
An Update on the Role of Ubiquitination in Melanoma Development and Therapies

 , ,
, ,
Abstract
:1. Introduction
2. Brief Overview of Melanoma and Its Treatments
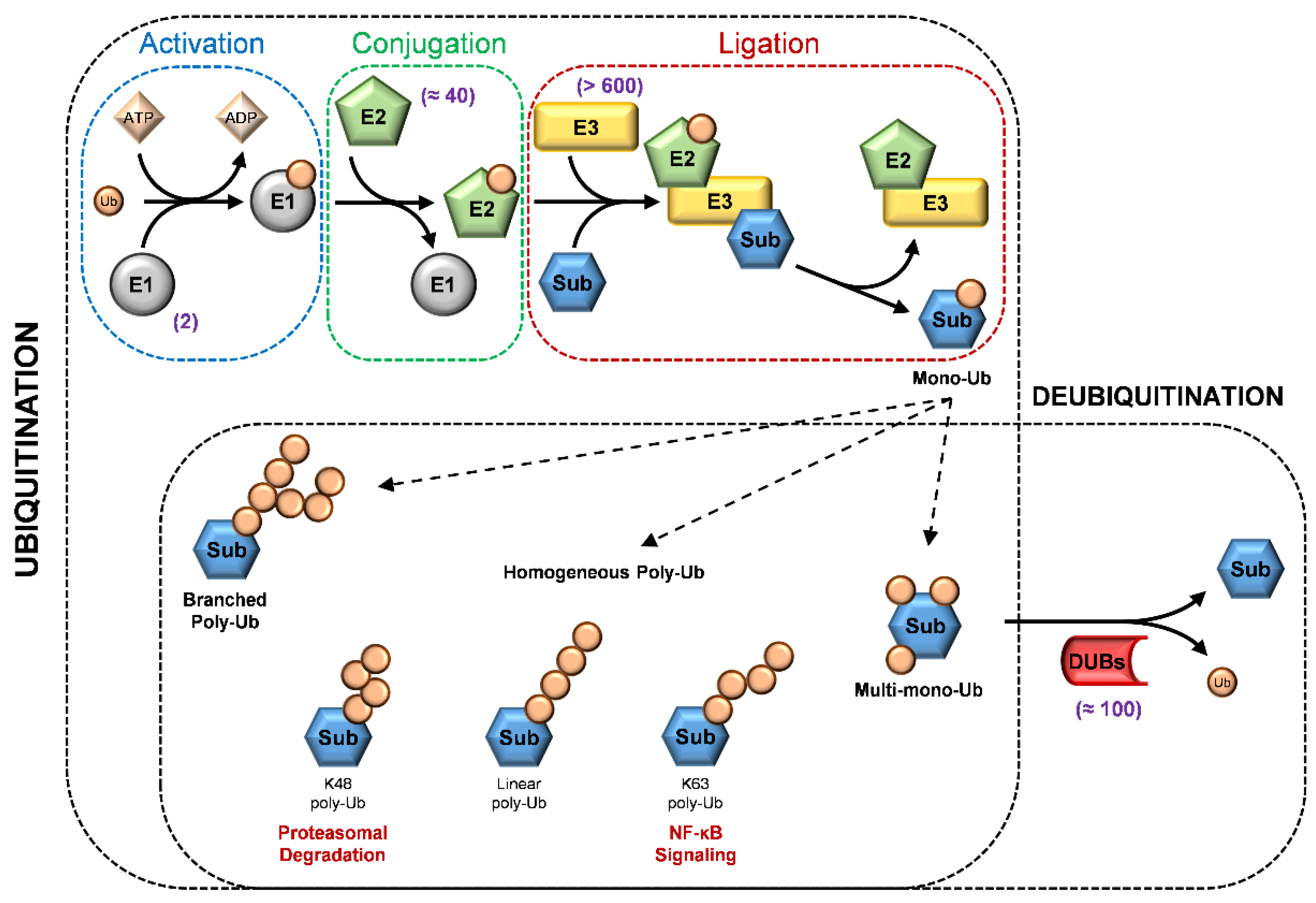
3. A Glimpse of Ubiquitination Processes
4. E2 Enzyme Involvement in Melanoma Progression

{kind=link}
{kind=link}
| E2 Class | E2s | Roles in Melanoma | Refs | |
|---|---|---|---|---|
| I |  | UBE2N UBE2I UBE2B | Overexpressed, proliferation and malignancy Proliferation, apoptosis evasion, sumoylation of MITF Overexpressed, progression and pathogenesis | [39] [44,45,46,47] [50,51] |
| II |  | UBE2C | Overexpressed, associated with poor prognosis | [40,41] |
| III |  | UBE2T UBE2S | Overexpressed, biological role to be determined Proliferation, cell survival, tumor growth, EMT | [35] [49] |
| IV |  |
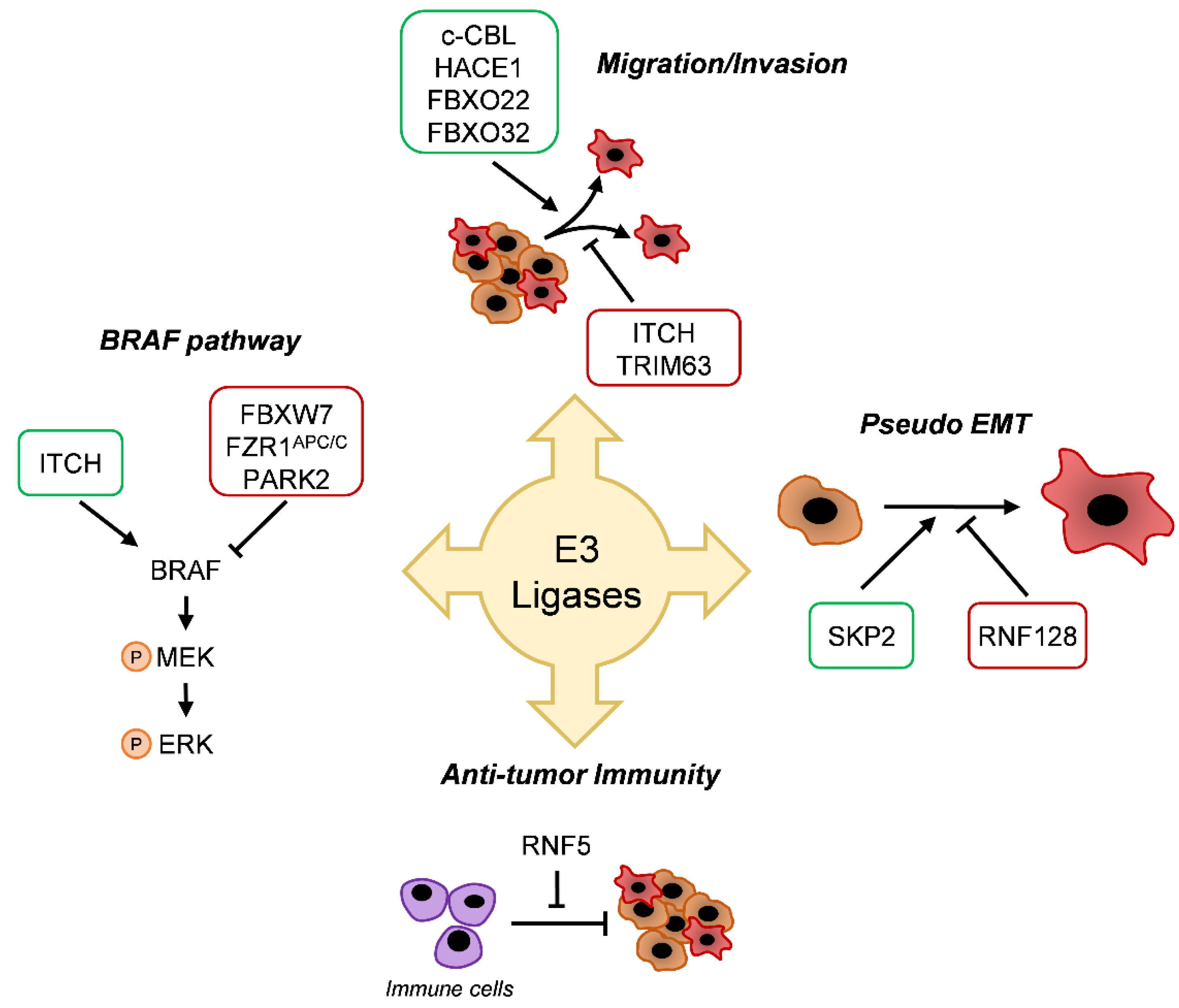
5. E3 Enzyme Involvement in Melanoma
5.1. BRAF Pathway
5.2. Migration/Invasion
5.3. Differentiation
5.4. Antitumor Immunity
6. Deubiquitination and Melanoma
6.1. Tumor Suppressors
6.2. Tumor Promoters
7. Ubiquitination and Resistance
7.1. Drugs Resistance
7.2. Resistance to ICTs
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Senturk, E.; Manfredi, J.J. Mdm2 and tumorigenesis: Evolving theories and unsolved mysteries. Genes Cancer 2012, 3, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Guo, W.; Li, C. Ubiquitination in melanoma pathogenesis and treatment. Cancer Med. 2017, 6, 1362–1377. [Google Scholar] [CrossRef]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- LoRusso, P.M.; Schalper, K.; Sosman, J. Targeted therapy and immunotherapy: Emerging biomarkers in metastatic melanoma. Pigment Cell Melanoma Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet. Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Satija, Y.K.; Bhardwaj, A.; Das, S. A portrayal of E3 ubiquitin ligases and deubiquitylases in cancer. Int. J. cancer 2013, 133, 2759–2768. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Chau, V.; Tobias, J.W.; Bachmair, A.; Marriott, D.; Ecker, D.J.; Gonda, D.K.; Varshavsky, A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 1989, 243, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Fushman, D. Polyubiquitin chains: Polymeric protein signals. Curr. Opin. Chem. Biol. 2004, 8, 610–616. [Google Scholar] [CrossRef]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef]
- Margottin-Goguet, F.; Hsu, J.Y.; Loktev, A.; Hsieh, H.M.; Reimann, J.D.R.; Jackson, P.K. Prophase destruction of Emi1 by the SCF(betaTrCP/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev. Cell 2003, 4, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Walsh, M.C.; Lee, J.; Choi, Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol. Rev. 2015, 266, 72–92. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front. Pharmacol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, K.D. DUBs at a glance. J. Cell Sci. 2009, 122, 2325–2329. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, S.Y.; Spiegelman, V.S.; Kumar, K.G.S. The many faces of beta-TrCP E3 ubiquitin ligases: Reflections in the magic mirror of cancer. Oncogene 2004, 23, 2028–2036. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Suresh Kumar, K.G.; Yu, D.; Molton, S.A.; McMahon, M.; Herlyn, M.; Thomas-Tikhonenko, A.; Fuchs, S.Y. Oncogenic BRAF regulates beta-Trcp expression and NF-kappaB activity in human melanoma cells. Oncogene 2007, 26, 1954–1958. [Google Scholar] [CrossRef] [Green Version]
- Santra, M.K.; Wajapeyee, N.; Green, M.R. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 2009, 459, 722–725. [Google Scholar] [CrossRef]
