
Genetic Cardiomyopathies: The Lesson Learned from hiPSCs

Abstract
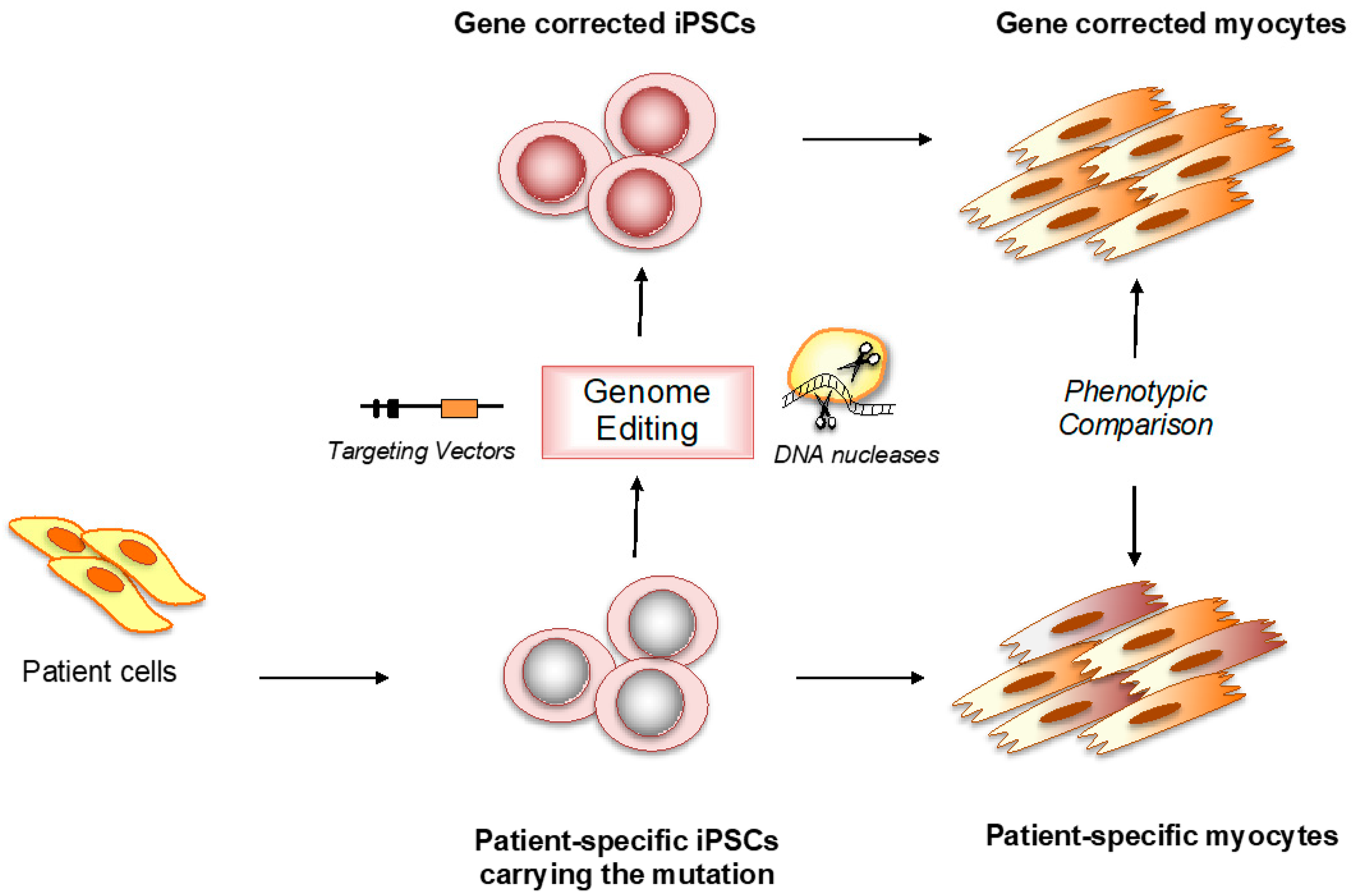
:1. Introduction
2. Hypertrophic Cardiomyopathy
3. Dilated Cardiomyopathy
4. Arrhythmogenic Cardiomyopathy
5. Left Ventricular Noncompaction Cardiomyopathy
6. Conclusions

{kind=link}
| Disease | Gene | Mutation | Cell Type | In Vitro Phenotype | Gene Editing Technique | Pharmacological Approach | References |
|---|---|---|---|---|---|---|---|
| HCM | MYH7 | R663H | patient hiPSCs | Cell enlargement, contractile arrhythmia, dysregulation of Ca2+ cycling and elevation of intracellular Ca2+ concentrations. | - | Verapamil, and other drugs | Lan F. et al. 2013 [13] |
| HCM | MYH7 | R442G | patient hiPSCs | Disruption of sarcomeric architecture, reduced Ca2+ transient, increased APD, arrhythmias. | - | Verapamil, and other drugs | Han L. et al. 2014 [14] |
| HCM | MYH7 | E848G | patient hiPSCs | Cell enlargement, myofibril disarray and contractile dysfunction using a nanopatterning approach. | - | - | Pioner J.M. et al. 2016 [15] |
| HCM/DCM | MYH7 | E848G | patient hiPSCs | Reduced contractile function as single cells and engineered heart tissues; disrupted MYH7 S2 and cMyBP-C C1C2 interaction. | CRISPR/Cas9 | - | Yang K.C. et al. 2018 [16] |
| HCM | MYH7 | R453C | control hiPSCs | Hypertrophy, multi-nucleation, sarcomeric disarray, higher metabolic respiration activity, abnormalities in calcium handling, arrhythmias, and contraction force. | CRISPR/Cas9 | Ranolazine (partial rescue) Omecamtiv mecarbil (no rescue) | Mosqueira D. et al. 2018 [17] |
| HCM | MYH7 | R403Q | patient hiPSCs | Increased maximal contraction and relaxation velocities and powers, increased maximal force on microppaterning devices. | MYH7 shRNA and antisense oligonucleotide | - | Dainis a. et al. 2020 [18] |
| HCM | MYH7 | R453C | patient hiPSCs | Calcium transient arrhythmias and intracellular calcium overload. Upregulation of CALM1, CASQ2 and CAMK2D, and downregulation of IRF8. Increased nuclear traslocation of NFATc1 and MEF2C. | CRISPR/Cas9 | Combination of dantrolene and ranolazine | Bhagwan J.R. et al. 2020 [19] |
| HCM | ACTC1 | E99K | patient hiPSCs | Calcium transient arrhythmias and intracellular calcium overload. Downregulation of CALM1, CASQ2 and CAMK2D, and upregulation of IRF8. Reduced nuclear traslocation of NFATc1 and MEF2C. | CRISPR/Cas9 | Combination of dantrolene and ranolazine; mavacamten | Bhagwan J.R. et al. 2020 [19] |
| HCM | MYBPC3 | G999_Q1004del | patient hiPSCs | Cardiomyocyte hypertrophy and intracellular myofibrillar disarray, strongly enhanced by Endothelin-1. | - | - | Tanaka A. et al.2014 [23] |
| HCM | MYBPC3 | W792Vfs+40X | patient hiPSCs | Greater surface area. | - | - | Dambrot C. et al. 2014 [24] |
| HCM | MYBPC3 | W792Vfs+40X | patient hiPSCs | Lower contractile force, under the presence of thyroid hormone, insulin growth factor-1, and dexamethasone. | - | - | Birket M.J. et al. 2015 [25] |
| HCM | MYBPC3 | G1061X | patient hiPSCs | Increased cell size and hypertrophic markers; EADs and DADs (higher thean in the TPM1-mut line). | - | - | Ojala M. et al 2016 [26] |
| HCM | TPM1 | D175N | patient hiPSCs | EADs and DADs; abnormal Ca2+ transients. Higher multinucleation, Ca2+ arrhythmias, longer APD than the in MYBPC3-mut line. | - | - | Ojala M. et al 2016 [26] |
| HCM | MYBPC3 | IVS26+1G/A | disease hESC | Sarcomere disarray, hypertrophy and impaired calcium impulse propagation, transient haploinsufficiency MYPBC3 during differentiation. | MYBPC3 AAV gene therapy | - | Da Rocha A.M et al [27] |
| HCM | MYBPC3 | V454Cfs+21X | patient hiPSCs | Increased cell size, cMyBPC haploinsufficiency, upregulation of BNP, MYH7 and other markers of hypertrophy. | MYBPC3 AAV gene therapy | - | Prondzynski M et al. 2017 [28] |
| HCM | MYL3 and MYBPC3 | MYL3-A57G and MYBPC3-V321M | patient and ctrl hiPSCs | Upregulation oh hypertrophy genes and proarrhythmic electrophysiological phenotype. | CRISPR/Cas9 | - | Ma N. et al. 2018 [35] |
| HCM/DCM | ALPK3 | W1264X | patient hiPSCs/hESCs | Disorganized sarcomere structures and intercalated discs, extended field potential duration, and increased irregular Ca2+ transients. | CRISPR/Cas9 knock out of ALPK3 | - | Phelan D.G. et al 2016 [36] |
| HCM | PRKAG2 | N488I | patient hiPSCs | Activating PRKAG2 mutation increase AMPK activity, glycogen accumulation, and AKT signaling resulting in hypertrophy with increased twitch force in 3D cardiac microtissues. | TALEN | - | Hinson J.T. at el. 2016 [37] |
| HCM | PRKAG2 | R302Q | patient hiPSCs | Abnormal firing patterns, DADs, triggered arrhythmias, and augmented beat rate variability. Increased glycogen storage. | CRISPR/Cas9 | - | Jehuda B.R et al. [38] |
| HCM | MYH7, MYBPC3, TNNT2 | MYH7-R663H MYBPC3-V321M MYBPC3-V219L MYBPC3-IVS27+1G>A TNNT2-R92W TNNT2-R92W | patient/control hiPSCs | Elevated diastolic intracellular calcium levels and enhanced myofilament calcium sensitivity. | CRISPR/Cas9 | Verapamil, diltiazem and late sodium channel blockers (ranolazine, electlazine) | Wu H. et al. 2019 [39] |
