
Implication of Lipids in Calcified Aortic Valve Pathogenesis: Why Did Statins Fail?

 ,
,  , and
, and 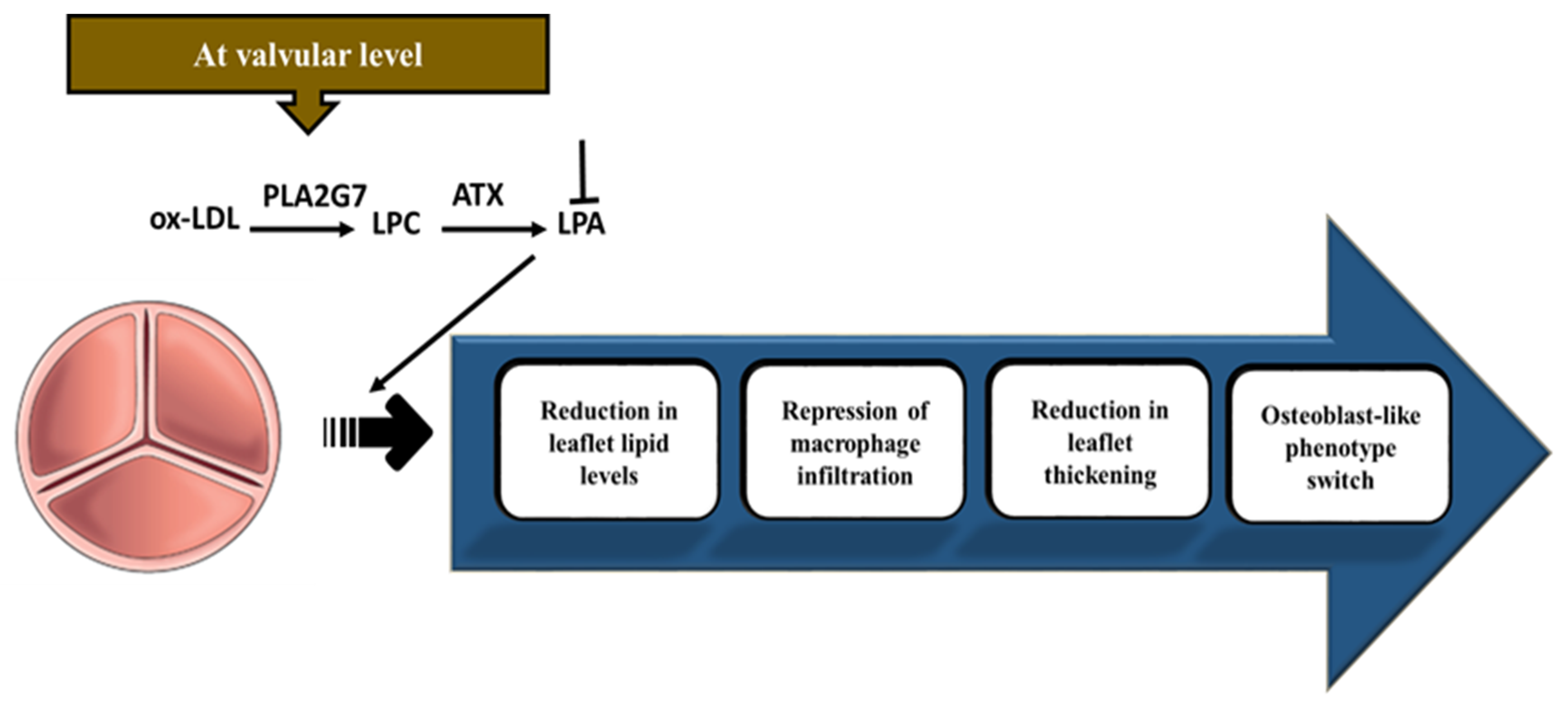{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction—Calcific Aortic Valve Disease (CAVD)
2. Role of Lipoprotein(a) and Oxidized Phospholipids in CAVD
2.1. The Implication of Nitric Oxide (NO) Activity in CAVD
2.2. The LPA Gene Locus and CAVD
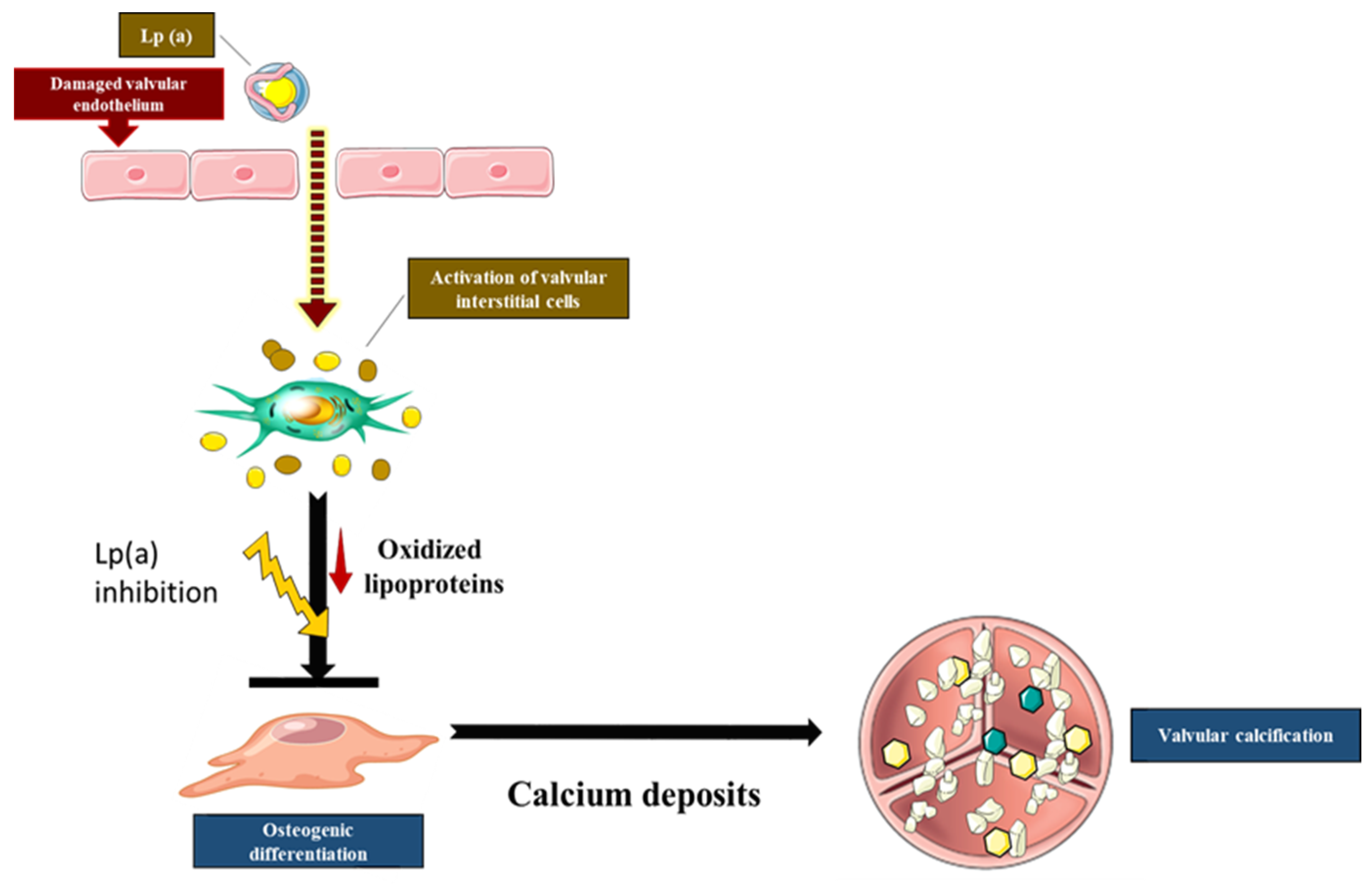
2.3. Lp(a), a Major Carrier of Oxidized Phospholipids (Ox-PL), Is a Risk Factor for Aortic Stenosis
2.4. Lipid Oxidation Promotes Calcific Aortic Stenosis
3. Autotaxin (ATX)–Lysophosphatidic Acid (LysoPA) Axis Mediates Mineralization of the Aortic Valve
4. The Implication of apoC-III in the Calcification of the Aortic Valve
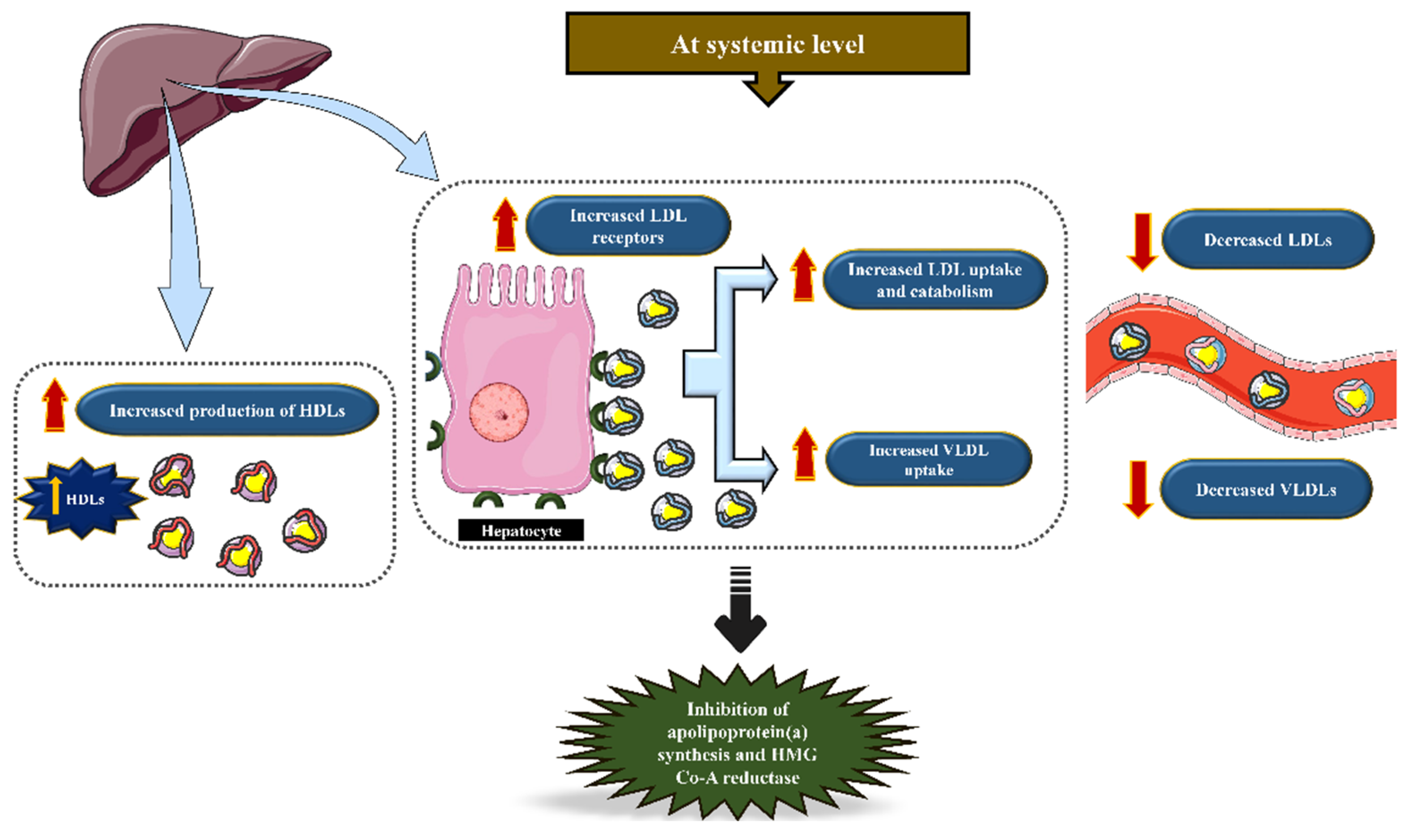
5. The Effect of Lipid-Lowering Therapy in Aortic Stenosis
5.1. Rationale of Statins
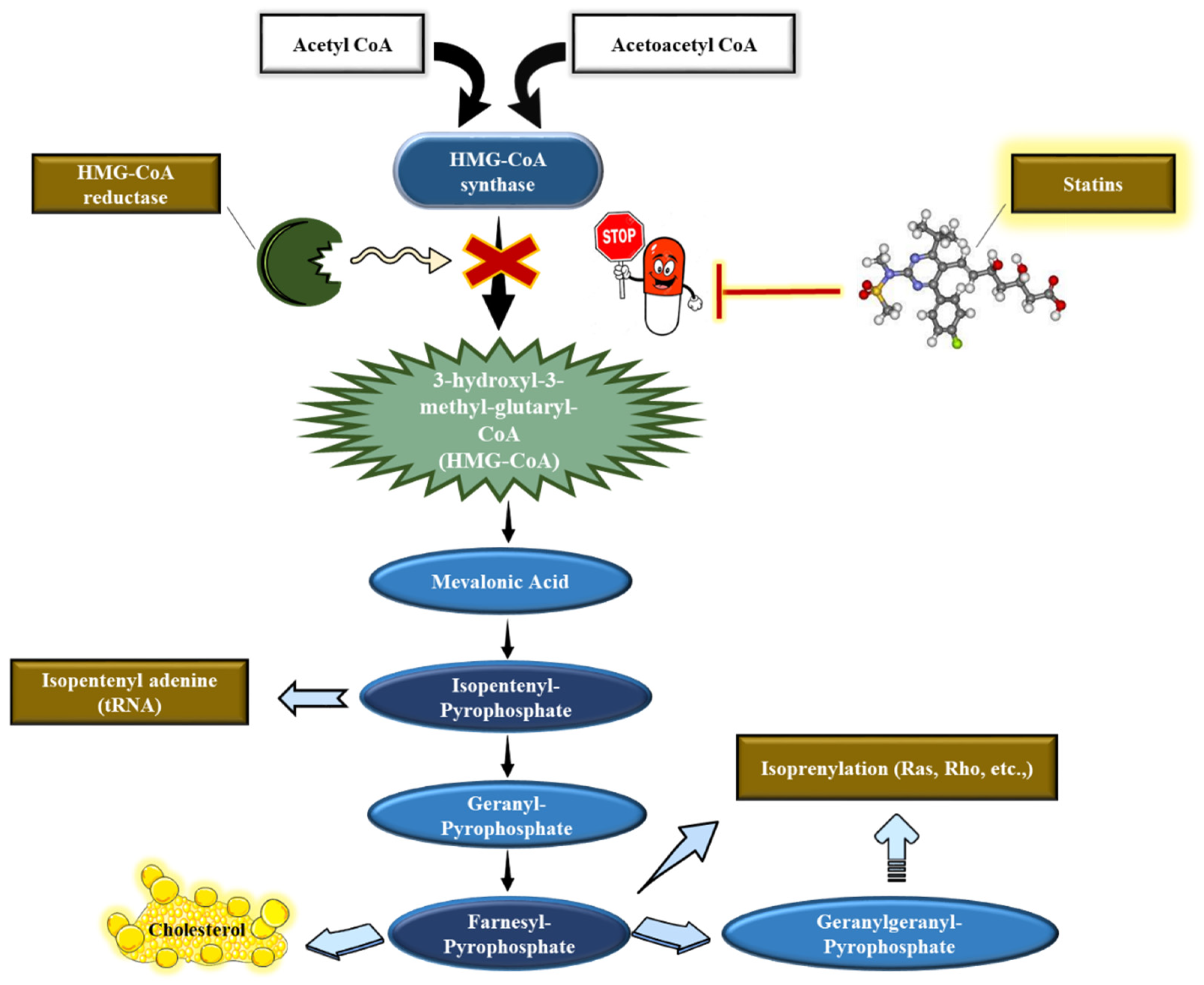
5.2. Statins’ Mechanism of Action
5.3. Statin-Mediated Lipid-Lowering Therapeutic Approaches to Target CAVD: Past, Present, and Future Prospects
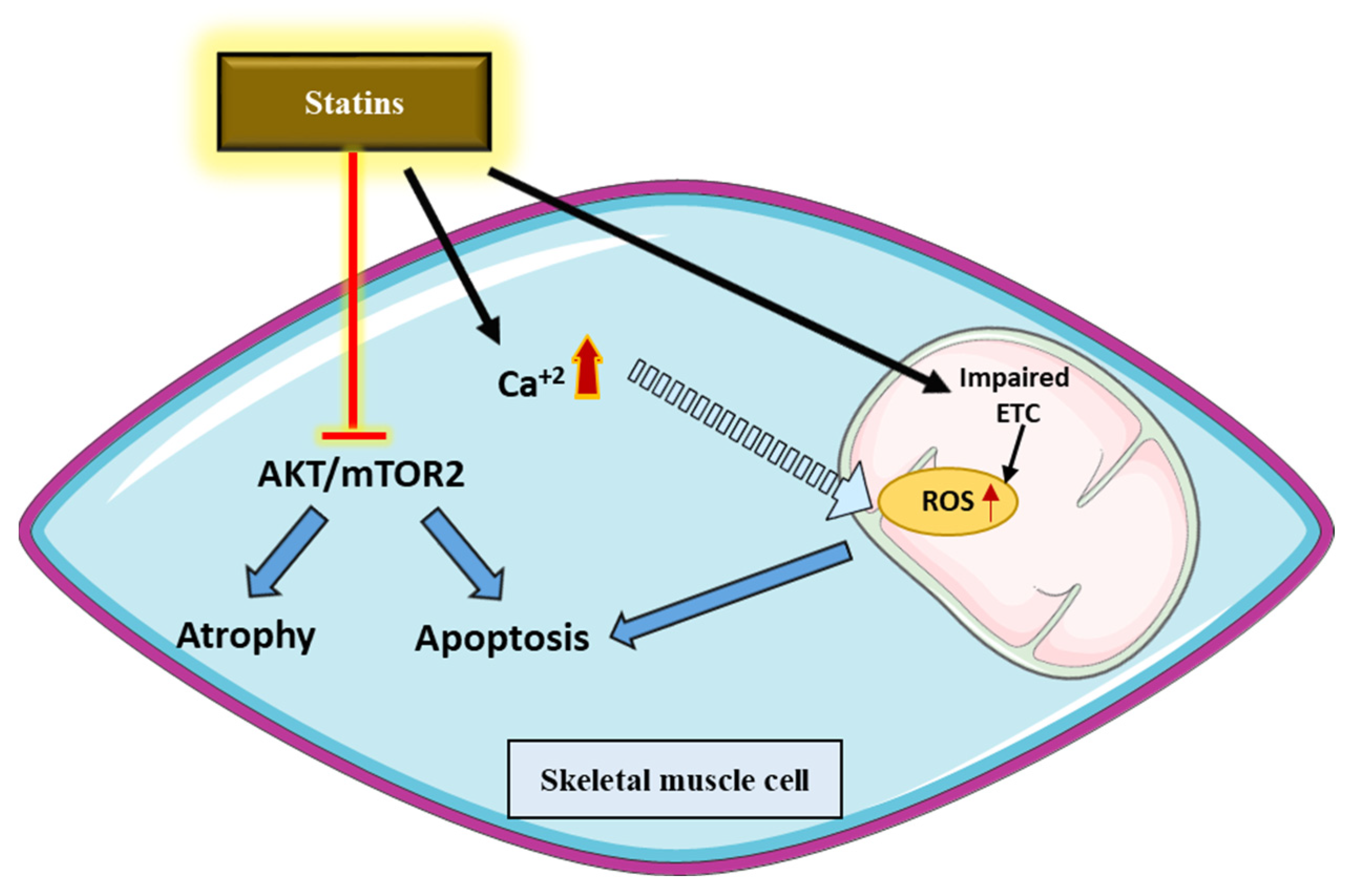
5.4. Statins’ Off-Target Effects

Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lindman, B.R.; Clavel, M.A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev. Dis. Primers 2016, 2, 16006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, P.; Boulanger, M.C. Basic Mechanisms of Calcific Aortic Valve Disease. Can. J. Cardiol. 2014, 30, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Bouchareb, R.; Storto, M.; Hussain, Y.; Gregg, A.; Marx, S.O.; Pitt, G.S. Increased Ca2+ influx through CaV1.2 drives aortic valve calcification. JCI Insight 2022, 7, e155569. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Chen, W.L.; Sider, K.L.; Yip, C.Y.; Simmons, C.A. beta-catenin mediates mechanically regulated, transforming growth factor-beta1-induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 590–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchareb, R.; Boulanger, M.C.; Fournier, D.; Pibarot, P.; Messaddeq, Y.; Mathieu, P. Mechanical strain induces the production of spheroid mineralized microparticles in the aortic valve through a RhoA/ROCK-dependent mechanism. J. Mol. Cell Cardiol. 2014, 67, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Seya, K.; Daitoku, K.; Motomura, S.; Fukuda, I.; Furukawa, K. Tumor necrosis factor-alpha accelerates the calcification of human aortic valve interstitial cells obtained from patients with calcific aortic valve stenosis via the BMP2-Dlx5 pathway. J. Pharmacol. Exp. Ther. 2011, 337, 16–23. [Google Scholar] [CrossRef]
