
Secondary Hemophagocytic Lymphohistiocytosis and Autoimmune Cytopenias: Case Description and Review of the Literature

Abstract
:1. Introduction
2. Patients and Methods
3. Results
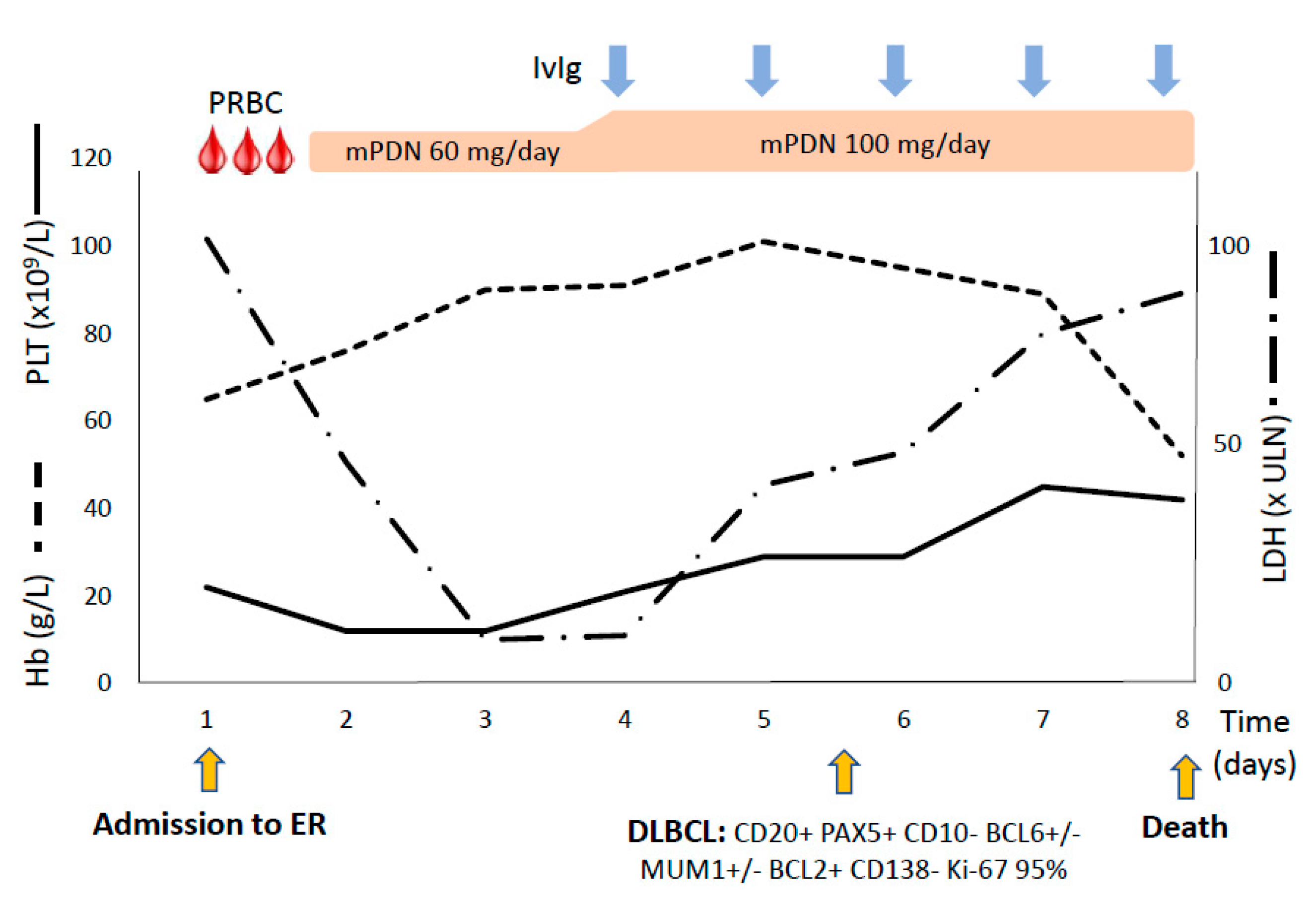
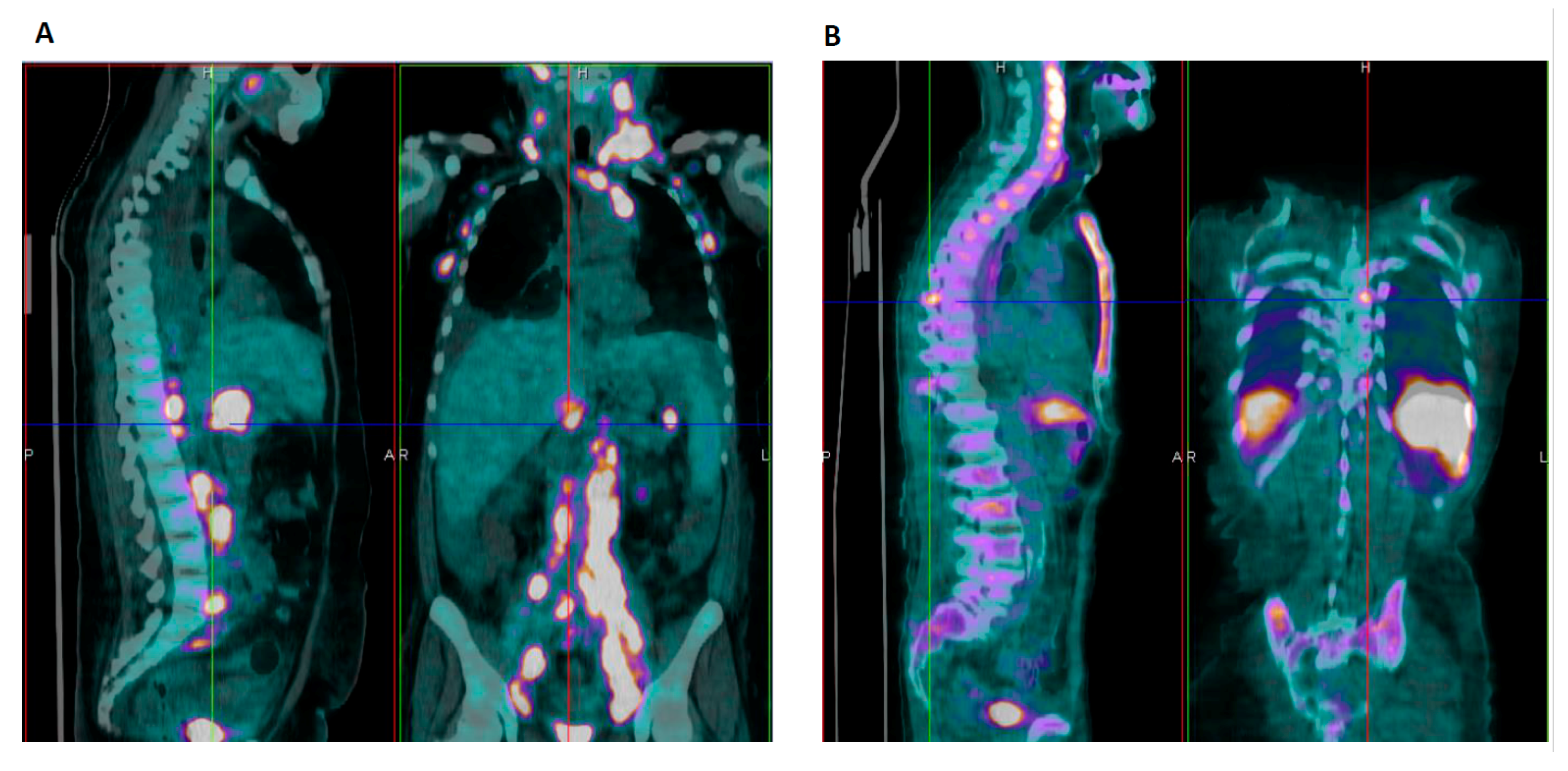
3.1. Case #1
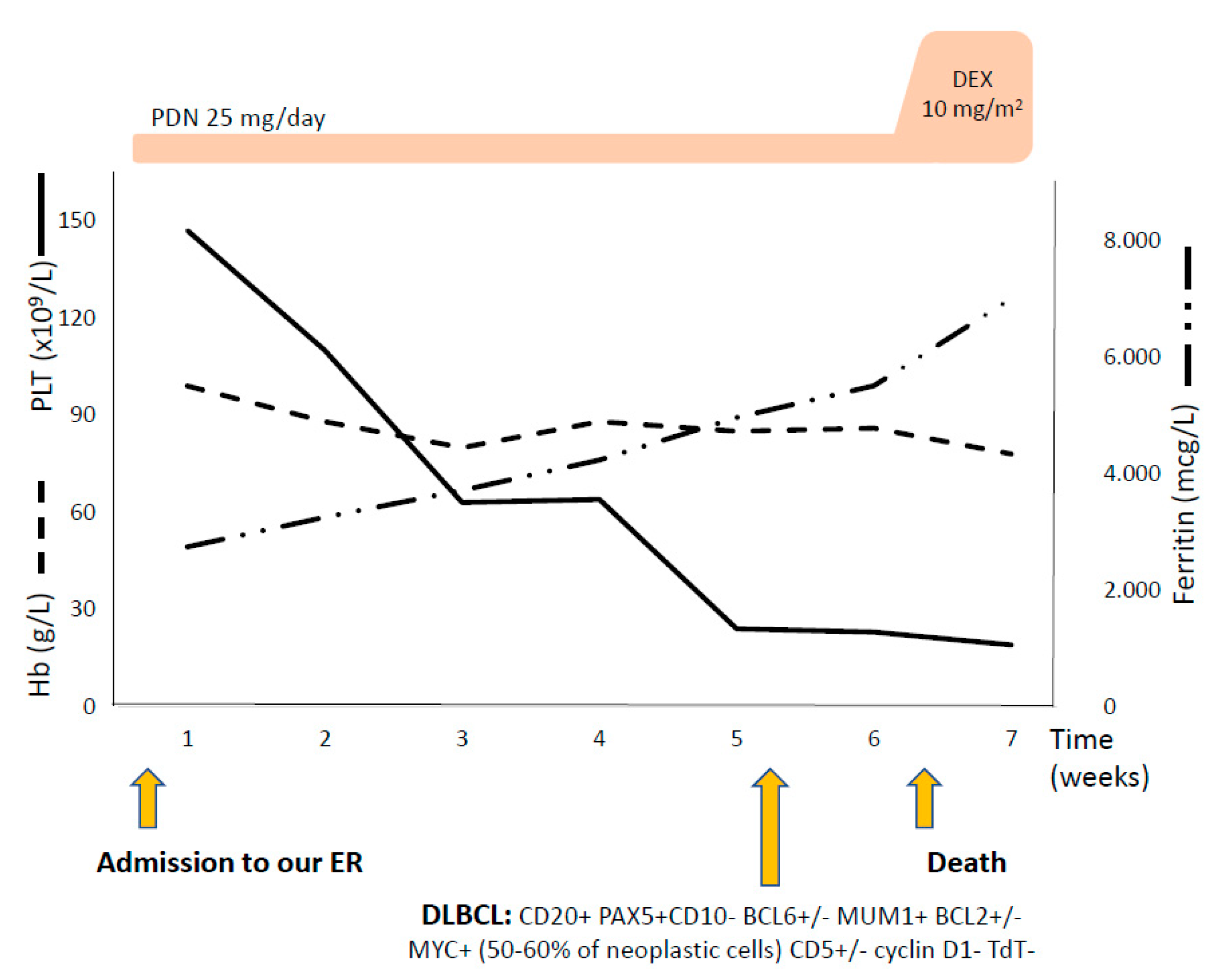
3.2. Case #2
4. Review of the Literature
4.1. Secondary HLH
4.2. Secondary AIC
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fardet, L.; Galicier, L.; Lambotte, O.; Marzac, C.; Aumont, C.; Chahwan, D.; Coppo, P.; Hejblum, G. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014, 66, 2613–2620. [Google Scholar] [CrossRef]
- Larroche, C. Hemophagocytic lymphohistiocytosis in adults: Diagnosis and treatment. Jt. Bone Spine 2012, 79, 356–361. [Google Scholar] [CrossRef]
- La Rosée, P.; Horne, A.; Hines, M.; Greenwood, T.B.; Machowicz, R.; Berliner, N.; Birndt, S.; Gil-Herrera, J.; Girschikofsky, M.; Jordan, M.B.; et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 2019, 133, 2465–2477. [Google Scholar]
- Janka, G.E.; Lehmberg, K. Hemophagocytic syndromes—An update. Blood Rev. 2014, 28, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Jäger, U.; Barcellini, W.; Broome, C.M.; Gertz, M.A.; Hill, A.; Hill, Q.A.; Jilma, B.; Kuter, D.J.; Michel, M.; Montillo, M.; et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev. 2020, 41, 100648. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W.; Fattizzo, B. The Changing Landscape of Autoimmune Hemolytic Anemia. Front. Immunol. 2020, 11, 946. [Google Scholar] [CrossRef] [PubMed]
- Fattizzo, B.; Giannotta, J.A.; Serpenti, F.; Barcellini, W. Difficult Cases of Autoimmune Hemolytic Anemia: A Challenge for the Internal Medicine Specialist. J. Clin. Med. 2020, 9, 3858. [Google Scholar] [CrossRef] [PubMed]
- Henter, J.I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Zaidi, F.; Aggarwal, A.; Gera, R. Recent advances in diagnostic and therapeutic guidelines for primary and secondary hemophagocytic lymphohistiocytosis. Blood Cells Mol. Dis. 2017, 64, 53–57. [Google Scholar] [CrossRef]
