
Antisense Oligonucleotides and Small Interfering RNA for the Treatment of Dyslipidemias

 , and
, and
Abstract
:
1. Introduction
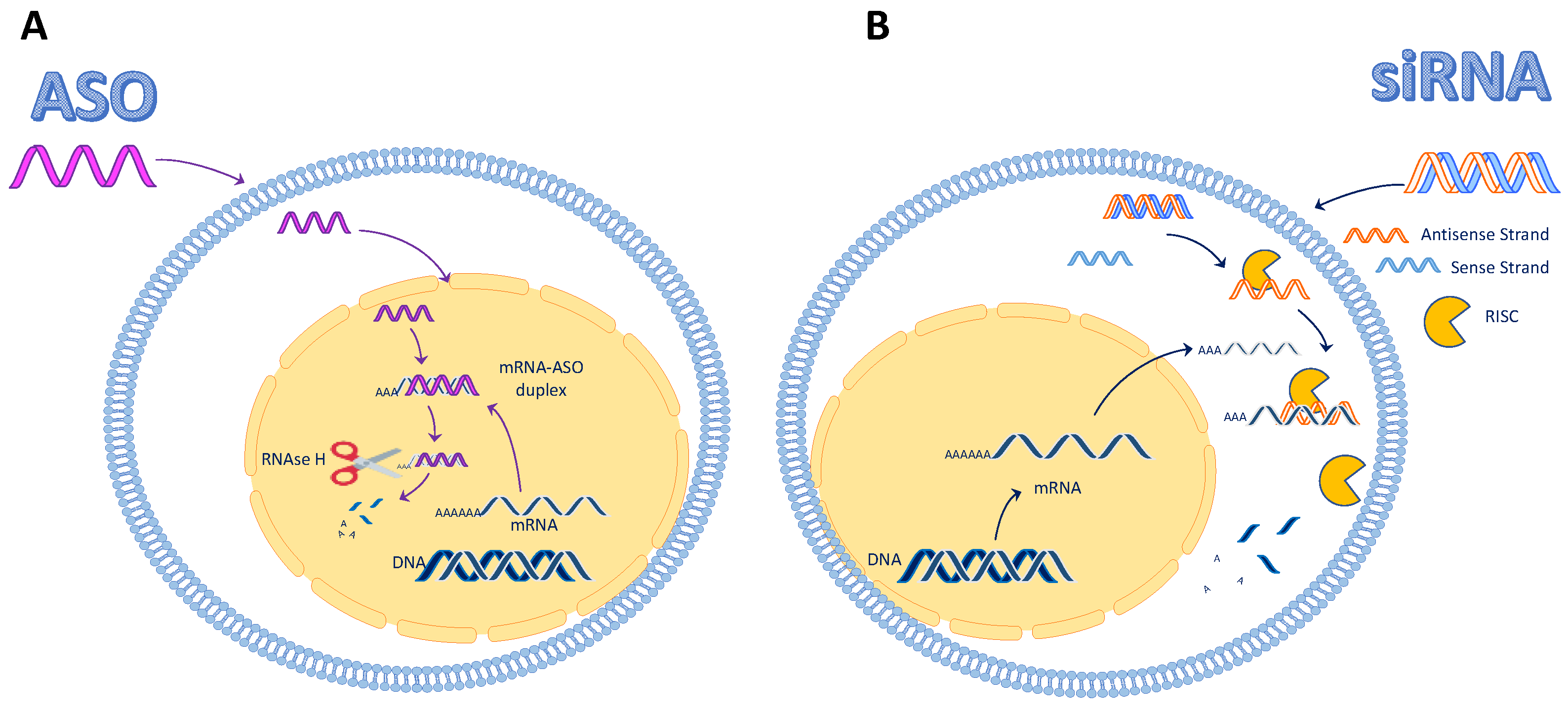
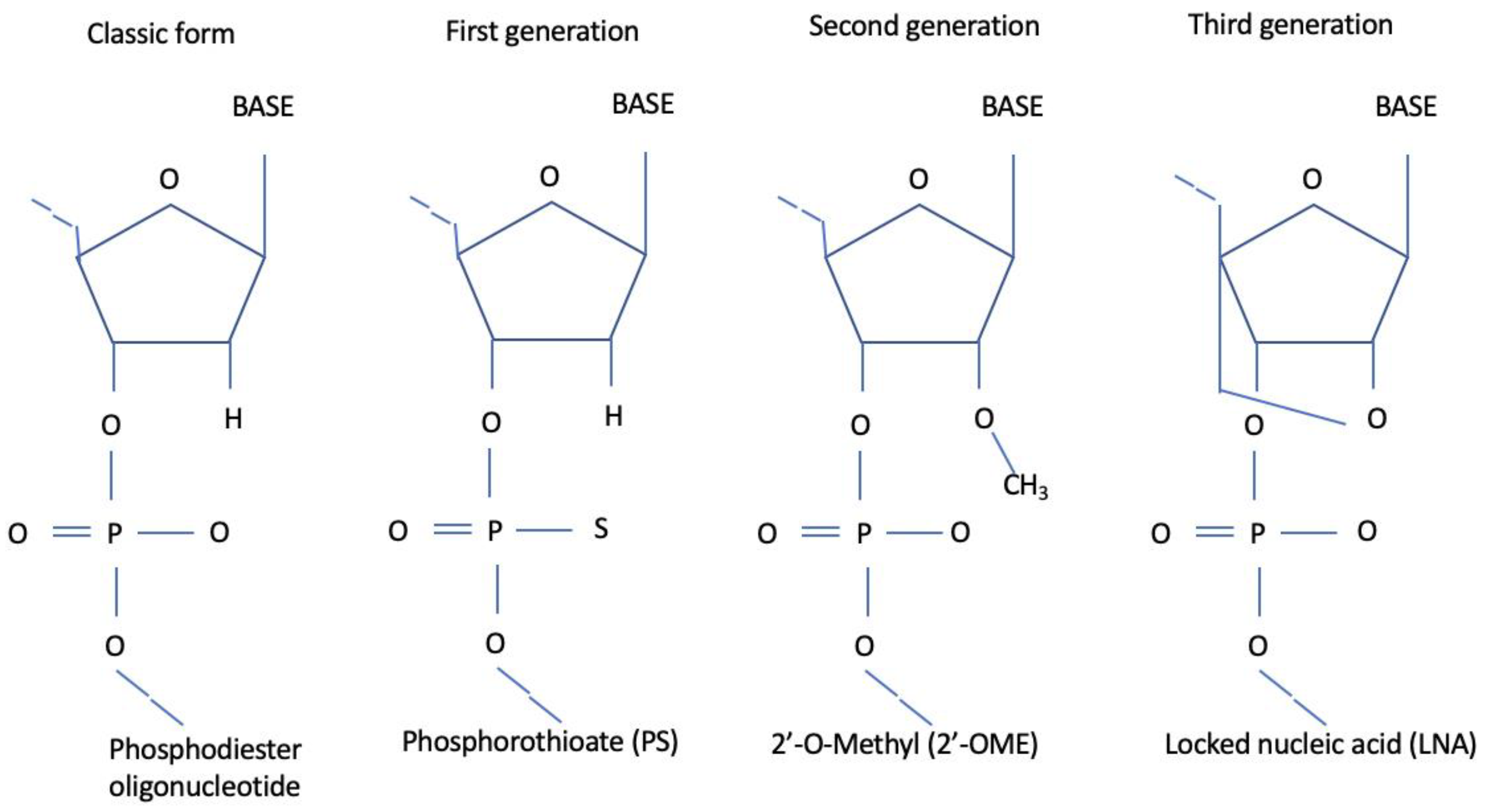
1.1. Antisense Oligonucleotides (ASO)
1.2. Short Interfering RNA (siRNA)
1.3. Specific Cell Target Delivery of ASOs or siRNAs In Vivo
1.4. ASO and siRNA in CVD
1.5. Inclisiran: A siRNA Directed against PCSK9
2. Discussion
ASOs vs. siRNAs: Who Is Getting the Upper Hand?