- Bouchareb, R.; Guauque-Olarte, S.; Snider, J.; Zaminski, D.; Anyanwu, A.; Stelzer, P.; Lebeche, D. Proteomic Architecture of Valvular Extracellular Matrix: FNDC1 and MXRA5 Are New Biomarkers of Aortic Stenosis. JACC Basic Transl. Sci. 2021, 6, 25–39. [Google Scholar] [CrossRef]
- Katsi, V.; Magkas, N.; Antonopoulos, A.; Trantalis, G.; Toutouzas, K.; Tousoulis, D. Aortic valve: Anatomy and structure and the role of vasculature in the degenerative process. Acta Cardiol. 2021, 76, 335–348. [Google Scholar] [CrossRef]
- Bouchareb, R.; Lebeche, D. Isolation of Mouse Interstitial Valve Cells to Study the Calcification of the Aortic Valve In Vitro. J. Vis. Exp. 2021, 171, e62419. [Google Scholar] [CrossRef]
- Dupuis, L.E.; McCulloch, D.R.; McGarity, J.D.; Bahan, A.; Wessels, A.; Weber, D.; Diminich, A.M.; Nelson, C.M.; Apte, S.S.; Kern, C.B. Altered versican cleavage in ADAMTS5 deficient mice; a novel etiology of myxomatous valve disease. Dev. Biol. 2011, 357, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Derbali, H.; Bosse, Y.; Cote, N.; Pibarot, P.; Audet, A.; Pepin, A.; Arsenault, B.; Couture, C.; Despres, J.P.; Mathieu, P. Increased biglycan in aortic valve stenosis leads to the overexpression of phospholipid transfer protein via Toll-like receptor 2. Am. J. Pathol. 2010, 176, 2638–2645. [Google Scholar] [CrossRef] [PubMed]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Boren, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef]
- Yu, B.; Hafiane, A.; Thanassoulis, G.; Ott, L.; Filwood, N.; Cerruti, M.; Gourgas, O.; Shum-Tim, D.; Al Kindi, H.; de Varennes, B.; et al. Lipoprotein(a) Induces Human Aortic Valve Interstitial Cell Calcification. JACC Basic Transl. Sci. 2017, 2, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.C.; Dahou, A.; Lepine, J.L.; Laflamme, M.H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin Derived from Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef]
- Nsaibia, M.J.; Mahmut, A.; Boulanger, M.C.; Arsenault, B.J.; Bouchareb, R.; Simard, S.; Witztum, J.L.; Clavel, M.A.; Pibarot, P.; Bosse, Y.; et al. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J. Intern. Med. 2016, 280, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Cowell, S.J.; Newby, D.E.; Prescott, R.J.; Bloomfield, P.; Reid, J.; Northridge, D.B.; Boon, N.A.; Scottish Aortic, S.; Lipid Lowering Trial, I.o.R.I. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N. Engl. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef]
