Kroep, J.R. The Anti-Tumor Effect of RANKL Inhibition in Malignant Solid Tumors—A Systematic Review. Cancer Treat. Rev. 2018, 62, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.R.; Rumney, R.M.H.; Schoof, E.M.; Perryman, L.; Høye, A.M.; Agrawal, A.; Bird, D.; Latif, N.A.; Forrest, H.; Evans, H.R.; et al. The Hypoxic Cancer Secretome Induces Pre-Metastatic Bone Lesions through Lysyl Oxidase. Nature 2015, 522, 106–110. [Google Scholar] [CrossRef]
- Gupta, A.; Cao, W.; Chellaiah, M.A. Integrin Avβ3 and CD44 Pathways in Metastatic Prostate Cancer Cells Support Osteoclastogenesis via a Runx2/Smad 5/Receptor Activator of NF-ΚB Ligand Signaling Axis. Mol. Cancer 2012, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.-N.; Zhang, F.; Tang, P.; Qi, X.-W.; Jiang, J. Hypoxia Induces RANK and RANKL Expression by Activating HIF-1α in Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2011, 408, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Palafox, M.; Ferrer, I.; Pellegrini, P.; Vila, S.; Hernandez-Ortega, S.; Urruticoechea, A.; Climent, F.; Soler, M.T.; Muñoz, P.; Viñals, F.; et al. RANK Induces Epithelial-Mesenchymal Transition and Stemness in Human Mammary Epithelial Cells and Promotes Tumorigenesis and Metastasis. Cancer Res. 2012, 72, 2879–2888. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Tsuda, M.; Takahashi, T.; Totsuka, Y.; Shindoh, M.; Ohba, Y. RANKL Expression Specifically Observed in Vivo Promotes Epithelial Mesenchymal Transition and Tumor Progression. Am. J. Pathol. 2011, 178, 2845–2856. [Google Scholar] [CrossRef] [Green Version]
- Renema, N.; Navet, B.; Heymann, M.-F.; Lezot, F.; Heymann, D. RANK-RANKL Signalling in Cancer. Biosci. Rep. 2016, 36, e00366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Aleza, C.; Nguyen, B.; Yoldi, G.; Ciscar, M.; Barranco, A.; Hernández-Jiménez, E.; Maetens, M.; Salgado, R.; Zafeiroglou, M.; Pellegrini, P.; et al. Inhibition of RANK Signaling in Breast Cancer Induces an Anti-Tumor Immune Response Orchestrated by CD8+ T Cells. Nat. Commun. 2020, 11, 6335. [Google Scholar] [CrossRef]
- Bonapace, L.; Coissieux, M.-M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 Inhibition Accelerates Breast Cancer Metastasis by Promoting Angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Batoon, L.; McCauley, L.K. Cross Talk Between Macrophages and Cancer Cells in the Bone Metastatic Environment. Front. Endocrinol. 2021, 12, 763846. [Google Scholar] [CrossRef] [PubMed]
- Coscia, M.; Quaglino, E.; Iezzi, M.; Curcio, C.; Pantaleoni, F.; Riganti, C.; Holen, I.; Mönkkönen, H.; Boccadoro, M.; Forni, G.; et al. Zoledronic Acid Repolarizes Tumour-Associated Macrophages and Inhibits Mammary Carcinogenesis by Targeting the Mevalonate Pathway. J. Cell Mol. Med. 2010, 14, 2803–2815. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, X.H.-F.; Massagué, J. Macrophage Binding to Receptor VCAM-1 Transmits Survival Signals in Breast Cancer Cells That Invade the Lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A Distinct Macrophage Population Mediates Metastatic Breast Cancer Cell Extravasation, Establishment and Growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Qian, B.-Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-Induced Chemokine Cascade Promotes Breast Cancer Metastasis by Enhancing Retention of Metastasis-Associated Macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Pukrop, T.; Dehghani, F.; Chuang, H.-N.; Lohaus, R.; Bayanga, K.; Heermann, S.; Regen, T.; Van Rossum, D.; Klemm, F.; Schulz, M.; et al. Microglia Promote Colonization of Brain Tissue by Breast Cancer Cells in a Wnt-Dependent Way. Glia 2010, 58, 1477–1489. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Cuiffo, B.G.; Karnoub, A.E. Mesenchymal Stem Cells in Tumor Development: Emerging Roles and Concepts. Cell Adh. Migr. 2012, 6, 220–230. [Google Scholar] [CrossRef]
- Garg, A.D.; Agostinis, P. Cell Death and Immunity in Cancer: From Danger Signals to Mimicry of Pathogen Defense Responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef] [PubMed]
- Romani, N.; Reider, D.; Heuer, M.; Ebner, S.; Kämpgen, E.; Eibl, B.; Niederwieser, D.; Schuler, G. Generation of Mature Dendritic Cells from Human Blood. An Improved Method with Special Regard to Clinical Applicability. J. Immunol. Methods 1996, 196, 137–151. [Google Scholar] [CrossRef]
- Ghirelli, C.; Reyal, F.; Jeanmougin, M.; Zollinger, R.; Sirven, P.; Michea, P.; Caux, C.; Bendriss-Vermare, N.; Donnadieu, M.-H.; Caly, M.; et al. Breast Cancer Cell-Derived GM-CSF Licenses Regulatory Th2 Induction by Plasmacytoid Predendritic Cells in Aggressive Disease Subtypes. Cancer Res. 2015, 75, 2775–2787. [Google Scholar] [CrossRef] [Green Version]
- Pinedo, H.M.; Buter, J.; Luykx-de Bakker, S.A.; Pohlmann, P.R.; van Hensbergen, Y.; Heideman, D.a.M.; van Diest, P.J.; de Gruijl, T.D.; van der Wall, E. Extended Neoadjuvant Chemotherapy in Locally Advanced Breast Cancer Combined with GM-CSF: Effect on Tumour-Draining Lymph Node Dendritic Cells. Eur. J. Cancer 2003, 39, 1061–1067. [Google Scholar] [CrossRef]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A Homologue of the TNF Receptor and Its Ligand Enhance T-Cell Growth and Dendritic-Cell Function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, G.; Vogel, P.; Neale, G.; Reizis, B.; Chi, H. Transforming Growth Factor Beta-Activated Kinase 1 (TAK1)-Dependent Checkpoint in the Survival of Dendritic Cells Promotes Immune Homeostasis and Function. Proc. Natl. Acad. Sci. USA 2012, 109, E343–E352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loser, K.; Mehling, A.; Loeser, S.; Apelt, J.; Kuhn, A.; Grabbe, S.; Schwarz, T.; Penninger, J.M.; Beissert, S. Epidermal RANKL Controls Regulatory T-Cell Numbers via Activation of Dendritic Cells. Nat. Med. 2006, 12, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Mancino, A.; Schioppa, T.; Larghi, P.; Pasqualini, F.; Nebuloni, M.; Chen, I.-H.; Sozzani, S.; Austyn, J.M.; Mantovani, A.; Sica, A. Divergent Effects of Hypoxia on Dendritic Cell Functions. Blood 2008, 112, 3723–3734. [Google Scholar] [CrossRef] [Green Version]
- Headley, M.B.; Bins, A.; Nip, A.; Roberts, E.W.; Looney, M.R.; Gerard, A.; Krummel, M.F. Visualization of Immediate Immune Responses to Pioneer Metastatic Cells in the Lung. Nature 2016, 531, 513–517. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in Cancer: Neutral No More. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Swierczak, A.; Mouchemore, K.A.; Hamilton, J.A.; Anderson, R.L. Neutrophils: Important Contributors to Tumor Progression and Metastasis. Cancer Metastasis Rev. 2015, 34, 735–751. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, D.P.; Correia, B.F.; Salvador, R.; de Sousa, N.; Jacinto, A.; Braga, S.; Cabral, M.G. Circulating Low Density Neutrophils of Breast Cancer Patients Are Associated with Their Worse Prognosis Due to the Impairment of T Cell Responses. Oncotarget 2021, 12, 2388–2403. [Google Scholar] [CrossRef]