| HCM | n.a. | - | patient/control hiPSCs | Chronic verapamil treatment down-regulates muscle contraction related genes. | - | Nifedipine, amlodipine, dilitiazem, and verapamil | Lam C.K. et al. 2019 [40] |
| DCM | TTN | W976R A22352fs P22582fs N22577fs T33520fs V6382fs | patient/control hiPSCs | Assembled into cardiac microtissues, 2A band mutations displayed higher degree of sarcomere insufficiency, impaired responses to mechanical and β-adrenergic stress, and attenuated growth factor and cell signaling activation, compared to I band mutations. | CRISPR/Cas9 | - | Hinson J.T. at el. 2015 [46] |
| DCM | TTN | S14450fs+4X | patient hiPSCs | Disorganized sarcomere structures and MYH7, MYH6 and ACTC1 downregulation. | Exon skipping | - | Gramlich M. et al 2015 [47] |
| DCM | TTN | S19628Ifs+1X | patient hiPSCs | Disorganized sarcomere, diminished inotropic and lusitropic responses to β-adrenergic stimulation with isoproterenol, increased [Ca2+]out and angiotensin-II, prolonged recovery in response to caffeine. | - | - | Shick R. et al. 2018 [48] |
| DCM | LMNA | R225X | patient hiPSCs | Accelerated nuclear senescence with increased nuclear bleb formation and micronucleation, as well as increased apoptosis on electrical stimulation. | - | U0126 and selumetinib (MEK1/2 inhibitors that block ERK1/2 pathway) | Siu C.W. et al. 2012 [52] |
| DCM | LMNA | K219T | patient hiPSCs | Reduced peak sodium current and diminished conduction velocity. Downregulated Nav1.5 channel expression and increased binding of Lamin A/C to the promoter of SCN5A. Binding of the PRC2 protein SUZ12 and deposition of the repressive histone mark H3K27me3 are increased at SCN5A. | CRISPR/Cas9 | - | Salvarani N. et al. 2019 [53] |
| DCM | LMNA | K117Efs+9X | patient/control hiPSCs | Calcium-handling dysfunction, leading to arrhythmic phenotype at the single-cell level. Aberrant activation of PDGF-signaling pathway. | CRISPR/Cas9 | Crenolanib and sunitinib | Lee J. et al. 2019 [54] |
| DCM | LMNA | S143P | patient hiPSCs | Increased sensitivity to hypoxia, bradyarrhythmia and increased occurrence of arrhythmias under β-adrenergic stimulation. | - | - | Shah D. et al. 2019 [55] |
| DCM | LMNA | R225X | patient hiPSCs | Prolonged and increased amplitude of field potential, increased APD, systolic hyperfunction and diastolic dysfunction, up-regulation of CACNA1C and down-regulation of KCNQ1. Chromatin compartment changes do not explain most gene expression alterations. | CRISPR/Cas9 | Inhibition of P/Q-type calcium channels | Bertero A. et al. 2019 [56] |
| DCM | TTN2 | R173W | patient hiPSCs | Abnormal Ca2+ handling, decreased contractility, myofibrillar disarray, which were exacerbated with β-adrenergic stimulation, upregulation of PDE3A/PDE2A. | Overexpression of SERCA2a | Metoprolol | Sun N. et al. 2012 [57] |
| DCM | TTN2 | R173W | patient hiPSCs | Increased nuclear translocation of TNNT2 and enhanced epigenetic activation of the phosphodiesterase genes PDE2A and PDE3A. | - | Treatment with the PDE2 and PDE3 pharmacological inhibitors BAY 60-7550 | Wu H. et al. 2015 [58] |
| DCM | TTN2 | R173W | patient hiPSCs | Altered myofibrillar architecture rescued by omecamtiv mecarbil. | - | Omecamtiv mecarbil | Broughton K.M. et al. 2016 [59] |
| DCM | TTN2 | R173W | patient hiPSCs | Destabilized molecular interactions of troponin with tropomyosin, and limited binding of PKA to local sarcomere microdomains, dysregulated local sarcomeric microdomains. | CRISPR/Cas9 | Small molecule-based activation of AMPK | Dai Y. et al. 2020 [60] |
| DCM/HCM | TTN2 and TTN | TNNT2-R173W, TTN-N22577fs TTN-c.67745delT | patient/control hiPSCs | Telomere shortening. | CRISPR/Cas9 | - | Chang A.C.Y. Et al. 2018 [61] |
| DCM | TNNT2 | R173W K210del | patient/control hiPSCs | Electrophysiological characterization of the several lines generated. | CRISPR/Cas9 | - | Lv W. et al 2018 [62] |
| DCM | TNNT2 | R173W | patient hiPSCs | The knockout strategy ameliorates the dilated cardiomyopathy phenotype in vitro. | TALEN | - | Karakikes I. et al. 2017 [63] |
| DCM | SPEG | E1680K | control hiPSCs | Aberrant calcium homeostasis, impaired contractility, and sarcomeric disorganization. | CRISPR/Cas9 | - | Levitas A. et al. 2020 [64] |
| DCM | DES | A285V | patient/control hiPSCs | Increased desmin aggregations, abnormal calcium handling and altered response to inotropic stress. | Overexpression | - | Tse H.F. et al. 2013 [65] |
| DCM | PLB | R14del | patient hiPSCs | Calcium handling abnormalities and myofibrillar disarray. | TALEN | - | Karakikes I. et al. 2015 [66] |
| DCM | PLB | R14del | patient hiPSCs | rAAV2-driven expression of PLBM rescues arrhythmic Ca2+ transients and alleviates decreased Ca2+ transport. | Overexpression | - | Stroik D.R. et al, 2020 [67] |
| DCM | PLB | R9C | control hiPSCs | Blunted β-agonist response both in 2D and 3D models, activation of a hypertrophic phenotype, altered metabolic state and profibrotic signaling. | CRISPR/Cas9 | - | Ceholski D.K. et al. 2018 [68] |