- Lee, E.K.; Lian, Z.; D’Andrea, K.; Letrero, R.; Sheng, W.; Liu, S.; Diehl, J.N.; Pytel, D.; Barbash, O.; Schuchter, L.; et al. The FBXO4 tumor suppressor functions as a barrier to BRAFV600E-dependent metastatic melanoma. Mol. Cell. Biol. 2013, 33, 4422–4433. [Google Scholar] [CrossRef] [Green Version]
- Valimberti, I.; Tiberti, M.; Lambrughi, M.; Sarcevic, B.; Papaleo, E. E2 superfamily of ubiquitin-conjugating enzymes: Constitutively active or activated through phosphorylation in the catalytic cleft. Sci. Rep. 2015, 5, 14849. [Google Scholar] [CrossRef]
- Hormaechea-Agulla, D.; Kim, Y.; Song, M.S.; Song, S.J. New Insights into the Role of E2s in the Pathogenesis of Diseases: Lessons Learned from UBE2O. Mol. Cells 2018, 41, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.M.; Okoye, I.; Chaleshtari, M.G.; Hazhirkarzar, B.; Mohamadnejad, J.; Azizi, G.; Hojjat-Farsangi, M.; Mohammadi, H.; Shotorbani, S.S.; Jadidi-Niaragh, F. E2 ubiquitin-conjugating enzymes in cancer: Implications for immunotherapeutic interventions. Clin. Chim. Acta. 2019, 498, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Gorlov, I.; Orlow, I.; Ringelberg, C.; Hernando, E.; Ernstoff, M.S.; Cheng, C.; Her, S.; Parker, J.S.; Thompson, C.L.; Gerstenblith, M.R.; et al. Identification of gene expression levels in primary melanoma associated with clinically meaningful characteristics. Melanoma Res. 2018, 28, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Park, J.-H.; Nishidate, T.; Kijima, K.; Hirata, K.; Nakamura, Y.; Katagiri, T. Ubiquitination and downregulation of BRCA1 by ubiquitin-conjugating enzyme E2T overexpression in human breast cancer cells. Cancer Res. 2009, 69, 8752–8760. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Xiao, L.; Cao, C.; Hua, S.; Wu, D. UBE2T promotes nasopharyngeal carcinoma cell proliferation, invasion, and metastasis by activating the AKT/GSK3β/β-catenin pathway. Oncotarget 2016, 7, 15161–15172. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, Y.; Yang, Z.; Liu, X.; Yang, P.; Wang, J.; Hu, K.; He, X.; Zhang, X.; Jing, H. High expression of UBE2T predicts poor prognosis and survival in multiple myeloma. Cancer Gene Ther. 2019, 26, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Dikshit, A.; Jin, Y.J.; Degan, S.; Hwang, J.; Foster, M.W.; Li, C.-Y.; Zhang, J.Y. UBE2N Promotes Melanoma Growth via MEK/FRA1/SOX10 Signaling. Cancer Res. 2018, 78, 6462–6472. [Google Scholar] [CrossRef] [Green Version]
- Kraft, S.; Moore, J.B.; Muzikansky, A.; Scott, K.L.; Duncan, L.M. Differential UBE2C and HOXA1 expression in melanocytic nevi and melanoma. J. Cutan. Pathol. 2017, 44, 843–850. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, J.; Pan, B.; Ma, G.; Liu, L. UBE2C overexpression in melanoma and its essential role in G2/M transition. J. Cancer 2019, 10, 2176–2184. [Google Scholar] [CrossRef] [Green Version]
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef]
- Mo, Y.Y.; Moschos, S.J. Targeting Ubc9 for cancer therapy. Expert Opin. Ther. Targets 2005, 9, 1203–1216. [Google Scholar] [CrossRef]
- Moschos, S.J.; Mo, Y.-Y. Role of SUMO/Ubc9 in DNA damage repair and tumorigenesis. J. Mol. Histol. 2006, 37, 309–319. [Google Scholar] [CrossRef]
- Moschos, S.J.; Smith, A.P.; Mandic, M.; Athanassiou, C.; Watson-Hurst, K.; Jukic, D.M.; Edington, H.D.; Kirkwood, J.M.; Becker, D. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: Identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene 2007, 26, 4216–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Gong, L.; Haddad, M.M.; Bischof, O.; Campisi, J.; Yeh, E.T.H.; Medrano, E.E. Regulation of microphthalmia-associated transcription factor MITF protein levels by association with the ubiquitin-conjugating enzyme hUBC9. Exp. Cell Res. 2000, 255, 135–143. [Google Scholar] [CrossRef]