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mc Namara, K.; Alzubaidi, H.; Jackson, J.K. Cardiovascular disease as a leading cause of death: How are pharmacists getting involved? Integr. Pharm. Res. Pract. 2019, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, R.; Pathak, K. Statins therapy: A review on conventional and novel formulation approaches. J. Pharm. Pharmacol. 2011, 63, 983–998. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Théroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef] [Green Version]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- HPS3/TIMI55–Reveal Collaborative Group; Bowman, L.; Hopewell, J.C.; Chenm, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Huggett, B.; Paisner, K. The commercial tipping point. Nat. Biotechnol. 2017, 35, 708–709. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Mansoor, M.; Melendez, A.J. Advances in antisense oligonucleotide development for target identification, validation, and as novel therapeutics. Gene Regul. Syst. Bio. 2008, 2, 275–295. [Google Scholar] [CrossRef]
- Smith, C.I.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef]
- McKay, R.A.; Miraglia, L.J.; Cummins, L.L.; Owens, S.R.; Sasmor, H.; Dean, N.M. Characterization of a Potent and Specific Class of Antisense Oligonucleotide Inhibitor of Human Protein Kinase C-α Expression. J. Biol. Chem. 1999, 274, 1715–1722. [Google Scholar] [CrossRef] [Green Version]
- Geary, R.S.; Watanabe, T.A.; Truong, L.; Freier, S.; Lesnik, E.A.; Sioufi, N.B.; Sasmor, H.; Manoharan, M.; Levin, A.A. Pharmacokinetic properties of 2′-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J. Pharmacol. Exp. Ther. 2001, 296, 890–897. [Google Scholar]
- Hamm, S.; Latz, E.; Hangel, D.; Müller, T.; Yu, P.; Golenbock, D.; Sparwasser, T.; Wagner, H.; Bauer, S. Alternating 2′-O-ribose methylation is a universal approach for generating non-stimulatory siRNA by acting as TLR7 antagonist. Immunobiology 2010, 215, 559–569. [Google Scholar] [CrossRef]
- Siomi, H.; Siomi, M.C. On the road to reading the RNA-interference code. Nature 2009, 457, 396–404. [Google Scholar] [CrossRef]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs: Table 1. Genes Dev. 2006, 20, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Iaconetti, C.; Gareri, C.; Polimeni, A.; Indolfi, C. Non-Coding RNAs: The “dark matter” of cardiovascular pathophysiology. Int. J. Mol. Sci. 2013, 14, 19987–20018. [Google Scholar] [CrossRef] [Green Version]
- Buhler, M.; Moazed, D. Transcription and RNAi in heterochromatic gene silencing. Nat. Struct. Mol. Biol. 2007, 14, 1041–1048. [Google Scholar] [CrossRef]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Kuwajima, S.; Braich, R.; Rajeev, K.G.; Pena, J.; Tuschl, T.; Manoharan, M.; Stoffel, M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007, 35, 2885–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, J.C.; Rossi, J.J. RNA-Based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Bramsen, J.B.; Kjems, J. Engineering small interfering RNAs by strategic chemical modification. Methods Mol. Biol. 2013, 942, 87–109. [Google Scholar] [PubMed]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Corey, D.R. Chemical modification: The key to clinical application of RNA interference? J. Clin. Investig. 2007, 117, 3615–3622. [Google Scholar] [CrossRef]
- Judge, A.D.; Bola, G.; Lee, A.C.; MacLachlan, I. Design of Noninflammatory Synthetic siRNA Mediating Potent Gene Silencing in Vivo. Mol. Ther. 2006, 13, 494–505. [Google Scholar] [CrossRef]
- Judge, A.; MacLachlan, I. Overcoming the innate immune response to small interfering RNA. Hum. Gene Ther. 2008, 19, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Kortylewski, M.; Swiderski, P.; Herrmann, A.; Wang, L.; Kowolik, C.; Kujawski, M.; Lee, H.; Scuto, A.; Liu, Y.; Yang, C.; et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 2009, 27, 925–932. [Google Scholar] [CrossRef] [Green Version]