| DCM | RBM20 | R636 | patient hiPSCs | Abnormal Ca2+ handling, disruption of sarcomeric architecture. | - | - | Wyles S.P. et al. 2016 [69] |
| DCM | RBM20 | S635A | patient hiPSCs | Abnormal sarcomeric structure and altered calcium handling, abnormal active force generation and passive stress in engineered heart muscles, reduced titin N2B-isoform expression. | - | - | Streckfuss-Bömeke K. et al. 2017 [70] |
| DCM | BAG3 | knock-out | control hiPSCs | Myofibrillar disarray and contractile dysfunction increased by bortezomib. | CRISPR/Cas9 and TALEN | - | Judge L.M. et al. 2017 [72] |
| DCM | BAG3 | R477H | control hiPSCs | Fiber length and alignment declined markedly in mutated and knock-out lines following proteasome inhibition. | CRISPR/Cas9 | - | McDermott-Roe C. et al. 2019 [73] |
| DCM | TPM1 and VCL | TPM1-E33K VCL-N220Kfs+20X | patient hiPSCs | Reduced contractility and abnormal sarcomeric structure. | CRISPR/Cas9 | - | Deacon D.C. et al. 2019 [74] |
| ACM | PKP2 | L614P | patient hiPSCs | Reduced density of plakophilin-2 and plakoglobin proteins. Larger cells with darker lipid droplets under adipogenic medium. | - | - | Ma D. et al. 2013 [76] |
| ACM | PKP2 | A324fs+11X T50Sfs+60X | patient hiPSCs | Reduced levels of desmosomal protein PKP2, plakoglobin and connexin-43, prolonged field potential rise time, widened and distorted desmosomes, increased apoptosis. Findings enhanced under adipogenic medium. | - | - | Caspi O. et al. 2013 [77] |
| ACM | PKP2 | G828Gfs+102X K672fs+11X | patient hiPSCs | Abnormal plakoglobin nuclear translocation; reduced β-catenin activity; exaggerated lipogenesis (PPARγ activation) and apoptosis. | - | - | Kim C. et al. 2013 [78] |
| ACM | PKP2 and MYH10 | PKP2-V587Afs+68X MYH10-R577X | patient hiPSCs | RhoA signaling downstream of cell-cell junctions regulates pathological myocyte-to-adipocyte conversion by controlling MRTF-A cellular localization and PPARγ activation. | - | - | Dorn T. et al. 2018 [79] |
| ACM | PKP2 | G784= K672fs | patient hiPSCs | Coactivation of PPARα and γ resulted in lipid accumulation; apoptosis, altered sodium current and calcium handling. | - | - | Wen J.Y. et al. 2015 [80] |
| ACM | PKP2 | G784= | patient hiPSCs | Testosterone worsened and estradiol improved cardiomyocyte apoptosis and lipogenesis. | - | - | Akdis D. et al 2017 [81] |
| ACM | PKP2 | Q617X 2013delC | patient hiPSCs | Marked reduction in immunoreactive signals for plakoglobin, Cx43, SAP97, and Nav1.5; reversed after exposure to SB216763. | - | SB216763 | Asimaki A. et al 2014 [82] |
| ACM | DSG2 | G638R | patient hiPSCs | Smaller amplitude and maximal upstroke velocity of action potential due to decreased peak sodium current, resting potential and APD was not changed. | - | - | El-Battrawy, I. et al 2018 [83] |
| ACM | DSG2 | G638R | patient hiPSCs | Increased expression of NDPK-B, via activating SK4 channels, contributing to arrhythmogenesis. | - | - | Buljubasic F. et al 2020 [84] |
| ACM | SCN5A | R1898H | patient hiPSCs | Reduced sodium current, Nav1.5 and N-Cadherin clusters at junctional sites. | CRISPR/Cas9 | - | Riele A.S.T. et al. 2017 [85] |
| ACM | DSP | R451G | patient hiPSCs | Augmented calpain-mediated degradation of desmoplakin, depressed levels of desmoplakin in the absence of abnormal electrical propagation. | - | - | Ng R. et al 2019 [86] |
| ACM | TMEM43 | S358L | control hiPSCs | Slower rising and decay phases of the Ca2+ transient under isoproterenol, increased contraction duration, time to peak, and relaxation time, accompanied by decreased contraction amplitude. | CRISPR/Cas9 | CHIR99021 (GSK3β inhibitor) | Padrón-Barthe L. et al. 2019 [87] |
| ACM | OBSCN | L5218fs | patient hiPSCs | Accumulation of lipids, increased pleomorphism, irregular Z-bands, and increased L type calcium currents, decreased expression of OBSCN and its anchor protein, Ank1.5, and other desmosomal proteins, increased expression of N-Cadherin, PPARγ, C/EBPα, and FABP4. | - | - | Chen P. et al. 2020 [88] |
| LVNC | TBX20 PRDM16 | TBX20-Y317X TBX20-T262M | patient/control hiPSCs | Proliferation defect at a single cell level, upregulation of genes in TGFb pathway, TBX20 controls TGF beta signaling modifiers (including PRDM16). | CRISPR/Cas9 | TGF-β receptor-1 inhibitors | Kodo K. et al. 2016 [92] |
| LVNC | GATA4 | G296S | patient hiPSCs | Impaired contractility, calcium handling, and metabolic activity, disrupted TBX5 recruitment to cardiac super-enhancers. | CRISPR/Cas9 | - | Ang Y.S. et al 2016 [93] |
| LVNC | MYH7, MKL2, NKX2-5 | MYH7-L387F MKL2-Q670H NKX2-5 A119S | patient hiPSCs | Cell detachment and down-regulation of genes associated with cell adhesion and extracellular matrix deposition, up-regulation of cell cycle, cardiac developmental and trabeculae genes. | - | - | Gifford C.A. et al. 2019 [94] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Ben Gal, T.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, P.W.; Holmström, A.; Wu, J.C. Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 2015, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-Specific Induced Pluripotent Stem-Cell Models for Long-QT Syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [Green Version]
