

Exploring the Complex Network of Heme-Triggered Effects on the Blood Coagulation System
 , and
, and 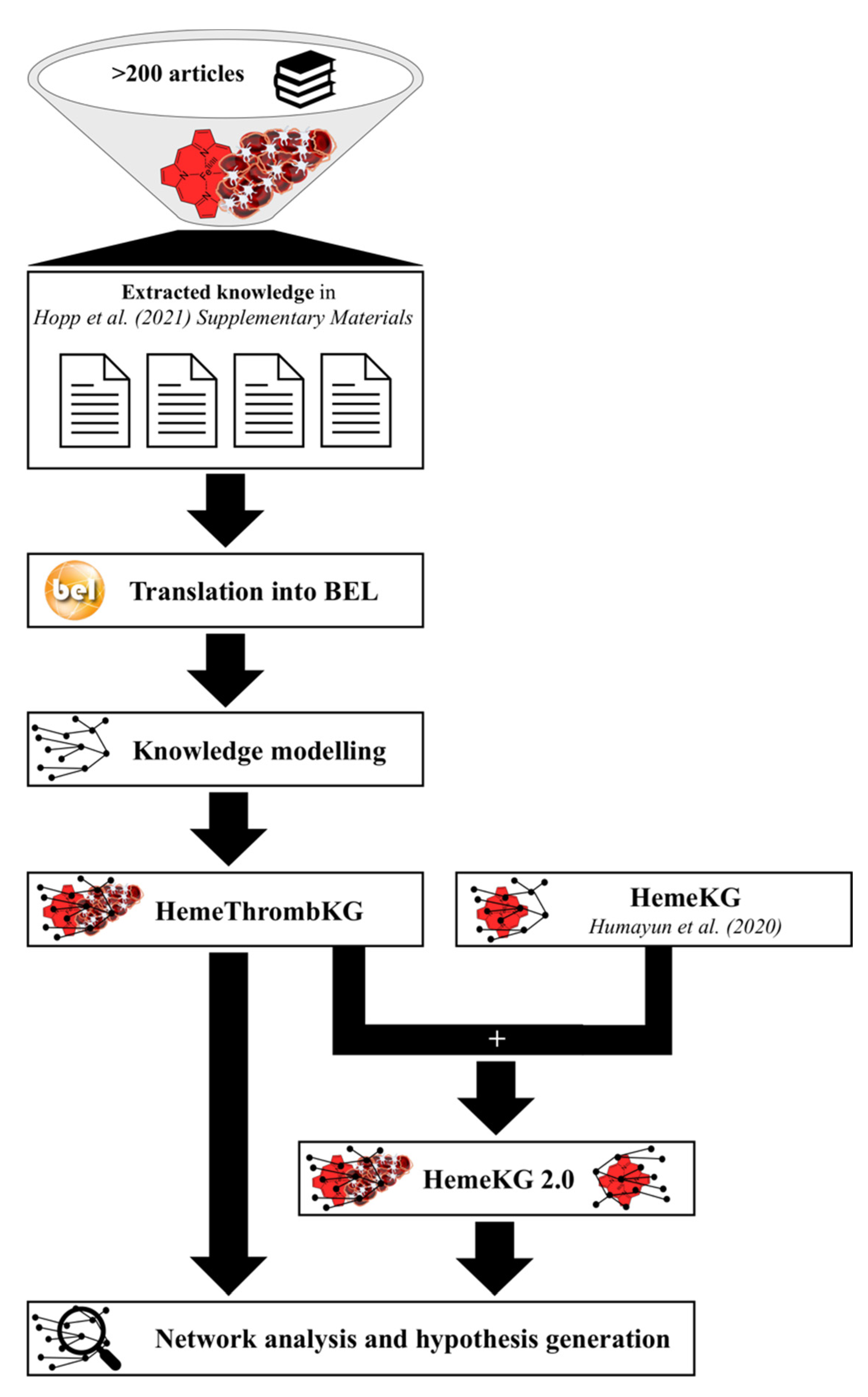{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Knowledge Modeling and Inclusion of the Knowledge Graph into HemeKG
2.2. Analysis of the Crosstalk of Heme with the Blood Coagulation System
3. Results
3.1. HemeThrombKG Illustrates the Knowledge about Heme’s Interference in the Blood Coagulation System
3.2. HemeThrombKG and HemeKG 2.0 Enable the Detailed Analysis of Heme-Triggered Thrombosis at the Molecular Level
3.2.1. Crosstalk of Heme and the Plasma Proteins of the Blood Coagulation System
3.2.2. Crosstalk of Heme within the Pathways of Platelet Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shet, A.S.; Lizarralde-Iragorri, M.A.; Naik, R.P. The Molecular Basis for the Prothrombotic State in Sickle Cell Disease. Haematologica 2020, 105, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- van Bijnen, S.T.A.; van Heerde, W.L.; Muus, P. Mechanisms and Clinical Implications of Thrombosis in Paroxysmal Nocturnal Hemoglobinuria. J. Thromb. Haemost. 2012, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Panch, S.R.; Montemayor-Garcia, C.; Klein, H.G. Hemolytic Transfusion Reactions. New Engl. J. Med. 2019, 381, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Delvasto-Nuñez, L.; Jongerius, I.; Zeerleder, S. It Takes Two to Thrombosis: Hemolysis and Complement. Blood Rev. 2021, 50, 100834. [Google Scholar] [CrossRef]
- Srisuwananukorn, A.; Raslan, R.; Zhang, X.; Shah, B.N.; Han, J.; Gowhari, M.; Molokie, R.E.; Gordeuk, V.R.; Saraf, S.L. Clinical, Laboratory, and Genetic Risk Factors for Thrombosis in Sickle Cell Disease. Blood Adv. 2020, 4, 1978–1986. [Google Scholar] [CrossRef]
- Naik, R.P.; Streiff, M.B.; Haywood, C.; Nelson, J.A.; Lanzkron, S. Venous Thromboembolism in Adults with Sickle Cell Disease: A Serious and under-Recognized Complication. Am. J. Med. 2013, 126, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Nouraie, M.; Lee, J.S.; Zhang, Y.; Kanias, T.; Zhao, X.; Xiong, Z.; Oriss, T.B.; Zeng, Q.; Kato, G.J.; Gibbs, J.S.R.; et al. The Relationship between the Severity of Hemolysis, Clinical Manifestations and Risk of Death in 415 Patients with Sickle Cell Anemia in the US and Europe. Haematologica 2013, 98, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Roumenina, L.T.; Rayes, J.; Lacroix-Desmazes, S.; Dimitrov, J.D. Heme: Modulator of Plasma Systems in Hemolytic Diseases. Trends Mol. Med. 2016, 22, 200–213. [Google Scholar] [CrossRef]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The Clinical Sequelae of Intravascular Hemolysis and Extracellular Plasma Hemoglobin. J. Am. Med Assoc. 2005, 293, 1653. [Google Scholar] [CrossRef]
- Samuel, P.P.; White, M.A.; Ou, W.C.; Case, D.A.; Phillips, G.N.; Olson, J.S. The Interplay between Molten Globules and Heme Disassociation Defines Human Hemoglobin Disassembly. Biophys. J. 2020, 118, 1381–1400. [Google Scholar] [CrossRef]