- Rossebo, A.B.; Pedersen, T.R.; Boman, K.; Brudi, P.; Chambers, J.B.; Egstrup, K.; Gerdts, E.; Gohlke-Barwolf, C.; Holme, I.; Kesaniemi, Y.A.; et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N. Engl. J. Med. 2008, 359, 1343–1356. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J.; Investigators, A. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: Results of the aortic stenosis progression observation: Measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Gordts, P.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Koh, K.K.; Quon, M.J.; Han, S.H.; Lee, Y.; Kim, S.J.; Shin, E.K. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J. Am. Coll. Cardiol. 2010, 55, 1209–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, M.F.; Sirtori, C.R.; Corsini, A.; Ezhov, M.; Sampietro, T.; Ruscica, M. Lipoprotein(a) Lowering-From Lipoprotein Apheresis to Antisense Oligonucleotide Approach. J. Clin. Med. 2020, 9, 2103. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Fata, V.L.; Liao, J.K. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J. Biol. Chem. 1997, 272, 31725–31729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langsted, A.; Nordestgaard, B.G.; Benn, M.; Tybjaerg-Hansen, A.; Kamstrup, P.R. PCSK9 R46L Loss-of-Function Mutation Reduces Lipoprotein(a), LDL Cholesterol, and Risk of Aortic Valve Stenosis. J. Clin. Endocrinol. Metab. 2016, 101, 3281–3287. [Google Scholar] [CrossRef]
- Kraler, S.; Garg, V.; Akhmedov, A. Calcific aortic valve disease: Novel insights into nitric oxide signalling. Eur. Heart J. 2022, 43, 1665–1667. [Google Scholar] [CrossRef]
- El Accaoui, R.N.; Gould, S.T.; Hajj, G.P.; Chu, Y.; Davis, M.K.; Kraft, D.C.; Lund, D.D.; Brooks, R.M.; Doshi, H.; Zimmerman, K.A.; et al. Aortic valve sclerosis in mice deficient in endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1302–H1313. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, U.; Manivannan, S.; Basu, M.; Ueyama, Y.; Blaser, M.C.; Cameron, E.; McDermott, M.R.; Lincoln, J.; Cole, S.E.; Wood, S.; et al. Nitric oxide prevents aortic valve calcification by S-nitrosylation of USP9X to activate NOTCH signaling. Sci. Adv. 2021, 7, eabe3706. [Google Scholar] [CrossRef]
- Gould, S.T.; Matherly, E.E.; Smith, J.N.; Heistad, D.D.; Anseth, K.S. The role of valvular endothelial cell paracrine signaling and matrix elasticity on valvular interstitial cell activation. Biomaterials 2014, 35, 3596–3606. [Google Scholar] [CrossRef] [Green Version]