- Carvajal-Vergara, X.; Sevilla, A.; D’Souza, S.L.; Ang, Y.-S.; Schaniel, C.; Lee, D.-F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nat. Cell Biol. 2010, 465, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Del Álamo, J.C.; Lemons, D.; Serrano, R.; Savchenko, A.; Cerignoli, F.; Bodmer, R.; Mercola, M. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 1717–1727. [Google Scholar] [CrossRef]
- Feyen, D.A.; McKeithan, W.L.; Bruyneel, A.A.; Spiering, S.; Hörmann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925. [Google Scholar] [CrossRef]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef] [PubMed]
- Goedel, A.; My, I.; Sinnecker, D.; Moretti, A. Perspectives and Challenges of Pluripotent Stem Cells in Cardiac Arrhythmia Research. Curr. Cardiol. Rep. 2017, 19, 23. [Google Scholar] [CrossRef]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 76, e159–e240. [Google Scholar] [CrossRef]
- Bottillo, I.; D’Angelantonio, D.; Caputo, V.; Paiardini, A.; Lipari, M.; De Bernardo, C.; Giannarelli, D.; Pizzuti, A.; Majore, S.; Castori, M.; et al. Molecular analysis of sarcomeric and non-sarcomeric genes in patients with hypertrophic cardiomyopathy. Gene 2016, 577, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal Calcium Handling Properties Underlie Familial Hypertrophic Cardiomyopathy Pathology in Patient-Specific Induced Pluripotent Stem Cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Li, Y.; Tchao, J.; Kaplan, A.D.; Lin, B.; Li, Y.; Mich-Basso, J.; Lis, A.; Hassan, N.; London, B.; et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 2014, 104, 258–269. [Google Scholar] [CrossRef]
- Pioner, J.M.; Racca, A.W.; Klaiman, J.M.; Yang, K.-C.; Guan, X.; Pabon, L.; Muskheli, V.; Zaunbrecher, R.; Macadangdang, J.; Jeong, M.Y.; et al. Isolation and Mechanical Measurements of Myofibrils from Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cell Rep. 2016, 6, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.-C.; Breitbart, A.; De Lange, W.J.; Hofsteen, P.; Futakuchi-Tsuchida, A.; Xu, J.; Schopf, C.; Razumova, M.V.; Jiao, A.; Boucek, R.; et al. Novel Adult-Onset Systolic Cardiomyopathy Due to MYH7 E848G Mutation in Patient-Derived Induced Pluripotent Stem Cells. JACC Basic Transl. Sci. 2018, 3, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Mosqueira, D.; Mannhardt, I.; Bhagwan, J.R.; Lis-Slimak, K.; Katili, P.; Scott, E.; Hassan, M.; Prondzynski, M.; Harmer, S.C.; Tinker, A.; et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. 2018, 39, 3879–3892. [Google Scholar] [CrossRef] [PubMed]
- Dainis, A.; Zaleta-Rivera, K.; Ribeiro, A.; Chang, A.C.H.; Shang, C.; Lan, F.; Burridge, P.W.; Liu, W.R.; Wu, J.C.; Chang, A.C.Y.; et al. Silencing of MYH7 ameliorates disease phenotypes in human iPSC-cardiomyocytes. Physiol. Genom. 2020, 52, 293–303. [Google Scholar] [CrossRef]
- Bhagwan, J.R.; Mosqueira, D.; Chairez-Cantu, K.; Mannhardt, I.; Bodbin, S.E.; Bakar, M.; Smith, J.G.; Denning, C. Isogenic models of hypertrophic cardiomyopathy unveil differential phenotypes and mechanism-driven therapeutics. J. Mol. Cell. Cardiol. 2020, 145, 43–53. [Google Scholar] [CrossRef]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Dauphin, C.; Jouk, P.S.; Da Costa, A.; Prieur, F.; Bresson, J.-L.; Faivre, L.; Eicher, J.-C.; et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur. J. Med Genet. 2010, 53, 261–267. [Google Scholar] [CrossRef]
- Kaski, J.P.; Syrris, P.; Esteban, M.T.T.; Jenkins, S.; Pantazis, A.; Deanfield, J.E.; McKenna, W.J.; Elliott, P.M. Prevalence of Sarcomere Protein Gene Mutations in Preadolescent Children With Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, J.; Daehmlow, S.; Wischke, S.; Senyuva, M.; Werner, U.; Raible, J.; Tanis, N.; Dyachenko, S.; Hummel, M.; Hetzer, R.; et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin. Genet. 2003, 64, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Yuasa, S.; Mearini, G.; Egashira, T.; Seki, T.; Kodaira, M.; Kusumoto, D.; Kuroda, Y.; Okata, S.; Suzuki, T.; et al. Endothelin-1 Induces Myofibrillar Disarray and Contractile Vector Variability in Hypertrophic Cardiomyopathy–Induced Pluripotent Stem Cell–Derived Cardiomyocytes. J. Am. Heart Assoc. 2014, 3, e001263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dambrot, C.; Braam, S.R.; Tertoolen, L.G.J.; Birket, M.; Atsma, D.E.; Mummery, C.L. Serum supplemented culture medium masks hypertrophic phenotypes in human pluripotent stem cell derived cardiomyocytes. J. Cell. Mol. Med. 2014, 18, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Birket, M.J.; Ribeiro, M.C.; Kosmidis, G.; Ward, D.; Leitoguinho, A.R.; van de Pol, V.; Dambrot, C.; Devalla, H.D.; Davis, R.P.; Mastroberardino, P.G.; et al. Contractile Defect Caused by Mutation in MYBPC3 Revealed under Conditions Optimized for Human PSC-Cardiomyocyte Function. Cell Rep. 2015, 13, 733–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojala, M.; Prajapati, C.; Pölönen, R.