- Cheli, Y.; Giuliano, S.; Guiliano, S.; Botton, T.; Rocchi, S.; Hofman, V.; Hofman, P.; Bahadoran, P.; Bertolotto, C.; Ballotti, R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene 2011, 30, 2307–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; De Lichy, M.; Bille, K.; Dessen, P.; D’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, Y.; Ma, Y.; Zhang, X.; Li, Z.; Yu, W.; Zhu, M.; Wang, J.; Xu, Y.; Xu, A. Comprehensive Investigation into the Role of Ubiquitin-Conjugating Enzyme E2S in Melanoma Development. J. Invest. Dermatol. 2021, 141, 374–384. [Google Scholar] [CrossRef]
- Gajan, A.; Martin, C.E.; Kim, S.; Joshi, M.; Michelhaugh, S.K.; Sloma, I.; Mittal, S.; Firestine, S.; Shekhar, M.P.V. Alternative Splicing of RAD6B and Not RAD6A is Selectively Increased in Melanoma: Identification and Functional Characterization. Cells 2019, 8, 1375. [Google Scholar] [CrossRef] [Green Version]
- Sarma, A.; Gajan, A.; Kim, S.; Gurdziel, K.; Mao, G.; Nangia-Makker, P.; Shekhar, M.P.V. RAD6B loss disrupts expression of melanoma phenotype in part by inhibiting WNT/beta-catenin signaling. Am. J. Pathol. 2020, 112490. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Ji, A.; Qiu, G.; Feng, H.; Li, J.; Li, S.; Zou, Y.; Cui, Y.; Song, C.; He, H.; et al. FBW7 is associated with prognosis, inhibits malignancies and enhances temozolomide sensitivity in glioblastoma cells. Cancer Sci. 2018, 109, 1001–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, X.; Ye, M.; Jing, P.; Xiong, J.; Han, Z.; Kong, J.; Li, M.; Lai, X.; Chang, N.; et al. FBW7 loss promotes epithelial-to-mesenchymal transition in non-small cell lung cancer through the stabilization of Snail protein. Cancer Lett. 2018, 419, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Nihira, N.T.; Inuzuka, H.; Wei, W. Physiological functions of FBW7 in cancer and metabolism. Cell. Signal. 2018, 46, 15–22. [Google Scholar] [CrossRef]
- Aydin, I.T.; Melamed, R.D.; Adams, S.J.; Castillo-Martin, M.; Demir, A.; Bryk, D.; Brunner, G.; Cordon-Cardo, C.; Osman, I.; Rabadan, R.; et al. FBXW7 mutations in melanoma and a new therapeutic paradigm. J. Natl. Cancer Inst. 2014, 106, dju107. [Google Scholar] [CrossRef] [Green Version]
- Saei, A.; Palafox, M.; Benoukraf, T.; Kumari, N.; Jaynes, P.W.; Iyengar, P.V.; Muñoz-Couselo, E.; Nuciforo, P.; Cortés, J.; Nötzel, C.; et al. Loss of USP28-mediated BRAF degradation drives resistance to RAF cancer therapies. J. Exp. Med. 2018, 215, 1913–1928. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Chen, M.; Cao, J.; Dai, X.; Yin, Q.; Zhang, J.; Song, S.-J.; Lu, Y.; Liu, J.; Inuzuka, H.; et al. The APC/C E3 Ligase Complex Activator FZR1 Restricts BRAF Oncogenic Function. Cancer Discov. 2017, 7, 424–441. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Wyatt, C.J.; Han, T.; Smalley, K.S.M.; Wan, L. ITCH as a potential therapeutic target in human cancers. Semin. Cancer Biol. 2020, 1–14. [Google Scholar] [CrossRef]
- Yin, Q.; Han, T.; Fang, B.; Zhang, G.; Zhang, C.; Roberts, E.R.; Izumi, V.; Zheng, M.; Jiang, S.; Yin, X.; et al. K27-linked ubiquitination of BRAF by ITCH engages cytokine response to maintain MEK-ERK signaling. Nat. Commun. 2019, 10, 1870. [Google Scholar] [CrossRef]