- Machowicz, R.; Janka, G.; Wiktor-Jedrzejczak, W. Similar but not the same: Differential diagnosis of HLH and sepsis. Crit. Rev. Oncol. Hematol. 2017, 114, 1–12. [Google Scholar] [CrossRef]
- Martín, A.; Marques, L.; Soler-Palacín, P.; Caragol, I.; Hernandez, M.; Figueras, C.; Español, T. Visceral leishmaniasis associated hemophagocytic syndrome in patients with chronic granulomatous disease. Pediatric Infect. Dis. J. 2009, 28, 753–754. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brito-Zerón, P.; López-Guillermo, A.; Khamashta, M.A.; Bosch, X. Adult haemophagocytic syndrome. Lancet 2014, 383, 1503–1516. [Google Scholar] [CrossRef]
- Singh, P.K.; Kodati, R.; Rohilla, M.; Sharma, P. Hemophagocytic lymphohistiocytosis: A rare association with pulmonary cryptococcosis. BMJ Case Rep. 2019, 12, e230255. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, G.; Wazir, S.; Sachdeva, A.; Kumar, S. Primary dengue infection triggered haemophagocytic lymphohistiocytosis in a neonate. BMJ Case Rep. 2020, 13, e236881. [Google Scholar] [CrossRef] [PubMed]
- Retamozo, S.; Brito-Zerón, P.; Sisó-Almirall, A.; Flores-Chávez, A.; Soto-Cárdenas, M.J.; Ramos-Casals, M. Haemophagocytic syndrome and COVID-19. Clin. Rheumatol. 2021, 3, 1–12. [Google Scholar]
- Siddiqui, R.S.; Agladze, M.; Bashir, T. Hemophagocytic Lymphohistiocytosis as the Presenting Manifestation of Relapsed Classic Hodgkin’s Lymphoma in the Presence of Concurrent Human Immunodeficiency Virus, Genital Herpes, Epstein-Barr Virus and Mycobacterium Avium Complex Infection. Cureus 2020, 12, e11563. [Google Scholar] [CrossRef] [PubMed]
- Valentine, G.; Thomas, T.A.; Nguyen, T.; Lai, Y.C. Chronic granulomatous disease presenting as hemophagocytic lymphohistiocytosis: A case report. Pediatrics 2014, 134, e1727–e1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivatsav, S.; Mahalingam, S.; Ramineni, P.; Manya, S. Dengue and Plasmodium falciparum Coinfection with Secondary Hemophagocytic Lymphohistiocytosis in a 3-Year-Old Boy: A Clinical Conundrum. J. Pediatric Hematol. Oncol. 2020. [Google Scholar] [CrossRef]