- Andersen, C.B.F.; Stødkilde, K.; Sæderup, K.L.; Kuhlee, A.; Raunser, S.; Graversen, J.H.; Moestrup, S.K. Haptoglobin. Antioxid. Redox Signal. 2017, 26, 814–831. [Google Scholar] [CrossRef] [PubMed]
- Alayash, A.I.; Andersen, C.B.F.; Moestrup, S.K.; Bülow, L. Haptoglobin: The Hemoglobin Detoxifier in Plasma. Trends Biotechnol. 2013, 31, 2–3. [Google Scholar] [CrossRef]
- Kumar, S.; Bandyopadhyay, U. Free Heme Toxicity and Its Detoxification Systems in Human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Bozza, M.T. Red Alert: Labile Heme Is an Alarmin. Curr. Opin. Immunol. 2016, 38, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Noé, R.; Bozinovic, N.; Lecerf, M.; Lacroix-Desmazes, S.; Dimitrov, J.D. Use of Cysteine as a Spectroscopic Probe for Determination of Heme-Scavenging Capacity of Serum Proteins and Whole Human Serum. J. Pharm. Biomed. Anal. 2019, 172, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.S.; Govender, K.; Munoz, C.J.; Williams, A.T.; O’Boyle, Q.T.; Savla, C.; Cabrales, P.; Palmer, A.F. Purification and Analysis of a Protein Cocktail Capable of Scavenging Cell-free Hemoglobin, Heme, and Iron. Transfusion 2021, 61, 1894–1907. [Google Scholar] [CrossRef] [PubMed]
- Humayun, F.; Domingo-Fernández, D.; Paul George, A.A.; Hopp, M.-T.; Syllwasschy, B.F.; Detzel, M.S.; Hoyt, C.T.; Hofmann-Apitius, M.; Imhof, D. A Computational Approach for Mapping Heme Biology in the Context of Hemolytic Disorders. Front. Bioeng. Biotechnol. 2020, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graça-Souza, A.V.; Bozza, M.T. Characterization of Heme as Activator of Toll-like Receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [Green Version]
- Janciauskiene, S.; Vijayan, V.; Immenschuh, S. TLR4 Signaling by Heme and the Role of Heme-Binding Blood Proteins. Front. Immunol. 2020, 11, 1964. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M.; et al. Heme Triggers TLR4 Signaling Leading to Endothelial Cell Activation and Vaso-Occlusion in Murine Sickle Cell Disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef]
- Merle, N.S.; Paule, R.; Leon, J.; Daugan, M.; Robe-Rybkine, T.; Poillerat, V.; Torset, C.; Frémeaux-Bacchi, V.; Dimitrov, J.D.; Roumenina, L.T. P-Selectin Drives Complement Attack on Endothelium during Intravascular Hemolysis in TLR-4/Heme-dependent Manner. Proc. Natl. Acad. Sci. USA 2019, 116, 6280–6285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roumenina, L.T.; Chadebech, P.; Bodivit, G.; Vieira-Martins, P.; Grunenwald, A.; Boudhabhay, I.; Poillerat, V.; Pakdaman, S.; Kiger, L.; Jouard, A.; et al. Complement Activation in Sickle Cell Disease: Dependence on Cell Density, Hemolysis and Modulation by Hydroxyurea Therapy. Am. J. Hematol. 2020, 95, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frimat, M.; Tabarin, F.; Dimitrov, J.D.; Poitou, C.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement Activation by Heme as a Secondary Hit for Atypical Hemolytic Uremic Syndrome. Blood 2013, 122, 282–292. [Google Scholar] [CrossRef]