- Montagnani, V.; Maresca, L.; Apollo, A.; Pepe, S.; Carr, R.M.; Fernandez-Zapico, M.E.; Stecca, B. E3 ubiquitin ligase PARK2, an inhibitor of melanoma cell growth, is repressed by the oncogenic ERK1/2-ELK1 transcriptional axis. J. Biol. Chem. 2020, 295, 16058–16071. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, Y.; Xu, Y.; Tang, X. miR-10b promoted melanoma progression through Wnt/β-catenin pathway by repressing ITCH expression. Gene 2019, 710, 39–47. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, G.; Martinka, M.; Ho, V.; Li, G. Prognostic significance of Fbw7 in human melanoma and its role in cell migration. J. Investig. Dermatol. 2013, 133, 1794–1802. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Job, B.; Petit, V.; Gesbert, F.; Delmas, V.; Seberg, H.; Meurice, G.; Van Otterloo, E.; Dessen, P.; Robert, C.; et al. New Functional Signatures for Understanding Melanoma Biology from Tumor Cell Lineage-Specific Analysis. Cell Rep. 2015, 13, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Nihal, M.; Wood, G.S. c-CBL regulates melanoma proliferation, migration, invasion and the FAK-SRC-GRB2 nexus. Oncotarget 2016, 7, 53869–53880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrino, S.; Visvikis, O.; Doye, A.; Boyer, L.; Stefani, C.; Munro, P.; Bertoglio, J.; Gacon, G.; Mettouchi, A.; Lemichez, E. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev. Cell 2011, 21, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Goka, E.T.; Lippman, M.E. Loss of the E3 ubiquitin ligase HACE1 results in enhanced Rac1 signaling contributing to breast cancer progression. Oncogene 2015, 34, 5395–5405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.F.; Wu, Y.N.; Bai, Z.T.; Zhang, L.; Zhou, Q.; Li, X. Tumor-suppressive role of HACE1 in hepatocellular carcinoma and its clinical significance. Oncol. Rep. 2016, 36, 3427–3435. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhang, H.-S.; Zhang, Z.-G.; Sun, H.-L.; Liu, H.-Y.; Gou, X.-M.; Yu, X.-Y.; Huang, Y.-H. Loss of HACE1 promotes colorectal cancer cell migration via upregulation of YAP1. J. Cell. Physiol. 2019, 234, 9663–9672. [Google Scholar] [CrossRef]
- El-Hachem, N.; Habel, N.; Naiken, T.; Bzioueche, H.; Cheli, Y.; Beranger, G.E.; Jaune, E.; Rouaud, F.; Nottet, N.; Reinier, F.; et al. Uncovering and deciphering the pro-invasive role of HACE1 in melanoma cells. Cell Death Differ. 2018, 25, 2010–2022. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Chen, H.; Zhao, Y.; Zhang, X.; Liu, J.; Pan, Y.; Bai, J.; Zhang, H. Knockdown of FBXO22 inhibits melanoma cell migration, invasion and angiogenesis via the HIF-1α/VEGF pathway. Investig. New Drugs 2020, 38, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, Z.-Y.; Cui, J.; Yang, X.-M.; Jing, L.; Zhou, Y.; Chen, Z.-N.; Jiang, J.-L. F-Box Protein FBXO22 Mediates Polyubiquitination and Degradation of CD147 to Reverse Cisplatin Resistance of Tumor Cells. Int. J. Mol. Sci. 2017, 18, 212. [Google Scholar] [CrossRef] [Green Version]
- Habel, N.; El-Hachem, N.; Soysouvanh, F.; Hadhiri-Bzioueche, H.; Giuliano, S.; Nguyen, S.; Horák, P.; Gay, A.-S.; Debayle, D.; Nottet, N.; et al. FBXO32 links ubiquitination to epigenetic reprograming of melanoma cells. Cell Death Differ. 2021. [Google Scholar] [CrossRef]
- Li, F.Z.; Dhillon, A.S.; Anderson, R.L.; McArthur, G.; Ferrao, P.T. Phenotype Switching in Melanoma: Implications for Progression and Therapy. Front. Oncol. 2015, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Lin, M.; Liu, Y.; Wang, Z.W.; Zhu, X. Emerging role of F-box proteins in the regulation of epithelial-mesenchymal transition and stem cells in human cancers. Stem Cell Res. Ther. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Dai, J.; Xu, Z.; He, W.; Wang, X.; Zhu, Y.; Wang, H. Fbxw7 regulates renal cell carcinoma migration and invasion via suppression of the epithelial-mesenchymal transition. Oncol. Lett. 2018, 15, 3694–3702. [Google Scholar] [CrossRef]