- Posas-Mendoza, T.F.; McLeod, C.; Davis, W.; Zakem, J.; Quinet, R. Etiologies and management of haemophagocytic lymphohistiocytosis: Is it time for an updated protocol and targeted treatments? Rheumatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Kwak, A.; Jung, N.; Shim, Y.J.; Kim, H.S.; Lim, H.J.; Lee, J.M.; Heo, M.H.; Do, Y.R. A retrospective analysis of etiology and outcomes of hemophagocytic lymphohistiocytosis in children and adults. Yeungnam Univ. J. Med. 2020. [Google Scholar] [CrossRef]
- Sağlam, B.; Albayrak, M.; Acar, A.; Yıldız, A.; Maral, S.; Tığlıoğlu, M.; Battal, İ.; Şahin, E.N.; Kuş, A. Q fever as a rare cause of hemophagocytic lymphohistiocytosis: Case report. Transfus. Apher. Sci. 2020, 59, 102747. [Google Scholar] [CrossRef]
- Von Bahr Greenwood, T.; Holzgraefe, B.; Chiang, S.C.C.; Wang, Y.; Tesi, B.; Bryceson, Y.T.; Henter, J.I. Clinical and laboratory signs of haemophagocytic lymphohistiocytosis associated with pandemic influenza A (H1N1) infection in patients needing extracorporeal membrane oxygenation: A retrospective observational study. Eur. J. Anaesthesiol. 2020. [Google Scholar] [CrossRef]
- Soy, M.; Atagündüz, P.; Atagündüz, I.; Sucak, G.T. Hemophagocytic lymphohistiocytosis: A review inspired by the COVID-19 pandemic. Rheumatol. Int. 2021, 41, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.A.; Nery, J.; Trennepohl, J.; Pianovski, M.A. Hemophagocytic Lymphohistiocytosis after Initiation of Chemotherapy for Bilateral Adrenal Neuroblastoma. J. Pediatric Hematol. Oncol. 2016, 38, e13–e15. [Google Scholar] [CrossRef]
- Kuruvilla, N.; Rajendran, R.; Thomas, S.S.; Ali Km, I.; Kurian, S. An Unusual Presentation of Adult-Onset Still’s Disease as Hemophagocytic Lymphohistiocytosis in a Male Patient. Cureus 2020, 12, e11139. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Sato, M.; Uesugi, Y.; Nakata, A.; Sasaki, Y.; Shimada, S.; Watanuki, M.; Fujiwara, S.; Kawaguchi, Y.; Arai, N.; et al. Characteristics and predictors of post-transplant-associated hemophagocytic lymphohistiocytosis in adults. Int. J. Hematol. 2021. [Google Scholar] [CrossRef]