- Pawluczkowycz, A.W.; Lindorfer, M.A.; Waitumbi, J.N.; Taylor, R.P. Hematin Promotes Complement Alternative Pathway-Mediated Deposition of C3 Activation Fragments on Human Erythrocytes: Potential Implications for the Pathogenesis of Anemia in Malaria. J. Immunol. 2007, 179, 5543–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frimat, M.; Boudhabhay, I.; Roumenina, L.T. Hemolysis Derived Products Toxicity and Endothelium: Model of the Second Hit. Toxins 2019, 11, 660. [Google Scholar] [CrossRef] [Green Version]
- Hopp, M.-T.; Imhof, D. Linking Labile Heme with Thrombosis. J. Clin. Med. 2021, 10, 427. [Google Scholar] [CrossRef]
- Conran, N.; De Paula, E.V. Thromboinflammatory Mechanisms in Sickle Cell Disease—Challenging the Hemostatic Balance. Haematologica 2020, 105, 2380. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A Multifaceted Pathway Database Bridging Metabolomics to Other Omics Research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- Domingo-Fernández, D.; Mubeen, S.; Marín-Llaó, J.; Hoyt, C.T.; Hofmann-Apitius, M. PathMe: Merging and Exploring Mechanistic Pathway Knowledge. BMC Bioinform. 2019, 20, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyt, C.T.; Domingo-Fernández, D.; Aldisi, R.; Xu, L.; Kolpeja, K.; Spalek, S.; Wollert, E.; Bachman, J.; Gyori, B.M.; Greene, P.; et al. Re-Curation and Rational Enrichment of Knowledge Graphs in Biological Expression Language. Database 2019, 2019, baz068. [Google Scholar] [CrossRef] [PubMed]
- Huntley, R.P.; Binns, D.; Dimmer, E.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A User Tutorial for the Web-Based Gene Ontology Browser. Database 2009, 2009, bap010. [Google Scholar] [CrossRef] [PubMed]
- Hopp, M.T.; Domingo-Fernández, D.; Gadiya, Y.; Detzel, M.S.; Graf, R.; Schmalohr, B.F.; Kodamullil, A.T.; Imhof, D.; Hofmann-Apitius, M. Linking COVID-19 and Heme-Driven Pathophysiologies: A Combined Computational–Experimental Approach. Biomolecules 2021, 11, 644. [Google Scholar] [CrossRef]
- Becker, C.G.; Wagner, M.; Kaplan, A.P.; Silverberg, M.; Grady, R.W.; Liem, H.; Muller-Eberhard, U. Activation of Factor XII-Dependent Pathways in Human Plasma by Hematin and Protoporphyrin. J. Clin. Investig. 1985, 76, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Hopp, M.-T.; Alhanafi, N.; Paul George, A.A.; Hamedani, N.S.; Biswas, A.; Oldenburg, J.; Pötzsch, B.; Imhof, D. Molecular Insights and Functional Consequences of the Interaction of Heme with Activated Protein, C. Antioxid. Redox Signal. 2021, 34, 32–48. [Google Scholar] [CrossRef]
- Repessé, Y.; Dimitrov, J.D.; Peyron, I.; Moshai, E.F.; Kiger, L.; Dasgupta, S.; Delignat, S.; Marden, M.C.; Kaveri, S.V.; Lacroix-Desmazes, S. Heme Binds to Factor VIII and Inhibits Its Interaction with Activated Factor IX. J. Thromb. Haemost. 2012, 10, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Green, D.; Furby, F.H.; Berndt, M.C. The Interaction of the VIII/von Willebrand Factor Complex with Hematin. Thromb. Haemost. 1986, 56, 277–282. [Google Scholar]