- Casbon, A.-J.; Reynaud, D.; Park, C.; Khuc, E.; Gan, D.D.; Schepers, K.; Passegué, E.; Werb, Z. Invasive Breast Cancer Reprograms Early Myeloid Differentiation in the Bone Marrow to Generate Immunosuppressive Neutrophils. PNAS 2015, 112, E566–E575. [Google Scholar] [CrossRef]
- Zhang, W.; Shen, Y.; Huang, H.; Pan, S.; Jiang, J.; Chen, W.; Zhang, T.; Zhang, C.; Ni, C. A Rosetta Stone for Breast Cancer: Prognostic Value and Dynamic Regulation of Neutrophil in Tumor Microenvironment. Front. Immunol. 2020, 11, 1779. [Google Scholar] [CrossRef] [PubMed]
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T.; et al. Granulocyte-Colony Stimulating Factor Promotes Lung Metastasis through Mobilization of Ly6G+Ly6C+ Granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248–21255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.H.; Pickup, M.; Pang, Y.; Gorska, A.E.; Li, Z.; Chytil, A.; Geng, Y.; Gray, J.W.; Moses, H.L.; Yang, L. Gr-1+CD11b+ Myeloid Cells Tip the Balance of Immune Protection to Tumor Promotion in the Premetastatic Lung. Cancer Res. 2010, 70, 6139–6149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, A.; Sharma, S.; Wu, K.; Wu, S.-Y.; Xing, F.; Liu, Y.; Zhao, D.; Deshpande, R.P.; D’Agostino, R.B.; Watabe, K. Nicotine Promotes Breast Cancer Metastasis by Stimulating N2 Neutrophils and Generating Pre-Metastatic Niche in Lung. Nat. Commun. 2021, 12, 474. [Google Scholar] [CrossRef]
- Orditura, M.; Galizia, G.; Diana, A.; Saccone, C.; Cobellis, L.; Ventriglia, J.; Iovino, F.; Romano, C.; Morgillo, F.; Mosca, L.; et al. Neutrophil to Lymphocyte Ratio (NLR) for Prediction of Distant Metastasis-Free Survival (DMFS) in Early Breast Cancer: A Propensity Score-Matched Analysis. ESMO Open 2016, 1, e000038. [Google Scholar] [CrossRef] [Green Version]
- Templeton, A.J.; McNamara, M.G.; Šeruga, B.; Vera-Badillo, F.E.; Aneja, P.; Ocaña, A.; Leibowitz-Amit, R.; Sonpavde, G.; Knox, J.J.; Tran, B.; et al. Prognostic Role of Neutrophil-to-Lymphocyte Ratio in Solid Tumors: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2014, 106, dju124. [Google Scholar] [CrossRef] [Green Version]
- Ethier, J.-L.; Desautels, D.; Templeton, A.; Shah, P.S.; Amir, E. Prognostic Role of Neutrophil-to-Lymphocyte Ratio in Breast Cancer: A Systematic Review and Meta-Analysis. Breast Cancer Res. 2017, 19, 2. [Google Scholar] [CrossRef] [Green Version]
- Ueshima, C.; Kataoka, T.R.; Hirata, M.; Furuhata, A.; Suzuki, E.; Toi, M.; Tsuruyama, T.; Okayama, Y.; Haga, H. The Killer Cell Ig-like Receptor 2DL4 Expression in Human Mast Cells and Its Potential Role in Breast Cancer Invasion. Cancer Immunol. Res. 2015, 3, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Esposito, I.; Menicagli, M.; Funel, N.; Bergmann, F.; Boggi, U.; Mosca, F.; Bevilacqua, G.; Campani, D. Inflammatory Cells Contribute to the Generation of an Angiogenic Phenotype in Pancreatic Ductal Adenocarcinoma. J. Clin. Pathol. 2004, 57, 630–636. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.-W.; Yang, S.-Z.; Gao, J.; Pan, K.; Chen, J.-Y.; Wang, Y.-L.; Wei, L.-X.; Dong, J.-H. Prognostic Significance of Mast Cell Count Following Curative Resection for Pancreatic Ductal Adenocarcinoma. Surgery 2011, 149, 576–584. [Google Scholar] [CrossRef]