- Faitelson, Y.; Grunebaum, E. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders. Clin. Immunol. 2014, 155, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Bode, S.F.; Ammann, S.; Al-Herz, W.; Bataneant, M.; Dvorak, C.C.; Gehring, S.; Gennery, A.; Gilmour, K.C.; Gonzalez-Granado, L.I.; Groß-Wieltsch, U.; et al. Inborn Errors Working Party of the EBMT. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: Implications for differential diagnosis and pathogenesis. Haematologica 2015, 100, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Squire, J.D.; Vazquez, S.N.; Chan, A.; Smith, M.E.; Chellapandian, D.; Vose, L.; Teppa, B.; Hanson, I.C.; Chinn, I.K.; Forbes-Satter, L.; et al. Case Report: Secondary Hemophagocytic Lymphohistiocytosis with Disseminated Infection in Chronic Granulomatous Disease-A Serious Cause of Mortality. Front. Immunol. 2020, 11, 581475. [Google Scholar] [CrossRef] [PubMed]
- Vick, E.J.; Patel, K.; Prouet, P.; Martin, M.G. Proliferation through activation: Hemophagocytic lymphohistiocytosis in hematologic malignancy. Blood Adv. 2017, 1, 779–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudda, M.; Mann, C.; Heinz, J.; Schmidgen, I.; Weid, F.; Kühn, M.; Saloga, J.; Grabbe, S.; Loquai, C. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: A case report and review to the literature. Melanoma Res. 2021, 31, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, T.; Takata, H.; Sasaki, H.; Miyazaki, Y.; Ohtsuka, E.; Saburi, Y.; Ogata, M.; Shirao, K. Hemophagocytic lymphohistiocytosis following influenza vaccination in a patient with aplastic anemia undergoing allogeneic bone marrow stem cell transplantation. Int. J. Hematol. 2017, 105, 389–391. [Google Scholar] [CrossRef] [PubMed]
- González, M.J.; Franco, A.G.; Alvaro, C.G. Hemophagocytic lymphohistiocytosis secondary to Calmette-Guèrin bacilli infection. Eur. J. Intern. Med. 2008, 19, 150. [Google Scholar] [CrossRef] [PubMed]