- Khairuddin, N.; Gantier, M.P.; Blake, S.J.; Wu, S.Y.; Behlke, M.A.; Williams, B.R.; McMillan, N.A. siRNA-Induced immunostimulation through TLR7 promotes antitumoral activity against HPV-Driven tumors in vivo. Immunol. Cell Biol. 2012, 90, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliano, R.; Alam, M.R.; Dixit, V.; Kang, H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008, 36, 4158–4171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-Like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, M.; Judge, A.; MacLachlan, I. siRNA and Innate Immunity. Oligonucleotides 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807. [Google Scholar] [CrossRef] [Green Version]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Juliano, R.L.; Carver, K. Cellular uptake and intracellular trafficking of oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Wagenaar, T.R.; Tolstykh, T.; Shi, C.; Jiang, L.; Zhang, J.; Li, Z.; Yu, Q.; Qu, H.; Sun, F.; Cao, H.; et al. Identification of the endosomal sorting complex required for transport-I (ESCRT-I) as an important modulator of anti-miR uptake by cancer cells. Nucleic Acids Res. 2015, 43, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Ming, X.; Cao, C.; Laing, B.; Yuan, A.; Porter, M.A.; Hull-Ryde, E.A.; Maddry, J.; Suto, M.; Janzen, W.P.; et al. High-throughput screening identifies small molecules that enhance the pharmacological effects of oligonucleotides. Nucleic Acids Res. 2015, 43, 1987–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-C.; Mozumdar, S.; Huang, L. Lipid-based systemic delivery of siRNA. Adv. Drug Deliv. Rev. 2009, 61, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, A.; Dowdy, S.F. siRNA delivery using peptide transduction domains. Trends Pharm. Sci. 2009, 30, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.; Pais, A.; Vitorino, C. Targeted siRNA Delivery Using Lipid Nanoparticles. Drug Deliv. Syst. 2019, 2059, 259–283. [Google Scholar] [CrossRef]
- Wittrup, A.; Ai, A.; Liu, X.; Hamar, P.; Trifonova, R.; Charisse, K.; Manoharan, M.; Kirchhausen, T.; Lieberman, J. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 2015, 33, 870–876. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Jang, B.; Lee, Y.-R.; Suh, E.-Y.; Yoo, J.-S.; Lee, M.-J.; Lee, J.-Y.; Lee, H. The cutting-edge technologies of siRNA delivery and their application in clinical trials. Arch. Pharmacal Res. 2018, 41, 867–874. [Google Scholar] [CrossRef]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef]
- Merwin, J.R.; Noell, G.S.; Thomas, W.L.; Chiou, H.C.; DeRome, M.E.; McKee, T.D.; Spitalny, G.L.; Findeis, M.A. Targeted delivery of DNA using YEE(GalNAcAH)3, a synthetic glycopeptide ligand for the asialoglycoprotein receptor. Bioconjug Chem. 1994, 5, 612–620. [Google Scholar] [CrossRef]
- Zimmermann, T.S.; Karsten, V.; Chan, A.; Chiesa, J.; Boyce, M.; Bettencourt, B.R.; Hutabarat, R.; Nochur, S.; Vaishnaw, A.; Gollob, J. Clinical Proof of Concept for a Novel Hepatocyte-Targeting GalNAc-siRNA Conjugate. Mol. Ther. 2017, 25, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Musunuru, K. Angiopoietin-Like 3: From Discovery to Therapeutic Gene Editing. JACC Basic Transl. Sci. 2019, 4, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. Promis and Myocardial Infarction Genetics Consortium, I. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, N.S.; Dallinga-Thie, G.M.; Stroes, E.S.G. Targeting apoC-III and ANGPTL3 in the treatment of hypertriglyceridemia. Expert Rev. Cardiovasc. Ther. 2020, 18, 355–361. [Google Scholar] [CrossRef]