- Kraler, S.; Blaser, M.C.; Aikawa, E.; Camici, G.G.; Luscher, T.F. Calcific aortic valve disease: From molecular and cellular mechanisms to medical therapy. Eur. Heart J. 2022, 43, 683–697. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Beisiegel, U.; Henne-Bruns, D.; Eaton, D.L.; Lawn, R.M. Apolipoprotein(a) size heterogeneity is related to variable number of repeat sequences in its mRNA. Biochemistry 1990, 29, 640–644. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Boekholdt, S.M.; Dube, M.P.; Rheaume, E.; Wareham, N.J.; Khaw, K.T.; Sandhu, M.S.; Tardif, J.C. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: A prospective Mendelian randomization study and replication in a case-control cohort. Circ. Cardiovasc. Genet. 2014, 7, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, B.J.; Coffey, S.; Travis, R.C.; Prendergast, B.; Green, J.; Engert, J.C.; Lathrop, M.; Thanassoulis, G.; Clarke, R. A Replicated, Genome-Wide Significant Association of Aortic Stenosis with a Genetic Variant for Lipoprotein(a): Meta-Analysis of Published and Novel Data. Circulation 2017, 135, 1181–1183. [Google Scholar] [CrossRef] [PubMed]
- Enas, E.A.; Varkey, B.; Dharmarajan, T.S.; Pare, G.; Bahl, V.K. Lipoprotein(a): An independent, genetic, and causal factor for cardiovascular disease and acute myocardial infarction. Indian Heart J. 2019, 71, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, A.; Schiavo, A.; Moscarella, E.; Coletta, S.; Conte, M.; Gragnano, F.; Fimiani, F.; Monda, E.; Caiazza, M.; Limongelli, G.; et al. Lipoprotein(a): A genetic marker for cardiovascular disease and target for emerging therapies. J. Cardiovasc. Med. 2021, 22, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Chignon, A.; Bon-Baret, V.; Boulanger, M.C.; Bossé, Y.; Mathieu, P. Oxyphospholipids in Cardiovascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Kamstrup, P.R.; Hung, M.Y.; Witztum, J.L.; Tsimikas, S.; Nordestgaard, B.G. Oxidized Phospholipids and Risk of Calcific Aortic Valve Disease: The Copenhagen General Population Study. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1570–1578. [Google Scholar] [CrossRef] [Green Version]
- Nsaibia, M.J.; Boulanger, M.C.; Bouchareb, R.; Mkannez, G.; Le Quang, K.; Hadji, F.; Argaud, D.; Dahou, A.; Bosse, Y.; Koschinsky, M.L.; et al. OxLDL-derived lysophosphatidic acid promotes the progression of aortic valve stenosis through a LPAR1-RhoA-NF-kappaB pathway. Cardiovasc. Res. 2017, 113, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.D.; Reichenbach, D.D.; Marcovina, S.M.; Kuusisto, J.; Alpers, C.E.; Otto, C.M. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 523–532. [Google Scholar] [CrossRef]