- Hill, Q.A.; Stamps, R.; Massey, E.; Grainger, J.D.; Provan, D.; Hill, A.; British Society for Haematology Guidelines. Guidelines on the management of drug-induced immune and secondary autoimmune, hemolytic anaemia. Br. J. Haematol. 2017, 177, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannotta, J.A.; Fattizzo, B.; Cavallaro, F.; Barcellini, W. Infectious Complications in Autoimmune Hemolytic Anemia. J. Clin. Med. 2021, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W.; Giannotta, J.; Fattizzo, B. Autoimmune hemolytic anemia in adults: Primary risk factors and diagnostic procedures. Expert Rev. Hematol. 2020, 13, 585–597. [Google Scholar] [CrossRef]
- El Khoury, C.; Farhat, H. Severe acute anemia attributable to concomitant occurrence of AIHA with PRCA induced by parvovirus B19 infection. Blood 2018, 131, 1388. [Google Scholar] [CrossRef] [Green Version]
- Chiao, E.Y.; Engels, E.A.; Kramer, J.R.; Pietz, K.; Henderson, L.; Giordano, T.P.; Landgren, O. Risk of immune thrombocytopenic purpura and autoimmune hemolytic anemia among 120 908 US veterans with hepatitis C virus infection. Arch. Intern. Med. 2009, 169, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Visco, C.; Barcellini, W.; Maura, F.; Neri, A.; Cortelezzi, A.; Rodeghiero, F. Autoimmune cytopenias in chronic lymphocytic leukemia. Am. J. Hematol. 2014, 89, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Fattizzo, B.; Barcellini, W. Autoimmune Cytopenias in Chronic Lymphocytic Leukemia: Focus on Molecular Aspects. Front. Oncol. 2020, 9, 1435. [Google Scholar] [CrossRef]
- Gormezano, N.W.; Kern, D.; Pereira, O.L.; Esteves, G.C.; Sallum, A.M.; Aikawa, N.E.; Pereira, R.M.; Silva, C.A.; Bonfá, E. Autoimmune hemolytic anemia in systemic lupus erythematosus at diagnosis: Differences between pediatric and adult patients. Lupus 2017, 26, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, K. A case of systemic sclerosis complicated by autoimmune hemolytic anemia. Mod. Rheumatol. 2006, 16, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Yun, M.J.; Lee, S.T.; Lee, S.J.; Oh, S.Y.; Sohn, I. Evans syndrome following long-standing Hashimoto′s thyroiditis and successful treatment with rituximab. Korean J. Hematol. 2011, 46, 279–282. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.A.; Gliga, L.; Nagalla, S. Thyroid storm and warm autoimmune hemolytic anemia. Transfus. Apher. Sci. 2017, 56, 606–608. [Google Scholar] [CrossRef] [PubMed]