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. European Prospective Investigation into C and Nutrition-Cardiovascular Disease, C. Association of LPA Variants with Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018, 3, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Langsted, A.; Nordestgaard, B.G. Antisense Oligonucleotides Targeting Lipoprotein (a). Curr. Atheroscler. Rep. 2019, 21, 30. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Steffensen, R.; Nordestgaard, B.G. Genetically Elevated Lipoprotein(a) and Increased Risk of Myocardial Infarction. JAMA 2009, 301, 2331–2339. [Google Scholar] [CrossRef]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic Variants Associated with Lp (a) Lipoprotein Level and Coronary Disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef] [Green Version]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated Lipoprotein (a) and Risk of Aortic Valve Stenosis in the General Population. J. Am. Coll. Cardiol. 2013, 63, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerging Risk Factors Collaboration; Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; et al. Lipoprotein (a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009, 302, 412–423. [Google Scholar]
- Investigators, A.-H.; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- HPS2-Thrive Collaborative Group; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–312. [Google Scholar]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.P.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of Torcetrapib in Patients at High Risk for Coronary Events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- Kronenberg, F. Human Genetics and the Causal Role of Lipoprotein (a) for Various Diseases. Cardiovasc. Drugs Ther. 2016, 30, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S. A Test in Context: Lipoprotein (a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Stiekema, L.C.A.; Stroes, E.S.G.; Verweij, S.L.; Kassahun, H.; Chen, L.; Wasserman, S.M.; Sabatine, M.S.; Mani, V.; Fayad, Z.A. Persistent arterial wall inflammation in patients with elevated lipoprotein (a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur. Heart J. 2018, 40, 2775–2781. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Ceska, R.; et al. Lipoprotein (a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein (a) levels. Eur. Heart, J. 2019, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Viney, N.J.; Hughes, S.G.; Xia, S.; Witztum, J.L.; Tsimikas, S. Temporal variability in lipoprotein (a) levels in patients enrolled in the placebo arms of IONIS-APO(a)Rx and IONIS-APO(a)-LRx antisense oligonucleotide clinical trials. J. Clin. Lipidol. 2018, 12, 122–129.e2. [Google Scholar] [CrossRef] [PubMed]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.-C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein (a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Lopez, J.A.G.; Knusel, B.; Gencer, B.; Wang, H.; Wu, Y.; Kassahun, H.; Sabatine, M.S. Study Design and Rationale for the OCEAN(a)-DOSE (Olpasiran trials of Cardiovascular Events and LipoproteiN (a) reduction-DOSE Finding Study) Trial. Am. Heart J. 2022, 251, 61–69. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals with Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679. [Google Scholar] [CrossRef]
- Morita, S.-Y. Metabolism and Modification of Apolipoprotein B-Containing Lipoproteins Involved in Dyslipidemia and Atherosclerosis. Biol. Pharm. Bull. 2016, 39, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, M.D.; Fazio, S. Apolipoprotein B-Containing lipoproteins and atherosclerotic cardiovascular disease. F1000Research 2017, 6, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ference, B.A. Using Genetic Variants in the Targets of Lipid Lowering Therapies to Inform Drug Discovery and Development: Current and Future Treatment Options. Clin. Pharmacol. Ther. 2018, 105, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Macchi, C.; Sirtori, C.; Corsini, A.; Santos, R.; Watts, G.; Ruscica, M. A new dawn for managing dyslipidemias: The era of rna-based therapies. Pharmacol. Res. 2019, 150, 104413. [Google Scholar] [CrossRef] [PubMed]