-P.; Rajala, K.; Pekkanen-Mattila, M.; Rasku, J.; Larsson, K.; Aalto-Setälä, K. Mutation-Specific Phenotypes in hiPSC-Derived Cardiomyocytes Carrying Either Myosin-Binding Protein C Orα-Tropomyosin Mutation for Hypertrophic Cardiomyopathy. Stem Cells Int. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Rocha, A.M.; Guerrero-Serna, G.; Helms, A.; Luzod, C.; Mironov, S.; Russell, M.; Jalife, J.; Day, S.M.; Smith, G.D.; Herron, T.J. Deficient cMyBP-C protein expression during cardiomyocyte differentiation underlies human hypertrophic cardiomyopathy cellular phenotypes in disease specific human ES cell derived cardiomyocytes. J. Mol. Cell. Cardiol. 2016, 99, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Prondzynski, M.; Krämer, E.; Laufer, S.D.; Shibamiya, A.; Pless, O.; Flenner, F.; Müller, O.J.; Münch, J.; Redwood, C.; Hansen, A.; et al. Evaluation of MYBPC3 trans -Splicing and Gene Replacement as Therapeutic Options in Human iPSC-Derived Cardiomyocytes. Mol. Ther. Nucleic Acids 2017, 7, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Seeger, T.; Shrestha, R.; Lam, C.K.; Chen, C.; McKeithan, W.L.; Lau, E.; Wnorowski, A.; McMullen, G.; Greenhaw, M.; Lee, J.; et al. A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardiomyopathy via Chronic Activation of Nonsense-Mediated Decay. Circulation 2019, 139, 799–811. [Google Scholar] [CrossRef]
- Cohn, R.; Thakar, K.; Lowe, A.; Ladha, F.A.; Pettinato, A.M.; Romano, R.; Meredith, E.; Chen, Y.-S.; Atamanuk, K.; Huey, B.D.; et al. A Contraction Stress Model of Hypertrophic Cardiomyopathy due to Sarcomere Mutations. Stem Cell Rep. 2019, 12, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Kim, K.; Parikh, S.; Cadar, A.G.; Bersell, K.R.; He, H.; Pinto, J.R.; Kryshtal, D.O.; Knollmann, B.C. Hypertrophic cardiomyopathy-linked mutation in troponin T causes myofibrillar disarray and pro-arrhythmic action potential changes in human iPSC cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 114, 320–327. [Google Scholar] [CrossRef]
- Zhou, W.; Bos, J.M.; Ye, D.; Tester, D.J.; Hrstka, S.; Maleszewski, J.J.; Ommen, S.R.; Nishimura, R.A.; Schaff, H.V.; Kim, C.S.; et al. Induced Pluripotent Stem Cell–Derived Cardiomyocytes from a Patient with MYL2-R58Q-Mediated Apical Hypertrophic Cardiomyopathy Show Hypertrophy, Myofibrillar Disarray, and Calcium Perturbations. J. Cardiovasc. Transl. Res. 2019, 12, 394–403. [Google Scholar] [CrossRef]
- Smith, J.G.; Owen, T.; Bhagwan, J.R.; Mosqueira, D.; Scott, E.; Mannhardt, I.; Patel, A.; Barriales-Villa, R.; Monserrat, L.; Hansen, A.; et al. Isogenic Pairs of hiPSC-CMs with Hypertrophic Cardiomyopathy/LVNC-Associated ACTC1 E99K Mutation Unveil Differential Functional Deficits. Stem Cell Rep. 2018, 11, 1226–1243. [Google Scholar] [CrossRef] [Green Version]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horváth, A.; Di Mauro, V.; Koivumäki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Krämer, E.; et al. Disease modeling of a mutation in α-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, J.Z.; Itzhaki, I.; Zhang, S.L.; Chen, H.; Haddad, F.; Kitani, T.; Wilson, K.D.; Tian, L.; Shrestha, R.; et al. Determining the Pathogenicity of a Genomic Variant of Uncertain Significance Using CRISPR/Cas9 and Human-Induced Pluripotent Stem Cells. Circulation 2018, 138, 2666–2681. [Google Scholar] [CrossRef]
- Phelan, D.G.; Anderson, D.J.; Howden, S.E.; Wong, R.C.; Hickey, P.F.; Pope, K.; Wilson, G.R.; Pébay, A.; Davis, A.M.; Petrou, S.; et al. ALPK3-deficient cardiomyocytes generated from patient-derived induced pluripotent stem cells and mutant human embryonic stem cells display abnormal calcium handling and establish that ALPK3 deficiency underlies familial cardiomyopathy. Eur. Heart J. 2016, 37, 2586–2590. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.T.; Chopra, A.; Lowe, A.; Sheng, C.C.; Gupta, R.M.; Kuppusamy, R.; O’Sullivan, J.; Rowe, G.; Wakimoto, H.; Gorham, J.; et al. Integrative Analysis of PRKAG2 Cardiomyopathy iPS and Microtissue Models Identifies AMPK as a Regulator of Metabolism, Survival, and Fibrosis. Cell Rep. 2016, 17, 3292–3304. [Google Scholar] [CrossRef] [PubMed]