- Hunt, R.C.; Katneni, U.; Yalamanoglu, A.; Indig, F.E.; Ibla, J.C.; Kimchi-Sarfaty, C. Contribution of ADAMTS13-independent VWF Regulation in Sickle Cell Disease. J. Thromb. Haemost. 2022, 20, 2098–2108. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Chantrathammachart, P.; Wang, S.; Jonas, W.; Kirchhofer, D.; Gailani, D.; Gruber, A.; Kasthuri, R.; Key, N.S.; Mackman, N.; et al. Excess of Heme Induces Tissue Factor-Dependent Activation of Coagulation in Mice. Haematologica 2015, 100, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Setty, B.N.Y.; Betal, S.G.; Zhang, J.; Stuart, M.J. Heme Induces Endothelial Tissue Factor Expression: Potential Role in Hemostatic Activation in Patients with Hemolytic Anemia. J. Thromb. Haemost. 2008, 6, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Souza, G.R.; Fiusa, M.M.L.; Lanaro, C.; Colella, M.P.; Montalvao, S.A.L.; Saad, S.T.O.; Costa, F.F.; Traina, F.; Annichino-Bizzacchi, J.M.; de Paula, E.V. Coagulation Activation by Heme: Evidence from Global Hemostasis Assays. Blood 2014, 124, 455. [Google Scholar] [CrossRef]
- May, O.; Yatime, L.; Merle, N.S.; Delguste, F.; Howsam, M.; Daugan, M.V.; Paul-Constant, C.; Billamboz, M.; Ghinet, A.; Lancel, S.; et al. The Receptor for Advanced Glycation End Products Is a Sensor for Cell-free Heme. FEBS J. 2021, 288, 3448–3464. [Google Scholar] [CrossRef] [PubMed]
- Glueck, R.; Green, D.; Cohen, I.; Ts’ao, C. Hematin: Unique Effects on Hemostasis. Blood 1983, 61, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.; Reynolds, N.; Klein, J.; Kohl, H.; Ts’ao, C.H. The Inactivation of Hemostatic Factors by Hematin. J. Lab. Clin. Med. 1983, 102, 361–369. [Google Scholar] [PubMed]
- Neely, S.M.; Gardner, D.V.; Reynolds, N.; Green, D.; Ts’ao, C. Mechanism and Characteristics of Platelet Activation by Haematin. Br. J. Haematol. 1984, 58, 305–316. [Google Scholar] [CrossRef]
- Ke, Z.; Huang, Q. Haem-Assisted Dityrosine-Cross-Linking of Fibrinogen under Non-Thermal Plasma Exposure: One Important Mechanism of Facilitated Blood Coagulation. Sci. Rep. 2016, 6, 26982. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.; Zhang, Y.; Wu, T.; Wang, M.; Zhang, Y.; Li, R.; Wang, L.; Xue, Q.; Wang, S. Label-Free Detection of Fibrinogen Based on Fibrinogen-Enhanced Peroxidase Activity of Fibrinogen-Hemin Composite. Analyst 2018, 143, 725–730. [Google Scholar] [CrossRef]
- Bergmeier, W.; Hynes, R.O. Extracellular Matrix Proteins in Hemostasis and Thrombosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005132. [Google Scholar] [CrossRef] [Green Version]