- Elpek, G.O.; Gelen, T.; Aksoy, N.H.; Erdoğan, A.; Dertsiz, L.; Demircan, A.; Keleş, N. The Prognostic Relevance of Angiogenesis and Mast Cells in Squamous Cell Carcinoma of the Oesophagus. J. Clin. Pathol. 2001, 54, 940–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunder, C.A.; St John, A.L.; Abraham, S.N. Mast Cell Modulation of the Vascular and Lymphatic Endothelium. Blood 2011, 118, 5383–5393. [Google Scholar] [CrossRef] [PubMed]
- Krasnova, Y.; Putz, E.M.; Smyth, M.J.; Souza-Fonseca-Guimaraes, F. Bench to Bedside: NK Cells and Control of Metastasis. Clin. Immunol. 2017, 177, 50–59. [Google Scholar] [CrossRef]
- Feuerer, M.; Rocha, M.; Bai, L.; Umansky, V.; Solomayer, E.F.; Bastert, G.; Diel, I.J.; Schirrmacher, V. Enrichment of Memory T Cells and Other Profound Immunological Changes in the Bone Marrow from Untreated Breast Cancer Patients. Int. J. Cancer 2001, 92, 96–105. [Google Scholar] [CrossRef]
- Rezaeifard, S.; Talei, A.; Shariat, M.; Erfani, N. Tumor Infiltrating NK Cell (TINK) Subsets and Functional Molecules in Patients with Breast Cancer. Mol. Immunol. 2021, 136, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.L.; Guimaraes, J.C.; Auf der Maur, P.; De Silva, D.; Trefny, M.P.; Okamoto, R.; Bruno, S.; Schmidt, A.; Mertz, K.; Volkmann, K.; et al. Author Correction: Hepatic Stellate Cells Suppress NK Cell-Sustained Breast Cancer Dormancy. Nature 2021, 600, E7. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.R.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A Combinational Therapy of EGFR-CAR NK Cells and Oncolytic Herpes Simplex Virus 1 for Breast Cancer Brain Metastases. Oncotarget 2016, 7, 27764–27777. [Google Scholar] [CrossRef] [Green Version]
- Afshar-Kharghan, V. The Role of the Complement System in Cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [Green Version]
- Popeda, M.; Markiewicz, A.; Stokowy, T.; Szade, J.; Niemira, M.; Kretowski, A.; Bednarz-Knoll, N.; Zaczek, A.J. Reduced Expression of Innate Immunity-Related Genes in Lymph Node Metastases of Luminal Breast Cancer Patients. Sci. Rep. 2021, 11, 5097. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Q.; Li, T.; Liao, Q.; Zhao, Y. Role of the Complement System in the Tumor Microenvironment. Cancer Cell Int. 2019, 19, 300. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chintala, N.K.; Vadrevu, S.K.; Patel, J.; Karbowniczek, M.; Markiewski, M.M. Pulmonary Alveolar Macrophages Contribute to the Premetastatic Niche by Suppressing Antitumor T Cell Responses in the Lungs. J. Immunol. 2015, 194, 5529–5538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, T.; Yamamoto-Ibusuki, M.; Sueta, A.; Kubo, T.; Irie, A.; Kikuchi, K.; Kariu, T.; Iwase, H. Influence of the C5a-C5a Receptor System on Breast Cancer Progression and Patient Prognosis. Breast Cancer 2016, 23, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Akhir, F.N.M.; Noor, M.H.M.; Leong, K.W.K.; Nabizadeh, J.A.; Manthey, H.D.; Sonderegger, S.E.; Fung, J.N.T.; McGirr, C.E.; Shiels, I.A.; Mills, P.C.; et al. An Immunoregulatory Role for Complement Receptors in Murine Models of Breast Cancer. Antibodies 2021, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Qian, Y.; Weiser, P.; Zhou, X.; Lu, H.; Studelska, D.R.; Zhang, L. Glycosaminoglycans and Activated Contact System in Cancer Patient Plasmas. Prog. Mol. Biol. Transl. Sci. 2010, 93, 473–495. [Google Scholar] [CrossRef] [PubMed]