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994, 90, 844–853. [Google Scholar] [CrossRef] [Green Version]
- Gebhard, C.; Maafi, F.; Stahli, B.E.; Dang, J.; Nachar, W.; de Oliveira Moraes, A.B.; Kernaleguen, A.E.; Lavoie, V.; Mecteau, M.; Mihalache-Avram, T.; et al. Apolipoprotein A-I proteolysis in aortic valve stenosis: Role of cathepsin S. Basic Res. Cardiol. 2018, 113, 30. [Google Scholar] [CrossRef] [PubMed]
- Cote, C.; Pibarot, P.; Despres, J.P.; Mohty, D.; Cartier, A.; Arsenault, B.J.; Couture, C.; Mathieu, P. Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart 2008, 94, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Maatta, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Witztum, J.L. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Curr. Opin. Lipidol. 2008, 19, 369–377. [Google Scholar] [CrossRef]
- Mahmut, A.; Boulanger, M.C.; El Husseini, D.; Fournier, D.; Bouchareb, R.; Despres, J.P.; Pibarot, P.; Bosse, Y.; Mathieu, P. Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease: Implications for valve mineralization. J. Am. Coll. Cardiol. 2014, 63, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Mahmut, A.; Mahjoub, H.; Boulanger, M.C.; Fournier, D.; Despres, J.P.; Pibarot, P.; Mathieu, P. Lp-PLA2 is associated with structural valve degeneration of bioprostheses. Eur. J. Clin. Investig. 2014, 44, 136–145. [Google Scholar] [CrossRef]
- Capoulade, R.; Mahmut, A.; Tastet, L.; Arsenault, M.; Bedard, E.; Dumesnil, J.G.; Despres, J.P.; Larose, E.; Arsenault, B.J.; Bosse, Y.; et al. Impact of plasma Lp-PLA2 activity on the progression of aortic stenosis: The PROGRESSA study. JACC Cardiovasc. Imaging 2015, 8, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulou, M.; Fanidis, D.; Aidinis, V.; Chatzaki, E. ENPP2 Methylation in Health and Cancer. Int. J. Mol. Sci. 2021, 22, 11958. [Google Scholar] [CrossRef]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 2008, 1781, 513–518. [Google Scholar] [CrossRef]
- Bourgeois, R.; Devillers, R.; Perrot, N.; Despres, A.A.; Boulanger, M.C.; Mitchell, P.L.; Guertin, J.; Couture, P.; Boffa, M.B.; Scipione, C.A.; et al. Interaction of Autotaxin with Lipoprotein(a) in Patients with Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2020, 5, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Mkannez, G.; Gagne-Ouellet, V.; Jalloul Nsaibia, M.; Boulanger, M.C.; Rosa, M.; Argaud, D.; Hadji, F.; Gaudreault, N.; Rheaume, G.; Bouchard, L.; et al. DNA methylation of a PLPP3 MIR transposon-based enhancer promotes an osteogenic programme in calcific aortic valve