- Nieswandt, B.; Pleines, I.; Bender, M. Platelet Adhesion and Activation Mechanisms in Arterial Thrombosis and Ischaemic Stroke. J. Thromb. Haemost. 2011, 9, 92–104. [Google Scholar] [CrossRef]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling During Platelet Adhesion and Activation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Woollard, K.J.; Sturgeon, S.; Chin-Dusting, J.P.F.; Salem, H.H.; Jackson, S.P. Erythrocyte Hemolysis and Hemoglobin Oxidation Promote Ferric Chloride-Induced Vascular Injury. J. Biol. Chem. 2009, 284, 13110–13118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, S.; Tsukiji, N.; Otake, S.; Oishi, N.; Sasaki, T.; Shirai, T.; Yoshikawa, Y.; Takano, K.; Shinmori, H.; Inukai, T.; et al. Heme Activates Platelets and Exacerbates Rhabdomyolysis-Induced Acute Kidney Injury via CLEC-2 and GPVI/FcRγ. Blood Adv. 2021, 5, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M.; Ezumi, Y.; Arai, M.; Takayama, H. A Novel Association of Fc Receptor γ-Chain with Glycoprotein VI and Their Co-Expression as a Collagen Receptor in Human Platelets. J. Biol. Chem. 1997, 272, 23528–23531. [Google Scholar] [CrossRef] [Green Version]
- Bourne, J.H.; Colicchia, M.; Di, Y.; Martin, E.; Slater, A.; Roumenina, L.T.; Dimitrov, J.D.; Watson, S.P.; Rayes, J. Heme Induces Human and Mouse Platelet Activation through C-Type-Lectin-like Receptor-2. Haematologica 2020, 106, 626–629. [Google Scholar] [CrossRef]
- Porto, B.N.; Alves, L.S.; Fernández, P.L.; Dutra, T.P.; Figueiredo, R.T.; Graça-Souza, A.V.; Bozza, M.T. Heme Induces Neutrophil Migration and Reactive Oxygen Species Generation through Signaling Pathways Characteristic of Chemotactic Receptors. J. Biol. Chem. 2007, 282, 24430–24436. [Google Scholar] [CrossRef] [Green Version]
- Annarapu, G.K.; Nolfi-Donegan, D.; Reynolds, M.; Wang, Y.; Kohut, L.; Zuckerbraun, B.; Shiva, S. Heme Stimulates Platelet Mitochondrial Oxidant Production to Induce Targeted Granule Secretion. Redox Biol. 2021, 48, 102205. [Google Scholar] [CrossRef]
- Graça-Souza, A.V.; Arruda, M.A.B.; de Freitas, M.S.; Barja-Fidalgo, C.; Oliveira, P.L. Neutrophil Activation by Heme: Implications for Inflammatory Processes. Blood 2002, 99, 4160–4165. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, G.; Tóth, A.; Csősz, É.; Kalló, G.; Dankó, K.; Csernátony, Z.; Smith, A.; Gram, M.; Akerström, B.; Édes, I.; et al. Heme-Induced Oxidation of Cysteine Groups of Myofilament Proteins Leads to Contractile Dysfunction of Permeabilized Human Skeletal Muscle Fibres. Int. J. Mol. Sci. 2020, 21, 8172. [Google Scholar] [CrossRef]
- Yao, X.; Balamurugan, P.; Arvey, A.; Leslie, C.; Zhang, L. Heme Controls the Regulation of Protein Tyrosine Kinases Jak2 and Src. Biochem. Biophys. Res. Commun. 2010, 403, 30–35. [Google Scholar] [CrossRef] [Green Version]
- da Guarda, C.C.; Santiago, R.P.; Pitanga, T.N.; Santana, S.S.; Zanette, D.L.; Borges, V.M.; Goncalves, M.S. Heme Changes HIF-α, ENOS and Nitrite Production in HUVECs after Simvastatin, HU, and Ascorbic Acid Therapies. Microvasc. Res. 2016, 106, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.F.; Tsai, A.L.; Wu, K.K. Cysteine 184 of Endothelial Nitric Oxide Synthase Is Involved in Heme Coordination and Catalytic Activity. J. Biol. Chem. 1994, 269, 25062–25066. [Google Scholar] [CrossRef]