- Langouo Fontsa, M.; Aiello, M.M.; Migliori, E.; Scartozzi, M.; Lambertini, M.; Willard-Gallo, K.; Solinas, C. Thromboembolism and Immune Checkpoint Blockade in Cancer Patients: An Old Foe for New Research. Target. Oncol. 2022, 17, 497–505. [Google Scholar] [CrossRef]
- Taylor, M.; Rössler, J.; Geoerger, B.; Laplanche, A.; Hartmann, O.; Vassal, G.; Farace, F. High Levels of Circulating VEGFR2+ Bone Marrow-Derived Progenitor Cells Correlate with Metastatic Disease in Patients with Pediatric Solid Malignancies. Clin. Cancer Res. 2009, 15, 4561–4571. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Ward, M.M.; O’Loughlin, J.; Boeck, M.; Wiener, N.; Chuang, E.; Cigler, T.; Moore, A.; Donovan, D.; Lam, C.; et al. Incremental Increase in VEGFR1+ Hematopoietic Progenitor Cells and VEGFR2+ Endothelial Progenitor Cells Predicts Relapse and Lack of Tumor Response in Breast Cancer Patients. Breast Cancer Res. Treat. 2012, 132, 235–242. [Google Scholar] [CrossRef]
- Mehta, A.K.; Kadel, S.; Townsend, M.G.; Oliwa, M.; Guerriero, J.L. Macrophage Biology and Mechanisms of Immune Suppression in Breast Cancer. Front. Immunol. 2021, 12, 626. [Google Scholar] [CrossRef]
- Jiang, X. Harnessing the Immune System for the Treatment of Breast Cancer. J. Zhejiang Univ. Sci. B 2014, 15, 1–15. [Google Scholar] [CrossRef]
- Ye, Y.; Xu, C.; Chen, F.; Liu, Q.; Cheng, N. Targeting Innate Immunity in Breast Cancer Therapy: A Narrative Review. Front. Immunol. 2021, 12, 771201. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Quispe-Tintaya, W.; Jahangir, A.; Asafu-Adjei, D.; Ramos, I.; Sintim, H.O.; Zhou, J.; Hayakawa, Y.; Karaolis, D.K.R.; Gravekamp, C. STING Ligand C-Di-GMP Improves Cancer Vaccination against Metastatic Breast Cancer. Cancer Immunol. Res. 2014, 2, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meric-Bernstam, F.; Sweis, R.F.; Hodi, F.S.; Messersmith, W.A.; Andtbacka, R.H.I.; Ingham, M.; Lewis, N.; Chen, X.; Pelletier, M.; Chen, X.; et al. Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas. Clin. Cancer Res. 2022, 28, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-Stimulating Factor 1 Receptor (CSF1R) Inhibitors in Cancer Therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Gomez-Roca, C.A.; Cassier, P.A.; Italiano, A.; Cannarile, M.; Ries, C.; Brillouet, A.; Mueller, C.; Jegg, A.-M.; Meneses-Lorente, G.; Baehner, M.; et al. Phase I Study of RG7155, a Novel Anti-CSF1R Antibody, in Patients with Advanced/Metastatic Solid Tumors. JCO 2015, 33, 3005. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Patnaik, A.; Eisenberg, P.; Sachdev, J.; Weise, A.; Kaufman, D.R.; Aromin, I.; West, B.L.; Tong, S.; Ribas, A. 18TiP—Phase 1/2a Study of Double Immune Suppression Blockade by Combining a CSF1R Inhibitor (Pexidartinib/PLX3397) with an Anti PD-1 Antibody (Pembrolizumab) to Treat Advanced Melanoma and Other Solid Tumors. Ann. Oncol. 2015, 26, viii5. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Sun, R.; Liu, L.; Yang, D.; Xiang, Q.; Li, L.; Tang, J.; Qiu, Z.; Peng, W.; Wang, Y.; et al. Tumor Suppressor DRD2 Facilitates M1 Macrophages and Restricts NF-ΚB Signaling to Trigger Pyroptosis in Breast Cancer. Theranostics 2021, 11, 5214–5231. [Google Scholar] [CrossRef]