disease. Cardiovasc. Res. 2018, 114, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Models Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, R.; Wong, N.D.; Kronmal, R.; Takasu, J.; Shavelle, D.M.; Probstfield, J.L.; Bertoni, A.G.; Budoff, M.J.; O’Brien, K.D. Features of the metabolic syndrome and diabetes mellitus as predictors of aortic valve calcification in the Multi-Ethnic Study of Atherosclerosis. Circulation 2006, 113, 2113–2119. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Clavel, M.A.; Dumesnil, J.G.; Chan, K.L.; Teo, K.K.; Tam, J.W.; Cote, N.; Mathieu, P.; Despres, J.P.; Pibarot, P.; et al. Impact of metabolic syndrome on progression of aortic stenosis: Influence of age and statin therapy. J. Am. Coll. Cardiol. 2012, 60, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Norata, G.D.; Tsimikas, S.; Pirillo, A.; Catapano, A.L. Apolipoprotein C-III: From Pathophysiology to Pharmacology. Trends Pharmacol. Sci. 2015, 36, 675–687. [Google Scholar] [CrossRef]
- D’Erasmo, L.; Di Costanzo, A.; Gallo, A.; Bruckert, E.; Arca, M. ApoCIII: A multifaceted protein in cardiometabolic disease. Metabolism 2020, 113, 154395. [Google Scholar] [CrossRef]
- Pechlaner, R.; Tsimikas, S.; Yin, X.; Willeit, P.; Baig, F.; Santer, P.; Oberhollenzer, F.; Egger, G.; Witztum, J.L.; Alexander, V.J.; et al. Very-Low-Density Lipoprotein-Associated Apolipoproteins Predict Cardiovascular Events and Are Lowered by Inhibition of APOC-III. J. Am. Coll. Cardiol. 2017, 69, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, A.B.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjaerg-Hansen, A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N. Engl. J. Med. 2014, 371, 32–41. [Google Scholar] [CrossRef] [Green Version]
- TG and HDL Working Group of the Exome Sequencing Project; National Heart, Lung, and Blood Institute; Crosby, J.; Peloso, G.M.; Auer, P.L.; Crosslin, D.R.; Stitziel, N.O.; Lange, L.A.; Lu, Y.; Tang, Z.Z.; et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med. 2014, 371, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Torzewski, M.; Mayr, M.; Chan, K.L.; Mathieu, P.; Bosse, Y.; Dumesnil, J.G.; Tam, J.; Teo, K.K.; Burnap, S.A.; et al. ApoCIII-Lp(a) complexes in conjunction with Lp(a)-OxPL predict rapid progression of aortic stenosis. Heart 2020, 106, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Corsini, A.; Maggi, F.M.; Catapano, A.L. Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacol. Res. 1995, 31, 9–27. [Google Scholar] [CrossRef]
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389.