- Jehuda, B.R.; Eisen, B.; Shemer, Y.; Mekies, L.N.; Szantai, A.; Reiter, I.; Cui, H.; Guan, K.; Haron-Khun, S.; Freimark, D.; et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm. 2018, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, H.; Rhee, J.-W.; Zhang, J.Z.; Lam, C.K.; Sallam, K.; Chang, A.C.Y.; Ma, N.; Lee, J.; Zhang, H.; et al. Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur. Heart J. 2019, 40, 3685–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, C.K.; Tian, L.; Belbachir, N.; Wnorowski, A.; Shrestha, R.; Ma, N.; Kitani, T.; Rhee, J.-W.; Wu, J.C. Identifying the Transcriptome Signatures of Calcium Channel Blockers in Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes. Circ. Res. 2019, 125, 212–222. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Carrier, L. Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes—A systematic review. Pflügers Arch. Eur. J. Physiol. 2019, 471, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [CrossRef]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement from the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.; Vatta, M.; Ware, S.M. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Nat. Rev. Cardiol. 2019, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.T.; Chopra, A.K.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.M.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramlich, M.; Pane, L.S.; Zhou, Q.; Chen, Z.; Murgia, M.; Schötterl, S.; Goedel, A.; Metzger, K.; Brade, T.; Parrotta, E.; et al. Antisense-mediated exon skipping: A therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol. Med. 2015, 7, 562–576. [Google Scholar] [CrossRef]
- Schick, R.; Mekies, L.N.; Shemer, Y.; Eisen, B.; Hallas, T.; Ben Jehuda, R.; Ben-Ari, M.; Szantai, A.; Willi, L.; Shulman, R.; et al. Functional abnormalities in induced Pluripotent Stem Cell-derived cardiomyocytes generated from titin-mutated patients with dilated cardiomyopathy. PLoS ONE 2018, 13, e0205719. [Google Scholar] [CrossRef]
- Dittmer, A.T.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Crasto, S.; My, I.; Di Pasquale, E. The Broad Spectrum of LMNA Cardiac Diseases: From Molecular Mechanisms to Clinical Phenotype. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Ho, J.C.Y.; Zhou, T.; Lai, W.-H.; Huang, Y.; Chan, Y.-C.; Li, X.; Wong, N.L.Y.; Li, Y.; Au, K.-W.; Guo, D.; et al. Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging 2011, 3, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, C.-W.; Lee, Y.-K.; Ho, J.C.-Y.; Lai, W.-H.; Chan, Y.-C.; Ng, K.-M.; Wong, L.-Y.; Au, K.-W.; Lau, Y.-M.; Zhang, J.; et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 2012, 4, 803–822. [Google Scholar] [CrossRef] [Green Version]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Ferro, M.D.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Termglinchan, V.; Diecke, S.; Itzhaki, I.; Lam, C.K.; Garg, P.; Lau, E.; Greenhaw, M.; Seeger, T.; Wu, H.; et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nat. Cell Biol. 2019, 572, 335–340. [Google Scholar] [CrossRef]
- Shah, D.; Virtanen, L.; Prajapati, C.; Kiamehr, M.; Gullmets, J.; West, G.; Kreutzer, J.; Pekkanen-Mattila, M.; Heliö, T.; Kallio, P.; et al. Modeling of LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cells. Cells 2019, 8, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, A.; Fields, P.A.; Smith, A.S.; Leonard, A.; Beussman, K.; Sniadecki, N.J.; Kim, D.-H.; Tse, H.-F.; Pabon, L.; Shendure, J.; et al. Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. J. Cell Biol. 2019, 218, 2919–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-Specific Induced Pluripotent Stem Cells as a Model for Familial Dilated Cardiomyopathy. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Lee, J.; Vincent, L.G.; Wang, Q.; Gu, M.; Lan, F.; Churko, J.M.; Sallam, K.I.; Matsa, E.; Sharma, A.; et al. Epigenetic Regulation of Phosphodiesterases 2A and 3A Underlies Compromised β-Adrenergic Signaling in an iPSC Model of Dilated Cardiomyopathy. Cell Stem Cell 2015, 17, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broughton, K.M.; Jieli, L.; Sarmah, E.; Warren, C.M.; Elina, S.; Henze, M.P.; Sanchez-Freire, V.; Solaro, R.J.; Russell, B. A myosin activator improves actin assembly and sarcomere function of human-induced pluripotent stem cell-derived cardiomyocytes with a troponin T point mutation. Am. J. Physiol. Circ. Physiol. 2016, 311, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Amenov, A.; Ignatyeva, N.; Koschinski, A.; Xu, H.; Soong, P.L.; Tiburcy, M.; Linke, W.A.; Zaccolo, M.; Hasenfuss, G.; et al. Troponin destabilization impairs sarcomere-cytoskeleton interactions in iPSC-derived cardiomyocytes from dilated cardiomyopathy patients. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.C.Y.; Chang, A.C.H.; Kirillova, A.; Sasagawa, K.; Su, W.; Weber, G.; Lin, J.; Termglinchan, V.; Karakikes, I.; Seeger, T.; et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 9276–9281. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.; Qiao, L.; Petrenko, N.; Li, W.; Owens, A.T.; McDermott-Roe, C.; Musunuru, K. Functional Annotation of TNNT2 Variants of Uncertain Significance With Genome-Edited Cardiomyocytes. Circulation 2018, 138, 2852–2854. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Termglinchan, V.; Cepeda, D.A.; Lee, J.; Diecke, S.; Hendel, A.; Itzhaki, I.; Ameen, M.; Shrestha, R.; Wu, H.; et al. A Comprehensive TALEN-Based Knockout Library for Generating Human-Induced Pluripotent Stem Cell–Based Models for Cardiovascular Diseases. Circ. Res. 2017, 120, 1561–1571. [Google Scholar] [CrossRef]