- Fogacci, F.; Ferri, N.; Toth, P.P.; Ruscica, M.; Corsini, A.; Cicero, A.F.G. Efficacy and Safety of Mipomersen: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Drugs 2019, 79, 751–766. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Packard, C.J.; Boren, J. Emerging Evidence that ApoC-III Inhibitors Provide Novel Options to Reduce the Residual CVD. Curr. Atheroscler. Rep. 2019, 21, 27. [Google Scholar] [CrossRef] [Green Version]
- Ooi, E.M.M.; Barrett, P.H.R.; Chan, D.C.; Watts, G.F. Apolipoprotein C-III: Understanding an emerging cardiovascular risk factor. Clin. Sci. 2008, 114, 611–624. [Google Scholar] [CrossRef] [Green Version]
- Sarwar, N.; Danesh, J.; Eiriksdottir, G.; Sigurdsson, G.; Wareham, N.; Bingham, S.; Boekholdt, S.M.; Khaw, K.-T.; Gudnason, V. Triglycerides and the Risk of Coronary Heart Disease. Circulation 2007, 115, 450–458. [Google Scholar] [CrossRef]
- Jørgensen, A.; Frikke-Schmidt, R.; Nordestgaard, B.; Tybjærg-Hansen, A. Loss-of-function mutations in APOC3 and reduced risk of ischemic vascular disease. Atherosclerosis 2014, 235, e18. [Google Scholar] [CrossRef]
- Blood, I.; Crosby, J.; Peloso, G.M.; Auer, P.L.; Crosslin, D.R.; Stitziel, N.O.; Lange, L.A.; Lu, Y.; Tang, Z.Z.; Zhang, H.; et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med. 2014, 371, 22–31. [Google Scholar]
- Graham, M.J.; Lee, R.G.; Bell, T.A.; Fu, W.; Mullick, A.E.; Alexander, V.J.; Singleton, W.; Viney, N.; Geary, R.; Su, J.; et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ. Res. 2013, 112, 1479–1490. [Google Scholar] [CrossRef]
- Fogacci, F.; Norata, G.D.; Toth, P.P.; Arca, M.; Cicero, A.F.G. Efficacy and Safety of Volanesorsen (ISIS 304801): The Evidence from Phase 2 and 3 Clinical Trials. Curr. Atheroscler. Rep. 2020, 22, 18. [Google Scholar] [CrossRef] [PubMed]
- Arca, M.; Hsieh, A.; Soran, H.; Rosenblit, P.; O’Dea, L.; Stevenson, M. The effect of volanesorsen treatment on the burden associated with familial chylomicronemia syndrome: The results of the ReFOCUS study. Expert Rev. Cardiovasc. Ther. 2018, 16, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Alexander, V.J.; Baker, B.F.; Brisson, D.; Tremblay, K.; Singleton, W.; Geary, R.S.; Hughes, S.G.; Viney, N.J.; Graham, M.J.; et al. Antisense Inhibition of Apolipoprotein C-III in Patients with Hypertriglyceridemia. N. Engl. J. Med. 2015, 373, 438–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, V.J.; Xia, S.; Hurh, E.; Hughes, S.G.; O’Dea, L.; Geary, R.S.; Witztum, J.L.; Tsimikas, S. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur. Heart J. 2019, 40, 2785–2796. [Google Scholar] [CrossRef]
- Gouni-Berthold, I. The role of antisense oligonucleotide therapy against apolipoprotein-CIII in hypertriglyceridemia. Atheroscler. Suppl. 2017, 30, 19–27. [Google Scholar] [CrossRef]
- Tang, Y.; Li, H.; Chen, C. Non-Coding RNA-Associated Therapeutic Strategies in Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 889743. [Google Scholar] [CrossRef]
- Merćep, I.; Friščić, N.; Strikić, D.; Reiner, Z. Advantages and Disadvantages of Inclisiran: A Small Interfering Ribonucleic Acid Molecule Targeting PCSK9—A Narrative Review. Cardiovasc. Ther. 2022, 2022, 8129513. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2019, 41, 111–188. [Google Scholar] [CrossRef]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.L.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, K.K.; Stoekenbroek, R.M.; Kallend, D.; Leiter, L.A.; Landmesser, U.; Wright, R.S.; Wijngaard, P.; Kastelein, J.J.P. Effect of an siRNA Therapeutic Targeting PCSK9 on Atherogenic Lipoproteins: Prespecified Secondary End Points in ORION 1. Circulation 2018, 138, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.S.; Collins, M.G.; Stoekenbroek, R.M.; Robson, R.; Wijngaard, P.L.; Landmesser, U.; Leiter, L.A.; Kastelein, J.J.; Ray, K.K.; Kallend, D. Effects of Renal Impairment on the Pharmacokinetics, Efficacy, and Safety of Inclisiran: An Analysis of the ORION-7 and ORION-1 Studies. Mayo Clin. Proc. 2019, 95, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Kallend, D.; Stoekenbroek, R.; He, Y.; Smith, P.F.; Wijngaard, P. Pharmacokinetics and pharmacodynamics of inclisiran, a small interfering RNA therapy, in patients with hepatic impairment. J. Clin. Lipidol. 2022, 16, 208–219. [Google Scholar] [CrossRef]