- Chi, A.S.; Tarapore, R.S.; Hall, M.D.; Shonka, N.; Gardner, S.; Umemura, Y.; Sumrall, A.; Khatib, Z.; Mueller, S.; Kline, C.; et al. Pediatric and Adult H3 K27M-Mutant Diffuse Midline Glioma Treated with the Selective DRD2 Antagonist ONC201. J. Neurooncol. 2019, 145, 97–105. [Google Scholar] [CrossRef]
- Stein, M.N.; Malhotra, J.; Tarapore, R.S.; Malhotra, U.; Silk, A.W.; Chan, N.; Rodriguez, L.; Aisner, J.; Aiken, R.D.; Mayer, T.; et al. Safety and Enhanced Immunostimulatory Activity of the DRD2 Antagonist ONC201 in Advanced Solid Tumor Patients with Weekly Oral Administration. J. Immunother. Cancer 2019, 7, 136. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Li, B.; Chen, J.; Dang, J.; Chen, S.; Gunes, E.G.; Xu, B.; Tian, L.; Muend, S.; Raoof, M.; et al. Effect of Cabazitaxel on Macrophages Improves CD47-Targeted Immunotherapy for Triple-Negative Breast Cancer. J. Immunother. Cancer 2021, 9, e002022. [Google Scholar] [CrossRef]
- Yuan, J.; Shi, X.; Chen, C.; He, H.; Liu, L.; Wu, J.; Yan, H. High Expression of CD47 in Triple Negative Breast Cancer Is Associated with Epithelial-Mesenchymal Transition and Poor Prognosis. Oncol. Lett. 2019, 18, 3249–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galsky, M.D.; Dritselis, A.; Kirkpatrick, P.; Oh, W.K. Cabazitaxel. Nat. Rev. Drug Discov. 2010, 9, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Coward, J.; Mislang, A.R.A.; Cosman, R.; Nagrial, A.; Jin, X.; Li, B.; Wang, Z.M.; Kwek, K.Y.; Xia, D.; et al. Safety of AK117, an Anti-CD47 Monoclonal Antibody, in Patients with Advanced or Metastatic Solid Tumors in a Phase I Study. JCO 2021, 39, 2630. [Google Scholar] [CrossRef]
- Forghani, P.; Waller, E.K. Poly (I: C) Modulates the Immunosuppressive Activity of Myeloid-Derived Suppressor Cells in a Murine Model of Breast Cancer. Breast Cancer Res. Treat. 2015, 153, 21–30. [Google Scholar] [CrossRef]
- Yin, T.; He, S.; Wang, Y. Toll-like Receptor 7/8 Agonist, R848, Exhibits Antitumoral Effects in a Breast Cancer Model. Mol. Med. Rep. 2015, 12, 3515–3520. [Google Scholar] [CrossRef] [Green Version]
- Chien, A.J.; Soliman, H.H.; Ewing, C.A.; Boughey, J.C.; Campbell, M.J.; Rugo, H.S.; Wallace, A.M.; Albain, K.S.; Stringer-Reasor, E.M.; Church, A.L.; et al. Evaluation of Intra-Tumoral (IT) SD-101 and Pembrolizumab (Pb) in Combination with Paclitaxel (P) Followed by AC in High-Risk HER2-Negative (HER2-) Stage II/III Breast Cancer: Results from the I-SPY 2 Trial. JCO 2021, 39, 508. [Google Scholar] [CrossRef]
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic Strategies for the Costimulatory Molecule OX40 in T-Cell-Mediated Immunity. Acta Pharm. Sin. B 2020, 10, 414–433. [Google Scholar] [CrossRef]
- Levy, R. Intratumoral Injection of SD-101, an Immunostimulatory CpG Oligonucleotide, in Combination With BMS- 986178, an OX40 Agonist Antibody, in Advanced Solid Malignancies [CA012-014]. Available online: https://clinicaltrials.gov/ct2/show/NCT03831295 (accessed on 20 September 2022).