- Li, Q.; Murphy, M.; Ross, J.; Sheehan, C.; Carlson, J.A. Skp2 and p27kip1 expression in melanocytic nevi and melanoma: An inverse relationship. J. Cutan. Pathol. 2004, 31, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yamauchi, H. p27-Associated G1 arrest induced by hinokitiol in human malignant melanoma cells is mediated via down-regulation of pRb, Skp2 ubiquitin ligase, and impairment of Cdk2 function. Cancer Lett. 2009, 286, 240–249. [Google Scholar] [CrossRef]
- Katagiri, Y.; Hozumi, Y.; Kondo, S. Knockdown of Skp2 by siRNA inhibits melanoma cell growth in vitro and in vivo. J. Dermatol. Sci. 2006, 42, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Pan, H.; Wang, H.; Chai, P.; Ge, S.; Jia, R.; Fan, X. SKP2 targeted inhibition suppresses human uveal melanoma progression by blocking ubiquitylation of p27. Oncol. Targets. Ther. 2019, 12, 4297–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Shen, L.; Zheng, Y.; Cui, Y.; Feng, Z.; Liu, F.; Liu, J. A signal transduction pathway from TGF-β1 to SKP2 via Akt1 and c-Myc and its correlation with progression in human melanoma. J. Investig. Dermatol. 2014, 134, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.-Y.; Zhu, M.-X.; Yang, Y.-W.; Zhang, P.-F.; Yang, X.; Peng, R.; Gao, C.; Lu, J.-C.; Wang, L.; Deng, X.-Y.; et al. Downregulation of RNF128 activates Wnt/β-catenin signaling to induce cellular EMT and stemness via CD44 and CTTN ubiquitination in melanoma. J. Hematol. Oncol. 2019, 12, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Zhang, J.; Wu, Q.; Fang, H.; Shi, C.; Li, Z.; Lin, C.; Tang, D.; Wang, D. Intestinal microbiota: A new force in cancer immunotherapy. Cell Commun. Signal. 2020, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tinoco, R.; Elmén, L.; Segota, I.; Xian, Y.; Fujita, Y.; Sahu, A.; Zarecki, R.; Marie, K.; Feng, Y.; et al. Gut microbiota dependent anti-tumor immunity restricts melanoma growth in Rnf5 −/− mice. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, B.; Leach, C.A.; Goldenberg, S.J.; Francis, D.M.; Kodrasov, M.P.; Tian, X.; Shanks, J.; Sterner, D.E.; Bernal, A.; Mattern, M.R.; et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 2008, 17, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClurg, U.L.; Robson, C.N. Deubiquitinating enzymes as oncotargets. Oncotarget 2015, 6, 9657–9668. [Google Scholar] [CrossRef] [Green Version]
- Ventii, K.H.; Devi, N.S.; Friedrich, K.L.; Chernova, T.A.; Tighiouart, M.; Van Meir, E.G.; Wilkinson, K.D. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008, 68, 6953–6962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 2011, 43, 1018–1021. [Google Scholar] [CrossRef] [Green Version]
- Scheuermann, J.C.; de Ayala Alonso, A.G.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Müller, J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hébert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Webster, J.D.; Pham, T.H.; Wu, X.; Hughes, N.W.; Li, Z.; Totpal, K.; Lee, H.-J.; Calses, P.C.; Chaurushiya, M.S.; Stawiski, E.W.; et al. The tumor suppressor BAP1 cooperates with BRAFV600E to promote tumor formation in cutaneous melanoma. Pigment Cell Melanoma Res. 2019, 32, 269–279. [Google Scholar] [CrossRef]