- Kretschmer-Kazemi Far, R.; Sczakiel, G. The activity of siRNA in mammalian cells is related to structural target accessibility: A comparison with antisense oligonucleotides. Nucleic Acids Res. 2003, 31, 4417–4424. [Google Scholar] [CrossRef] [Green Version]
- Eizema, K.; Fechner, H.; Bezstarosti, K.; Schneider-Rasp, S.; van der Laarse, A.; Wang, H.; Schultheiss, H.P.; Poller, W.C.; Lamers, J.M. Adenovirus-Based phospholamban antisense expression as a novel approach to improve cardiac contractile dysfunction: Comparison of a constitutive viral versus an endothelin-1-responsive cardiac promoter. Circulation 2000, 101, 2193–2199. [Google Scholar] [CrossRef] [Green Version]
- Gareri, C.; De Rosa, S.; Indolfi, C. MicroRNAs for Restenosis and Thrombosis After Vascular Injury. Circ. Res. 2016, 118, 1170–1184. [Google Scholar] [CrossRef] [Green Version]
- Sabatino, J.; Wicik, Z.; De Rosa, S.; Eyileten, C.; Jakubik, D.; Spaccarotella, C.; Mongiardo, A.; Postula, M.; Indolfi, C. MicroRNAs fingerprint of bicuspid aortic valve. J. Mol. Cell. Cardiol. 2019, 134, 98–106. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Brisson, D. Gene-Based therapies in lipidology: Current status and future challenges. Curr. Opin. Lipidol. 2015, 26, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.-T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9–Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Small Molecules | Antibodies | Antisense Oligonucleotides (ASO) | Short Interfering RNA (siRNA) | |
|---|---|---|---|---|
| Structure | Organic compound | Protein | Single-stranded RNA | Double-stranded RNA |
| Mass (kDa) | <1 | ~150 | ~12 | ~21 |
| Mechanism of action | Blocks enzyme or receptor | Blocks protein | Blocks gene mRNA transcripts | Blocks gene mRNA transcripts |
| Potential for off-target adverse effects | High | Low | Low | Low |
| Immunogenicity | Low | High | High | High |
| Efficacy | 50% reduction in LDL | 60% reduction in LDL | 90% reduction in Lp(a) | 50% reduction in LDL |
| Drug response variability | High | High | Low | Low |
| Half-life | Days | Weeks | Months | >1 year |
| Administration route | Oral | Subcutaneous | Subcutaneous | Subcutaneous |
| Dosing frequency | Daily | Weekly to twice monthly | Monthly | Twice yearly |
| Target Protein | Molecule (Type) | LDL-C | TG | Lp(a) |
|---|---|---|---|---|
| ANGPTL 3 | AKCEA-ANGPTL3-LRX (ASO) | + | + | |
| ANGPTL 3 | ARO-ANG3 (SiRNA) | + | ||
| Lipoprotein(a) | ISIS-APO(a)Rx (ASO) | + | ||
| Lipoprotein(a) | IONIS-APO (a) LRx (ASO) | + | ||
| Lipoprotein(a) | Olpasiran (siRNA) | + | ||
| Lipoprotein(a) | SLN360 (siRNA) | + | ||
| Apolipoprotein B | Mipomersen (ASO) | + | + | + |
| Apolipoprotein C III | Volanesorsen (ASO) | + | ||
| Apolipoprotein C III | AKCEA-APOCIII-LRX (ASO) | + | ||
| Apolipoprotein C III | ARO-APOC31001 (siRNA) | + | ||
| PCSK9 | Inclisiran (siRNA) | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gareri, C.; Polimeni, A.; Giordano, S.; Tammè, L.; Curcio, A.; Indolfi, C. Antisense Oligonucleotides and Small Interfering RNA for the Treatment of Dyslipidemias. J. Clin. Med. 2022, 11, 3884. https://doi.org/10.3390/jcm11133884
Gareri C, Polimeni A, Giordano S, Tammè L, Curcio A, Indolfi C. Antisense Oligonucleotides and Small Interfering RNA for the Treatment of Dyslipidemias. Journal of Clinical Medicine. 2022; 11(13):3884. https://doi.org/10.3390/jcm11133884
Chicago/Turabian StyleGareri, Clarice, Alberto Polimeni, Salvatore Giordano, Laura Tammè, Antonio Curcio, and Ciro Indolfi. 2022. "Antisense Oligonucleotides and Small Interfering RNA for the Treatment of Dyslipidemias" Journal of Clinical Medicine 11, no. 13: 3884. https://doi.org/10.3390/jcm11133884