- Arruda, M.A.; Rossi, A.G.; de Freitas, M.S.; Barja-Fidalgo, C.; Graça-Souza, A.V. Heme Inhibits Human Neutrophil Apoptosis: Involvement of Phosphoinositide 3-Kinase, MAPK, and NF-ΚB. J. Immunol. 2004, 173, 2023–2030. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Mundada, L.; Stomel, J.M.; Liu, J.J.; Sun, J.; Yet, S.-F.; Fay, W.P. Induction of Heme Oxygenase-1 Expression Inhibits Platelet-Dependent Thrombosis. Antioxid. Redox Signal. 2004, 6, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; Gerrard, J.M.; Rao, G.H.; Mills, E.L.; White, J.G. Interaction of Arachidonic Acid and Heme Iron in the Synthesis of Prostaglandins. Adv. Prostaglandin Thromboxane Res. 1980, 6, 157–161. [Google Scholar]
- Green, D.; Ts’ao, C. Hematin: Effects on Hemostasis. J. Lab. Clin. Med. 1990, 115, 144–147. [Google Scholar]
- NaveenKumar, S.K.; SharathBabu, B.N.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S.; Mugesh, G. The Role of Reactive Oxygen Species and Ferroptosis in Heme-Mediated Activation of Human Platelets. ACS Chem. Biol. 2018, 13, 1996–2002. [Google Scholar] [CrossRef]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklósi, J.; Méhes, G.; Csonka, T.; Smith, A.; et al. Red Cells, Hemoglobin, Heme, Iron, and Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef]
- Hastings, J.; de Matos, P.; Dekker, A.; Ennis, M.; Harsha, B.; Kale, N.; Muthukrishnan, V.; Owen, G.; Turner, S.; Williams, M.; et al. The ChEBI Reference Database and Ontology for Biologically Relevant Chemistry: Enhancements for 2013. Nucleic Acids Res. 2012, 41, D456–D463. [Google Scholar] [CrossRef]
- Lipscomb, C.E. Medical Subject Headings (MeSH). Bull. Med Libr. Assoc. 2000, 88, 265–266. [Google Scholar]
- Hoyt, C.T.; Domingo-Fernández, D.; Hofmann-Apitius, M. BEL Commons: An Environment for Exploration and Analysis of Networks Encoded in Biological Expression Language. Database 2018, 2018, bay126. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mubeen, S.; Domingo-Fernández, D.; Díaz del Ser, S.; Solanki, D.M.; Kodamullil, A.T.; Hofmann-Apitius, M.; Hopp, M.-T.; Imhof, D. Exploring the Complex Network of Heme-Triggered Effects on the Blood Coagulation System. J. Clin. Med. 2022, 11, 5975. https://doi.org/10.3390/jcm11195975
Mubeen S, Domingo-Fernández D, Díaz del Ser S, Solanki DM, Kodamullil AT, Hofmann-Apitius M, Hopp M-T, Imhof D. Exploring the Complex Network of Heme-Triggered Effects on the Blood Coagulation System. Journal of Clinical Medicine. 2022; 11(19):5975. https://doi.org/10.3390/jcm11195975
Chicago/Turabian StyleMubeen, Sarah, Daniel Domingo-Fernández, Sara Díaz del Ser, Dhwani M. Solanki, Alpha T. Kodamullil, Martin Hofmann-Apitius, Marie-T. Hopp, and Diana Imhof. 2022. "Exploring the Complex Network of Heme-Triggered Effects on the Blood Coagulation System" Journal of Clinical Medicine 11, no. 19: 5975. https://doi.org/10.3390/jcm11195975