- He, M.; Chaurushiya, M.S.; Webster, J.D.; Kummerfeld, S.; Reja, R.; Chaudhuri, S.; Chen, Y.-J.; Modrusan, Z.; Haley, B.; Dugger, D.L.; et al. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase BAP1. Science 2019, 364, 283–285. [Google Scholar] [CrossRef]
- Lee, H.-J.; Pham, T.; Chang, M.T.; Barnes, D.; Cai, A.G.; Noubade, R.; Totpal, K.; Chen, X.; Tran, C.; Hagenbeek, T.; et al. The tumor suppressor BAP1 regulates the Hippo pathway in pancreatic ductal adenocarcinoma. Cancer Res. 2020, 80, 1656–1668. [Google Scholar] [CrossRef] [Green Version]
- Liu-Smith, F.; Lu, Y. Opposite Roles of BAP1 in Overall Survival of Uveal Melanoma and Cutaneous Melanoma. J. Clin. Med. 2020, 9, 411. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Taylor, M.; Miao, B.; Ji, Z.; Njauw, J.C.-N.; Jönsson, G.; Frederick, D.T.; Tsao, H. BAP1 has a survival role in cutaneous melanoma. J. Invest. Dermatol. 2015, 135, 1089–1097. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Goto, E.; Shibata, Y.; Kubota, Y.; Yamagata, A.; Goto-Ito, S.; Kubota, K.; Inoue, J.; Takekawa, M.; Tokunaga, F.; et al. Structures of CYLD USP with Met1- or Lys63-linked diubiquitin reveal mechanisms for dual specificity. Nat. Struct. Mol. Biol. 2015, 22, 222–229. [Google Scholar] [CrossRef]
- Massoumi, R.; Podda, M.; Fässler, R.; Paus, R. Cylindroma as tumor of hair follicle origin. J. Investig. Dermatol. 2006, 126, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Massoumi, R.; Kuphal, S.; Hellerbrand, C.; Haas, B.; Wild, P.; Spruss, T.; Pfeifer, A.; Fässler, R.; Bosserhoff, A.K. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J. Exp. Med. 2009, 206, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jel, M.M.; Schott, M.; Lamm, S.; Neuhuber, W.; Kuphal, S.; Bosserhoff, A.-K. Loss of CYLD accelerates melanoma development and progression in the Tg(Grm1) melanoma mouse model. Oncogenesis 2019, 8, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.-J.; Hsu, K.-C.; Lin, T.E.; Chang, W.-C.; Hung, J.-J. The role of ubiquitin-specific peptidases in cancer progression. J. Biomed. Sci. 2019, 26, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Ma, J.; Pei, T.; Zhao, T.; Guo, S.; Yi, X.; Liu, Y.; Wang, S.; Zhu, G.; Jian, Z.; et al. Up-regulated deubiquitinase USP4 plays an oncogenic role in melanoma. J. Cell. Mol. Med. 2018, 22, 2944–2954. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Levy-Cohen, G.; Blank, M. Molecular functions of NEDD4 E3 ubiquitin ligases in cancer. Biochim. Biophys. Acta—Rev. Cancer 2015, 1856, 91–106. [Google Scholar] [CrossRef]
- Aronchik, I.; Kundu, A.; Quirit, J.G.; Firestone, G.L. The antiproliferative response of indole-3-carbinol in human melanoma cells is triggered by an interaction with NEDD4-1 and disruption of wild-type PTEN degradation. Mol. Cancer Res. 2014, 12, 1621–1634. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Luo, M.; Zhang, K.; Zhang, J.; Gao, T.; Connell, D.O.; Yao, F.; Mu, C.; Cai, B.; Shang, Y.; et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020, 11, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Frederick, D.T.; Levesque, M.P.; Cooper, Z.A.; Feng, Y.; Krepler, C.; Brill, L.; Samuels, Y.; Hayward, N.K.; Perlina, A.; et al. Downregulation of the Ubiquitin Ligase RNF125 Underlies Resistance of Melanoma Cells to BRAF Inhibitors via JAK1 Deregulation. Cell Rep. 2015, 11, 1458–1473. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Li, C.C.; Pardo, J.; Chu, P.C.; Liao, C.X.; Huang, J.; Dong, J.G.; Zhou, X.; Huang, Q.; Huang, B.; et al. A novel E3 ubiquitin ligase TRAC-1 positively regulates T cell activation. J. Immunol. 2005, 174, 5288–5297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimoto, K.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avitan-Hersh, E.; Feng, Y.; Oknin Vaisman, A.; Abu Ahmad, Y.; Zohar, Y.; Zhang, T.; Lee, J.S.; Lazar, I.; Sheikh Khalil, S.; Feiler, Y.; et al. Regulation of eIF2α by RNF4 Promotes Melanoma Tumorigenesis and Therapy Resistance. J. Invest. Dermatol. 2020, 140, 2466–2477. [Google Scholar] [CrossRef]