- Hegazi, M.O.; Ahmed, S. Atypical clinical manifestations of graves′ disease: An analysis in depth. J. Thyroid Res. 2012, 2012, 768019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efe, C.; Wahlin, S.; Ozaslan, E.; Berlot, A.H.; Purnak, T.; Muratori, L.; Quarneti, C.; Yüksel, O.; Thiéfin, G.; Muratori, P. Autoimmune hepatitis/primary biliary cirrhosis overlap syndrome and associated extrahepatic autoimmune diseases. Eur. J. Gastroenterol. Hepatol. 2012, 24, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.B. The expanding spectrum of the autoimmune lymphoproliferative syndromes. Curr. Opin. Pediatrics 2013, 25, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Feuille, E.J.; Anooshiravani, N.; Sullivan, K.E.; Fuleihan, R.L.; Cunningham-Rundles, C. Autoimmune Cytopenias and Associated Conditions in CVID: A Report from the USIDNET Registry. J. Clin. Immunol. 2018, 38, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Goldfinger, D.; Yuan, S. Autoimmune hemolytic anemia in pediatric liver or combined liver and small bowel transplant patients: A case series and review of the literature. Transfusion 2012, 52, 48–54. [Google Scholar] [CrossRef]
- González-Vicent, M.; Sanz, J.; Fuster, J.L.; Cid, J.; de Heredia, C.D.; Morillo, D.; Fernández, J.M.; Pascual, A.; Badell, I.; Serrano, D.; et al. Autoimmune hemolytic anemia (AIHA) following allogeneic hematopoietic stem cell transplantation (HSCT): A retrospective analysis and a proposal of treatment on behalf of the Grupo Español De Trasplante de Medula Osea en Niños (GETMON) and the Grupo Español de Trasplante Hematopoyetico (GETH). Transfus. Med. Rev. 2018, 3, S0887–S7963. [Google Scholar]