- Chu, D.-T.; Bac, N.D.; Nguyen, K.-H.; Tien, N.L.B.; Thanh, V.V.; Nga, V.T.; Ngoc, V.T.N.; Anh Dao, D.T.; Hoan, L.N.; Hung, N.P.; et al. An Update on Anti-CD137 Antibodies in Immunotherapies for Cancer. Int. J. Mol. Sci. 2019, 20, 1822. [Google Scholar] [CrossRef] [Green Version]
- F-Star Therapeutics Limited. A Phase 1 Open-Label. Study to Evaluate the Safety and Antitumor Activity of FS120, an OX40/CD137 Bispecific Antibody, Alone and in Combination With Pembrolizumab, in Subjects with Advanced Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT04648202 (accessed on 22 September 2022).
- Solinas, C.; Aiello, M.; De Silva, P.; Gu-Trantien, C.; Migliori, E.; Willard-Gallo, K. Targeting PD-1 in Cancer: Biological Insights with a Focus on Breast Cancer. Crit. Rev. Oncol. Hematol. 2019, 142, 35–43. [Google Scholar] [CrossRef]
- De Silva, P.; Aiello, M.; Gu-Trantien, C.; Migliori, E.; Willard-Gallo, K.; Solinas, C. Targeting CTLA-4 in Cancer: Is It the Ideal Companion for PD-1 Blockade Immunotherapy Combinations? Int. J. Cancer 2021, 149, 31–41. [Google Scholar] [CrossRef]
- Solinas, C.; Migliori, E.; De Silva, P.; Willard-Gallo, K. LAG3: The Biological Processes That Motivate Targeting This Immune Checkpoint Molecule in Human Cancer. Cancers 2019, 11, 1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galván Morales, M.A.; Barrera Rodríguez, R.; Santiago Cruz, J.R.; Teran, L.M. Overview of New Treatments with Immunotherapy for Breast Cancer and a Proposal of a Combination Therapy. Molecules 2020, 25, 5686. [Google Scholar] [CrossRef] [PubMed]
- The Netherlands Cancer Institute. Monalizumab and Trastuzumab In Metastatic HER2-POSitive BreAst Cancer: MIMOSA-Trial. Available online: https://clinicaltrials.gov/ct2/show/results/NCT04307329 (accessed on 23 September 2022).
- Geurts, V.C.M.; Voorwerk, L.; Sikorska, K.; Dongen, M.G.V.; Kemper, I.; Mandjes, I.a.M.; Haanen, J.B.a.G.; Sonke, G.S.; Kok, M. 90TiP Monalizumab and Trastuzumab in Metastatic HER2-Positive Breast Cancer: MIMOSA-Trial. Ann. Oncol. 2021, 32, S59. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb. A Phase 1 Study of the Safety and Pharmacokinetics of Anti-KIR Monoclonal Antibody (Lirilumab, BMS-986015) in Combination With Anti-PD-1 Monoclonal Antibody (Nivolumab, BMS-936558) or in Combination With Nivolumab and Anti-CTLA-4 Monoclonal Antibody (Ipilimumab, BMS-734016) in Advanced and/or Metastatic Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03203876 (accessed on 23 September 2022).