- Nagler, A.; Vredevoogd, D.W.; Alon, M.; Cheng, P.F.; Trabish, S.; Kalaora, S.; Arafeh, R.; Goldin, V.; Levesque, M.P.; Peeper, D.S.; et al. A genome-wide CRISPR screen identifies FBXO42 involvement in resistance toward MEK inhibition in NRAS-mutant melanoma. Pigment Cell Melanoma Res. 2020, 33, 334–344. [Google Scholar] [CrossRef]
- Schülein-Völk, C.; Wolf, E.; Zhu, J.; Xu, W.; Taranets, L.; Hellmann, A.; Jänicke, L.A.; Diefenbacher, M.E.; Behrens, A.; Eilers, M.; et al. Dual regulation of Fbw7 function and oncogenic transformation by Usp28. Cell Rep. 2014, 9, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didier, R.; Mallavialle, A.; Ben Jouira, R.; Domdom, M.A.; Tichet, M.; Auberger, P.; Luciano, F.; Ohanna, M.; Tartare-Deckert, S.; Deckert, M. Targeting the Proteasome-Associated Deubiquitinating Enzyme USP14 Impairs Melanoma Cell Survival and Overcomes Resistance to MAPK-Targeting Therapies. Mol. Cancer Ther. 2018, 17, 1416–1429. [Google Scholar] [CrossRef] [Green Version]
- Vanneste, M.; Feddersen, C.R.; Varzavand, A.; Zhu, E.Y.; Foley, T.; Zhao, L.; Holt, K.H.; Milhem, M.; Piper, R.; Stipp, C.S.; et al. Functional Genomic Screening Independently Identifies CUL3 as a Mediator of Vemurafenib Resistance via Src-Rac1 Signaling Axis. Front. Oncol. 2020, 10, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Meng, X.; Liu, X.; Guo, X.; Jiang, S.; Chen, T.; Hu, Z.; Liu, H.; Bai, Y.; Xue, M.; Hu, R.; et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature 2018, 564, 130–135. [Google Scholar] [CrossRef]
- Saldana, M.; VanderVorst, K.; Berg, A.L.; Lee, H.; Carraway, K.L. Otubain 1: A non-canonical deubiquitinase with an emerging role in cancer. Endocr. Relat. Cancer 2019, 26, R1–R14. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yu, J.; Cheng, X.; Zhao, B.; Manyam, G.C.; Zhang, L.; Schluns, K.; Li, P.; Wang, J.; Sun, S.-C. The deubiquitinase Otub1 controls the activation of CD8+ T cells and NK cells by regulating IL-15-mediated priming. Nat. Immunol. 2019, 20, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Hockemeyer, K.; Dolgalev, I.; Poźniak, J.; Rambow, F.; Li, Y.; Feng, Y.; Tinoco, R.; Otero, D.C.; Zhang, T.; et al. Siah2 control of T-regulatory cells limits anti-tumor immunity. Nat. Commun. 2020, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sessions, E.H.; Zhang, F.; Ban, F.; Placencio-Hickok, V.; Ma, C.T.; Zeng, F.Y.; Pass, I.; Terry, D.B.; Cadwell, G.; et al. Identification and characterization of small molecule inhibitors of the ubiquitin ligases Siah1/2 in melanoma and prostate cancer cells. Cancer Lett. 2019, 449, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; De Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017, 549, 106–110. [Google Scholar] [CrossRef]
- Merin, N.; Kelly, K. Clinical Use of Proteasome Inhibitors in the Treatment of Multiple Myeloma. Pharmaceuticals 2014, 8, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.L. Carfilzomib: A second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. Ann. Pharmacother. 2013, 47, 56–62. [Google Scholar] [CrossRef]