- Levitas, A.; Muhammad, E.; Zhang, Y.; Gil, I.P.; Serrano, R.; Diaz, N.; Arafat, M.; Gavidia, A.A.; Kapiloff, M.S.; Mercola, M.; et al. A Novel Recessive Mutation in SPEG Causes Early Onset Dilated Cardiomyopathy. PLoS Genet. 2020, 16, e1009000. [Google Scholar] [CrossRef]
- Tse, H.-F.; Ho, J.C.Y.; Choi, S.-W.; Lee, Y.-K.; Butler, A.W.; Ng, K.-M.; Siu, C.-W.; Simpson, M.A.; Lai, W.-H.; Chan, Y.-C.; et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum. Mol. Genet. 2013, 22, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.-W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef] [PubMed]
- Stroik, D.R.; Ceholski, D.K.; Bidwell, P.A.; Mleczko, J.; Thanel, P.F.; Kamdar, F.; Autry, J.M.; Cornea, R.L.; Thomas, D.D. Viral expression of a SERCA2a-activating PLB mutant improves calcium cycling and synchronicity in dilated cardiomyopathic hiPSC-CMs. J. Mol. Cell. Cardiol. 2020, 138, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Ceholski, D.K.; Turnbull, I.C.; Kong, C.-W.; Koplev, S.; Mayourian, J.; Gorski, P.A.; Stillitano, F.; Skodras, A.A.; Nonnenmacher, M.; Cohen, N.; et al. Functional and transcriptomic insights into pathogenesis of R9C phospholamban mutation using human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 119, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Wyles, S.P.; Li, X.; Hrstka, S.C.; Reyes, S.; Oommen, S.; Beraldi, R.; Edwards, J.; Terzic, A.; Olson, T.M.; Nelson, T.J. Modeling structural and functional deficiencies ofRBM20familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum. Mol. Genet. 2016, 25, 254–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streckfuss-Bömeke, K.; Tiburcy, M.; Fomin, A.; Luo, X.; Li, W.; Fischer, C.; Özcelik, C.; Perrot, A.; Sossalla, S.; Haas, J.; et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2017, 113, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, A.; Fields, P.A.; Ramani, V.; Bonora, G.; Yardimci, G.G.; Reinecke, H.; Pabon, L.; Noble, W.S.; Shendure, J.; Murry, C.E. Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Judge, L.M.; Perez-Bermejo, J.A.; Truong, A.; Ribeiro, A.J.; Yoo, J.C.; Jensen, C.L.; Mandegar, M.A.; Huebsch, N.; Kaake, R.M.; So, P.-L.; et al. A BAG3 chaperone complex maintains cardiomyocyte function during proteotoxic stress. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- McDermott-Roe, C.; Lv, W.; Maximova, T.; Wada, S.; Bukowy, J.; Marquez, M.; Lai, S.; Shehu, A.; Benjamin, I.; Geurts, A.; et al. Investigation of a dilated cardiomyopathy-associated variant in BAG3 using genome-edited iPSC-derived cardiomyocytes. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Deacon, D.C.; Happe, C.L.; Chen, C.; Tedeschi, N.; Manso, A.M.; Li, T.; Dalton, N.D.; Peng, Q.; Farah, E.N.; Gu, Y.; et al. Combinatorial interactions of genetic variants in human cardiomyopathy. Nat. Biomed. Eng. 2019, 3, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Ma, D.; Wei, H.; Lu, J.; Ho, S.; Zhang, G.; Sun, X.; Oh, Y.; Tan, S.H.; Ng, M.L.; Shim, W.; et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 34, 1122–1133. [Google Scholar] [CrossRef] [Green Version]
- Caspi, O.; Huber, I.; Gepstein, A.; Arbel, G.; Maizels, L.; Boulos, M.; Gepstein, L. Modeling of Arrhythmogenic Right Ventricular Cardiomyopathy With Human Induced Pluripotent Stem Cells. Circ. Cardiovasc. Genet. 2013, 6, 557–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nat. Cell Biol. 2013, 494, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Dorn, T.; Kornherr, J.; I Parrotta, E.; Zawada, D.; Ayetey, H.; Santamaria, G.; Iop, L.; Mastantuono, E.; Sinnecker, D.; Goedel, A.; et al. Interplay of cell–cell contacts and RhoA/ MRTF -A signaling regulates cardiomyocyte identity. EMBO J. 2018, 37, e98133. [Google Scholar] [CrossRef]
- Wen, J.-Y.; Wei, C.-Y.; Shah, K.; Wong, J.; Wang, C.; Chen, H.-S.V. Maturation-Based Model of Arrhythmogenic Right Ventricular Dysplasia Using Patient-Specific Induced Pluripotent Stem Cells. Circ. J. 2015, 79, 1402–1408. [Google Scholar] [CrossRef] [Green Version]
- Akdis, D.; Saguner, A.M.; Shah, K.; Wei, C.; Medeiros-Domingo, A.; Von Eckardstein, A.; Lüscher, T.F.; Brunckhorst, C.; Chen, H.V.; Duru, F. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: From a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur. Heart J. 2017, 38, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Arndt, A.K.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.-H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. Europace 2018, 20, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Buljubasic, F.; El-Battrawy, I.; Lan, H.; Lomada, S.K.; Chatterjee, A.; Zhao, Z.; Li, X.; Zhong, R.; Xu, Q.; Huang, M.; et al. Nucleoside Diphosphate Kinase B Contributes to Arrhythmogenesis in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes from a Patient with Arrhythmogenic Right Ventricular Cardiomyopathy. J. Clin. Med. 2020, 9, 486. [Google Scholar] [CrossRef] [Green Version]