- Rosenson, R.S. Statins in atherosclerosis: Lipid-lowering agents with antioxidant capabilities. Atherosclerosis 2004, 173, 1–12. [Google Scholar] [CrossRef]
- Halcox, J.P.; Deanfield, J.E. Beyond the laboratory: Clinical implications for statin pleiotropy. Circulation 2004, 109, II42–II48. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, M.; Liao, J.K. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1712–1719. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Liao, J.K. Isoprenoids as mediators of the biological effects of statins. J. Clin. Investig. 2002, 110, 285–288. [Google Scholar] [CrossRef]
- Van Aelst, L.; D’Souza-Schorey, C. Rho GTPases and signaling networks. Genes Dev. 1997, 11, 2295–2322. [Google Scholar] [CrossRef] [Green Version]
- Carabello, B.A. The SEAS Trial. Curr. Cardiol. Rep. 2010, 12, 122–124. [Google Scholar] [CrossRef]
- Teo, K.K.; Corsi, D.J.; Tam, J.W.; Dumesnil, J.G.; Chan, K.L. Lipid lowering on progression of mild to moderate aortic stenosis: Meta-analysis of the randomized placebo-controlled clinical trials on 2344 patients. Can. J. Cardiol. 2011, 27, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.A.; Otto, C.M.; Bonow, R.O.; Carabello, B.A.; Erwin, J.P., 3rd; Guyton, R.A.; O’Gara, P.T.; Ruiz, C.E.; Skubas, N.J.; Sorajja, P.; et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: Executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2438–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahanian, A.; Alfieri, O.; Andreotti, F.; Antunes, M.J.; Barón-Esquivias, G.; Baumgartner, H.; Borger, M.A.; Carrel, T.P.; De Bonis, M.; Evangelista, A.; et al. Joint task force on the management of valvular heart disease of the European society of cardiology (ESC); European association for cardio-thoracic surgery (EACTS). guidelines on the management of valvular heart disease (version 2012). Eur. Heart J. 2012, 33, 2451–2496. [Google Scholar] [PubMed] [Green Version]
- Capoulade, R.; Yeang, C.; Chan, K.L.; Pibarot, P.; Tsimikas, S. Association of Mild to Moderate Aortic Valve Stenosis Progression with Higher Lipoprotein(a) and Oxidized Phospholipid Levels: Secondary Analysis of a Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 1212–1217. [Google Scholar] [CrossRef]
- Poggio, P.; Songia, P.; Cavallotti, L.; Barbieri, S.S.; Zanotti, I.; Arsenault, B.J.; Valerio, V.; Ferri, N.; Capoulade, R.; Camera, M. PCSK9 Involvement in Aortic Valve Calcification. J. Am. Coll. Cardiol. 2018, 72, 3225–3227. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Ruscica, M.; Greco, M.F.; Ferri, N.; Corsini, A. Lipoprotein(a) and PCSK9 inhibition: Clinical evidence. Eur. Heart J. Suppl. 2020, 22, L53–L56. [Google Scholar] [CrossRef]
- De Luca, L.; Corsini, A.; Uguccioni, M.; Colivicchi, F. Statins plus ezetimibe in the era of proprotein convertase subtilisin/kexin type 9 inhibitors. Kardiol. Pol. 2020, 78, 850–860. [Google Scholar] [CrossRef]
- Bergmark, B.A.; O’Donoghue, M.L.; Murphy, S.A.; Kuder, J.F.; Ezhov, M.V.; Ceška, R.; Gouni-Berthold, I.; Jensen, H.K.; Tokgozoglu, S.L.; Mach, F.; et al. An Exploratory Analysis of Proprotein Convertase Subtilisin/Kexin Type 9 Inhibition and Aortic Stenosis in the FOURIER Trial. JAMA Cardiol. 2020, 5, 709–713. [Google Scholar] [CrossRef]
- Perrot, N.; Valerio, V.; Moschetta, D.; Boekholdt, S.M.; Dina, C.; Chen, H.Y.; Abner, E.; Martinsson, A.; Manikpurage, H.D.; Rigade, S.; et al. Genetic and In Vitro Inhibition of PCSK9 and Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2020, 5, 649–661. [Google Scholar] [CrossRef]
- Galtier, F.; Mura, T.; Raynaud de Mauverger, E.; Chevassus, H.; Farret, A.; Gagnol, J.P.; Costa, F.; Dupuy, A.; Petit, P.; Cristol, J.P.; et al. Effect of a high dose of simvastatin on muscle mitochondrial metabolism and calcium signaling in healthy volunteers. Toxicol. Appl. Pharmacol. 2012, 263, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, P.; Bordenave, S.; Vermaelen, M.; Roels, B.; Vassort, G.; Mercier, J.; Raynaud, E.; Lacampagne, A. Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem. Biophys. Res. Commun. 2005, 338, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, P.; Mercier, J.; Vassort, G.; Lacampagne, A. Simvastatin triggers mitochondria-induced Ca2+ signaling alteration in skeletal muscle. Biochem. Biophys. Res. Commun. 