- Richardson, P.G.; Zweegman, S.; O’Donnell, E.K.; Laubach, J.P.; Raje, N.; Voorhees, P.; Ferrari, R.H.; Skacel, T.; Kumar, S.K.; Lonial, S. Ixazomib for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2018, 19, 1949–1968. [Google Scholar] [CrossRef] [PubMed]
- Markovic, S.N.; Geyer, S.M.; Dawkins, F.; Sharfman, W.; Albertini, M.; Maples, W.; Fracasso, P.M.; Fitch, T.; Lorusso, P.; Adjei, A.A.; et al. A phase II study of bortezomib in the treatment of metastatic malignant melanoma. Cancer 2005, 103, 2584–2589. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Pavlick, A.C.; Boasberg, P.; Thompson, J.A.; Mulligan, G.; Pickard, M.D.; Faessel, H.; Dezube, B.J.; Hamid, O. A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Investig. New Drugs 2016, 34, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; Wang, X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. [Google Scholar] [CrossRef]
- Pawlak, W.Z.; Legha, S.S. Phase II study of thalidomide in patients with metastatic melanoma. Melanoma Res. 2004, 14, 57–62. [Google Scholar] [CrossRef]
- Glaspy, J.; Atkins, M.B.; Richards, J.M.; Agarwala, S.S.; O’Day, S.; Knight, R.D.; Jungnelius, J.U.; Bedikian, A.Y. Results of a multicenter, randomized, double-blind, dose-evaluating phase 2/3 study of lenalidomide in the treatment of metastatic malignant melanoma. Cancer 2009, 115, 5228–5236. [Google Scholar] [CrossRef] [Green Version]
- Altun, M.; Kramer, H.B.; Willems, L.I.; McDermott, J.L.; Leach, C.A.; Goldenberg, S.J.; Kumar, K.G.S.; Konietzny, R.; Fischer, R.; Kogan, E.; et al. Activity-Based Chemical Proteomics Accelerates Inhibitor Development for Deubiquitylating Enzymes. Chem. Biol. 2011, 18, 1401–1412. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Vogelmann, A.; Robaa, D.; Sippl, W.; Jung, M. Proteolysis targeting chimeras (PROTACs) for epigenetics research. Curr. Opin. Chem. Biol. 2020, 57, 8–16. [Google Scholar] [CrossRef] [PubMed]


| Family | Gene | Substrate | Pathway | Function in melanoma | Refs | |
|---|---|---|---|---|---|---|
| UCH | BAP1 | Histone H2A | DNA double-strand break repair | Cell differentiation | Tumor suppressor | [83,84,85,86,87,88,89] |
| CYLD | BCL-3 | N-cadherin expression | Proliferation, migration, invasion and lymph angiogenesis | Tumor suppressor | [93] | |
| - | Angiogenesis | [95] | ||||
| USP | USP4 | - | EMT | Migration and invasion | Tumor promoter | [97] |
| USP10/13 | p53 | p53 | Proliferation | Tumor promoter | [98,99] | |
| USP14 | Proteasome substrates | UPS | Proliferation | Resistance promotion | [109] | |
| USP28 | Fbw7 | MAPK | BRAF inhibitor resistance | Resistance prevention | [108] | |
| OTU | OTUB1 | UBE2N | DNA double-strand breaks repair | Initiation and progression | Tumor promoter | [113] |
| AKT | CD8 T cells and NK cells activation | Immune cell activation | [114] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soysouvanh, F.; Giuliano, S.; Habel, N.; El-Hachem, N.; Pisibon, C.; Bertolotto, C.; Ballotti, R. An Update on the Role of Ubiquitination in Melanoma Development and Therapies. J. Clin. Med. 2021, 10, 1133. https://doi.org/10.3390/jcm10051133
Soysouvanh F, Giuliano S, Habel N, El-Hachem N, Pisibon C, Bertolotto C, Ballotti R. An Update on the Role of Ubiquitination in Melanoma Development and Therapies. Journal of Clinical Medicine. 2021; 10(5):1133. https://doi.org/10.3390/jcm10051133
Chicago/Turabian StyleSoysouvanh, Frédéric, Serena Giuliano, Nadia Habel, Najla El-Hachem, Céline Pisibon, Corine Bertolotto, and Robert Ballotti. 2021. "An Update on the Role of Ubiquitination in Melanoma Development and Therapies" Journal of Clinical Medicine 10, no. 5: 1133. https://doi.org/10.3390/jcm10051133