- Riele, A.S.T.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Rothenberg, E.; Sobreira, N.L.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient mutations linked to arrhythmogenic cardiomyopathy enhance calpain-mediated desmoplakin degradation. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Padrón-Barthe, L.; Villalba-Orero, M.; Gómez-Salinero, J.M.; Domínguez, F.; Román, M.; Larrasa-Alonso, J.; Ortiz-Sánchez, P.; Martínez, F.; López-Olañeta, M.; Bonzón-Kulichenko, E.; et al. Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3β. Circulation 2019, 140, 1188–1204. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Xiao, Y.; Wang, Y.; Zheng, Z.; Chen, L.; Yang, X.; Li, J.; Wu, W.; Zhang, S. Intracellular calcium current disorder and disease phenotype in OBSCN mutant iPSC-based cardiomyocytes in arrhythmogenic right ventricular cardiomyopathy. Theranostics 2020, 10, 11215–11229. [Google Scholar] [CrossRef]
- Van Waning, J.I.; Caliskan, K.; Hoedemaekers, Y.M.; Van Spaendonck-Zwarts, K.Y.; Baas, A.F.; Boekholdt, S.M.; Van Melle, J.P.; Teske, A.J.; Asselbergs, F.W.; Backx, P.C.M.; et al. Genetics, Clinical Features, and Long-Term Outcome of Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 711–722. [Google Scholar] [CrossRef]
- Hoedemaekers, Y.M.; Caliskan, K.; Michels, M.; Frohn-Mulder, I.; Van Der Smagt, J.J.; Phefferkorn, J.E.; Wessels, M.W.; Cate, F.J.T.; Sijbrands, E.J.; Dooijes, D.; et al. The Importance of Genetic Counseling, DNA Diagnostics, and Cardiologic Family Screening in Left Ventricular Noncompaction Cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Klaassen, S.; Probst, S.; Oechslin, E.; Gerull, B.; Krings, G.; Schuler, P.; Greutmann, M.; Hürlimann, D.; Yegitbasi, M.; Pons, L.; et al. Mutations in Sarcomere Protein Genes in Left Ventricular Noncompaction. Circulation 2008, 117, 2893–2901. [Google Scholar] [CrossRef] [PubMed]
- Kodo, K.; Ong, S.-G.; Jahanbani, F.; Termglinchan, V.; Hirono, K.; InanlooRahatloo, K.; Ebert, A.D.; Shukla, P.; Abilez, O.J.; Churko, J.M.; et al. iPSC-derived cardiomyocytes reveal abnormal TGF-β signalling in left ventricular non-compaction cardiomyopathy. Nat. Cell Biol. 2016, 18, 1031–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, Y.-S.; Rivas, R.N.; Ribeiro, A.J.; Srivas, R.; Rivera, J.; Stone, N.R.; Pratt, K.; Mohamed, T.M.; Fu, J.-D.; Spencer, C.I.; et al. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell 2016, 167, 1734–1749. [Google Scholar] [CrossRef] [Green Version]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; De Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Birket, M.J.; Ribeiro, M.C.; Verkerk, A.O.; Ward, D.; Leitoguinho, A.R.; Hartogh, S.C.D.; Orlova, V.V.; Devalla, H.D.; Schwach, V.; Bellin, M.; et al. Expansion and patterning of cardiovascular progenitors derived from human pluripotent stem cells. Nat. Biotechnol. 2015, 33, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nat. Cell Biol. 2018, 556, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, E.; Meraviglia, V.; Campostrini, G.; Cochrane, A.; Cao, X.; van Helden, R.W.; Garcia, A.K.; Mircea, M.; Kostidis, S.; Davis, R.P.; et al. Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 2020, 26, 862–879. [Google Scholar] [CrossRef]
- Mikryukov, A.A.; Mazine, A.; Wei, B.; Yang, D.; Miao, Y.; Gu, M.; Keller, G.M. BMP10 Signaling Promotes the Development of Endocardial Cells from Human Pluripotent Stem Cell-Derived Cardiovascular Progenitors. Cell Stem Cell 2021, 28, 96–111. [Google Scholar] [CrossRef]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017, 21, 179–194. [Google Scholar] [CrossRef]
- Protze, I.S.; Liu, J.; Nussinovitch, U.; Ohana, L.; Backx, J.L.L.O.P.H.; Gepstein, U.N.L.; Keller, S.I.P.G.M. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat. Biotechnol. 2017, 35, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Rafatian, N.; Feric, N.T.; Cox, B.J.; Aschar-Sobbi, R.; Wang, E.Y.; Aggarwal, P.; Zhang, B.; Conant, G.; Ronaldson-Bouchard, K.; et al. A Platform for Generation of Chamber-Specific Cardiac Tissues and Disease Modeling. Cell 2019, 176, 913–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drakhlis, L.; Biswanath, S.; Farr, C.-M.; Lupanow, V.; Teske, J.; Ritzenhoff, K.; Franke, A.; Manstein, F.; Bolesani, E.; Kempf, H.; et al. Human heart-forming organoids recapitulate early heart and foregut development. Nat. Biotechnol. 2021, 1–10. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
My, I.; Di Pasquale, E. Genetic Cardiomyopathies: The Lesson Learned from hiPSCs. J. Clin. Med. 2021, 10, 1149. https://doi.org/10.3390/jcm10051149
My I, Di Pasquale E. Genetic Cardiomyopathies: The Lesson Learned from hiPSCs. Journal of Clinical Medicine. 2021; 10(5):1149. https://doi.org/10.3390/jcm10051149
Chicago/Turabian StyleMy, Ilaria, and Elisa Di Pasquale. 2021. "Genetic Cardiomyopathies: The Lesson Learned from hiPSCs" Journal of Clinical Medicine 10, no. 5: 1149. https://doi.org/10.3390/jcm10051149