- Pellegrino, B.; Tommasi, C.; Cursio, O.E.; Musolino, A.; Migliori, E.; De Silva, P.; Senevirathne, T.H.; Schena, M.; Scartozzi, M.; Farci, D.; et al. A Review of Immune Checkpoint Blockade in Breast Cancer. Semin. Oncol. 2021, 48, 208–225. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Metastatic Site | Marker and Cytokines | Distribution and Function in Healthy Tissue | Role in BC Metastases | Ref. |
|---|---|---|---|---|
| Nodes | CD168 | Macrophages of marginal and medullary sinuses | ↓CD163+ TAMs and ↑Foxp3+ Treg in metastatic SLN | [43] |
| CD68 | Monocyte lineage and resident macrophages | high CD68/(CD3+CD20+) ratio in BC primary tumors is associated with shorter RFS | [9] | |
| CD68+/stabilin-1+ cells in BC stroma are directly correlated with the number of metastatic nodes | [44] | |||
| CD163 | Resident macrophages | M2 marker; high levels are associated with bad prognosis | [45] | |
| Bone | CD137 | Circulating monocytes | Increases monocyte adhesion and induces osteoclast differentiation, favoring BC cell bone homing | [21] |
| CCL2 | Mediates the recruitment of monocytes and T lymphocytes | Induces macrophage-mediated bone destruction and tumor cell proliferation through VEGF-induced pathways | [57] | |
| CXCL12 | On different tissues (brain, thymus, lung, liver) with chemotactic properties | Facilitates BC cells bone homing | [58] | |
| Lung | VCAM-1 | On endothelial cells stimulated with cytokines; it binds to α4β1-integrin expressed by monocytes | VCAM-1/α4β1-integrin interaction is determinant for tumor cell extravasation and migration through the pulmonary endothelium | [60] |
| CD11b/CCR2+/VEGFR1+ | Monocytes | Combined expression induced by cytokines, facilitates lung metastatic involvement | [61] | |
| CCL3 | Lung-resident macrophages | In vivo model, its deletion or depletion of its receptor CCR1 in MAMs, reduce the number of lung metastasis foci | [62] |
| Target Cells | Compound | Effect | Administration | Trial Phase | Ref. |
|---|---|---|---|---|---|
| MDSCs | STING agonist | Increases the production of IL-12 with induction of the T lymphocyte response | IT | I | [113] |
| TAMs | ARRY-382 (CSF-1R inhibitor) | Reduces T-lymphocyte-suppressive TAM infiltrates | PO | I | [114] |
| Emactuzumab (anti-CSF-1R) | IV | I | [115] | ||
| Pexidartinib (CSF-1R inhibitor) | In combination with pembrolizumab, it inhibits M2 TAM polarization and restores the immune response against tumor cells | PO | I/II | [116] | |
| DRD2 antagonist | Increases M1 TAM polarization | PO | I | [119] | |
| CD47 antagonist | Induction of M1 TAM polarization and PrCR | IV | I | [123] | |
| NKs | TLR9 agonist | In combination with anti-OX40, it enhances the activity of immune cells against tumor cells | IT | I | [128] |
| FS120 (anti-OX40/CD137) | As single agents or in combination with pembrolizumab, it activates the cytotoxicity of CD8+ T lymphocytes and NKs and reprograms Tregs | IV | I | [130] | |
| Monalizumab (anti-NKG2A) | In combination with trastuzumab in metastatic HER2-positive BC, it improves ADCC and overcomes trastuzumab resistance. In addition, it can promote anti-tumor immunity by unleashing NK cells and CD8+ T lymphocytes | IV | II | [135,136] | |
| Lirilumab (anti-KIR) | In combination with nivolumab, it enhances NK cytotoxicity | IV | I | [137] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tommasi, C.; Pellegrino, B.; Diana, A.; Palafox Sancez, M.; Orditura, M.; Scartozzi, M.; Musolino, A.; Solinas, C. The Innate Immune Microenvironment in Metastatic Breast Cancer. J. Clin. Med. 2022, 11, 5986. https://doi.org/10.3390/jcm11205986
Tommasi C, Pellegrino B, Diana A, Palafox Sancez M, Orditura M, Scartozzi M, Musolino A, Solinas C. The Innate Immune Microenvironment in Metastatic Breast Cancer. Journal of Clinical Medicine. 2022; 11(20):5986. https://doi.org/10.3390/jcm11205986
Chicago/Turabian StyleTommasi, Chiara, Benedetta Pellegrino, Anna Diana, Marta Palafox Sancez, Michele Orditura, Mario Scartozzi, Antonino Musolino, and Cinzia Solinas. 2022. "The Innate Immune Microenvironment in Metastatic Breast Cancer" Journal of Clinical Medicine 11, no. 20: 5986. https://doi.org/10.3390/jcm11205986