2005, 329, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Lotteau, S.; Ivarsson, N.; Yang, Z.; Restagno, D.; Colyer, J.; Hopkins, P.; Weightman, A.; Himori, K.; Yamada, T.; Bruton, J.; et al. A Mechanism for Statin-Induced Susceptibility to Myopathy. JACC Basic Transl. Sci. 2019, 4, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Bonifacio, A.; Sanvee, G.M.; Bouitbir, J.; Krahenbuhl, S. The AKT/mTOR signaling pathway plays a key role in statin-induced myotoxicity. Biochim. Biophys. Acta 2015, 1853, 1841–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouitbir, J.; Charles, A.L.; Echaniz-Laguna, A.; Kindo, M.; Daussin, F.; Auwerx, J.; Piquard, F.; Geny, B.; Zoll, J. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: A ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur. Heart J. 2012, 33, 1397–1407. [Google Scholar] [CrossRef]
- Bonifacio, A.; Mullen, P.J.; Mityko, I.S.; Navegantes, L.C.; Bouitbir, J.; Krahenbuhl, S. Simvastatin induces mitochondrial dysfunction and increased atrogin-1 expression in H9c2 cardiomyocytes and mice in vivo. Arch. Toxicol. 2016, 90, 203–215. [Google Scholar] [CrossRef]
- Bouitbir, J.; Singh, F.; Charles, A.L.; Schlagowski, A.I.; Bonifacio, A.; Echaniz-Laguna, A.; Geny, B.; Krahenbuhl, S.; Zoll, J. Statins Trigger Mitochondrial Reactive Oxygen Species-Induced Apoptosis in Glycolytic Skeletal Muscle. Antioxid. Redox Signal. 2016, 24, 84–98. [Google Scholar] [CrossRef]
- Sanvee, G.M.; Hitzfeld, L.; Bouitbir, J.; Krahenbuhl, S. mTORC2 is an important target for simvastatin-associated toxicity in C2C12 cells and mouse skeletal muscle—Roles of Rap1 geranylgeranylation and mitochondrial dysfunction. Biochem. Pharmacol. 2021, 192, 114750. [Google Scholar] [CrossRef]
- Mullen, P.J.; Zahno, A.; Lindinger, P.; Maseneni, S.; Felser, A.; Krahenbuhl, S.; Brecht, K. Susceptibility to simvastatin-induced toxicity is partly determined by mitochondrial respiration and phosphorylation state of Akt. Biochim. Biophys. Acta 2011, 1813, 2079–2087. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Pradhan, A.; MacFadyen, J.G.; Libby, P.; Glynn, R.J. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: An analysis from the JUPITER trial. Lancet 2012, 380, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Crandall, J.P.; Mather, K.; Rajpathak, S.N.; Goldberg, R.B.; Watson, K.; Foo, S.; Ratner, R.; Barrett-Connor, E.; Temprosa, M. Statin use and risk of developing diabetes: Results from the Diabetes Prevention Program. BMJ Open Diabetes Res. Care 2017, 5, e000438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaluri, N.; Modi, S.; Kokkola, T. Simvastatin induces insulin resistance in L6 skeletal muscle myotubes by suppressing insulin signaling, GLUT4 expression and GSK-3beta phosphorylation. Biochem. Biophys. Res. Commun. 2016, 480, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Zhong, Z.; Wang, F.; Xu, J.; Xu, F.; Kong, W.; Ling, Z.; Shu, N.; Li, Y.; Wu, T.; et al. Atorvastatin impaired glucose metabolism in C2C12 cells partly via inhibiting cholesterol-dependent glucose transporter 4 translocation. Biochem. Pharmacol. 2018, 150, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Bouitbir, J.; Sanvee, G.M.; Panajatovic, M.V.; Singh, F.; Krahenbuhl, S. Mechanisms of statin-associated skeletal muscle-associated symptoms. Pharmacol. Res. 2020, 154, 104201. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nsaibia, M.J.; Devendran, A.; Goubaa, E.; Bouitbir, J.; Capoulade, R.; Bouchareb, R. Implication of Lipids in Calcified Aortic Valve Pathogenesis: Why Did Statins Fail? J. Clin. Med. 2022, 11, 3331. https://doi.org/10.3390/jcm11123331
Nsaibia MJ, Devendran A, Goubaa E, Bouitbir J, Capoulade R, Bouchareb R. Implication of Lipids in Calcified Aortic Valve Pathogenesis: Why Did Statins Fail? Journal of Clinical Medicine. 2022; 11(12):3331. https://doi.org/10.3390/jcm11123331
Chicago/Turabian StyleNsaibia, Mohamed J., Anichavezhi Devendran, Eshak Goubaa, Jamal Bouitbir, Romain Capoulade, and Rihab Bouchareb. 2022. "Implication of Lipids in Calcified Aortic Valve Pathogenesis: Why Did Statins Fail?" Journal of Clinical Medicine 11, no. 12: 3331. https://doi.org/10.3390/jcm11123331