- Kruizinga, M.D.; van Tol, M.J.D.; Bekker, V.; Netelenbos, T.; Smiers, F.J.; Bresters, D.; Jansen-Hoogendijk, A.M.; van Ostaijen-Ten Dam, M.M.; Kollen, W.J.W.; Zwaginga, J.J.; et al. Risk Factors, Treatment, and Immune Dysregulation in Autoimmune Cytopenia after Allogeneic Hematopoietic Stem Cell Transplantation in Pediatric Patients. Biol. Blood Marrow Transplant. 2018, 24, 772–778. [Google Scholar] [CrossRef] [Green Version]
- Skeate, R.; Singh, C.; Cooley, S.; Geller, M.; Northouse, J.; Welbig, J.; Slungaard, A.; Miller, J.; McKenna, D. Hemolytic anemia due to passenger lymphocyte syndrome in solid malignancy patients treated with allogeneic natural killer cell products. Transfusion 2013, 53, 419–423. [Google Scholar] [CrossRef]
- Barcellini, W.; Fattizzo, B.; Zaninoni, A. Management of refractory autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: Current perspectives. J. Blood Med. 2019, 10, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Klein, N.P.; Ray, P.; Carpenter, D.; Hansen, J.; Lewis, E.; Fireman, B.; Black, S.; Galindo, C.; Schmidt, J.; Baxter, R. Rates of autoimmune diseases in Kaiser Permanente for use in vaccine adverse event safety studies. Vaccine 2010, 28, 1062–1068. [Google Scholar] [CrossRef]
- Barcellini, W.; Fattizzo, B. Clinical Applications of Hemolytic Markers in the Differential Diagnosis and Management of Hemolytic Anemia. Dis. Markers 2015, 2015, 635670. [Google Scholar] [CrossRef] [Green Version]
- Rivière, S.; Galicier, L.; Coppo, P.; Marzac, C.; Aumont, C.; Lambotte, O.; Fardet, L. Reactive hemophagocytic syndrome in adults: A retrospective analysis of 162 patients. Am. J. Med. 2014, 127, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Comstock, P.; Campo, M.; Gorovets, D.; Mullally, A.; Bodio, K.; Arnason, J.; Berliner, N. Haemophagocytic lymphohistiocytosis in adults: A multicentre case series over 7 years. Br. J. Haematol. 2016, 172, 412–419. [Google Scholar] [CrossRef]
- Arca, M.; Fardet, L.; Galicier, L.; Rivière, S.; Marzac, C.; Aumont, C.; Lambotte, O.; Coppo, P. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: Impact of triggering disease and early treatment with etoposide. Br. J. Haematol. 2015, 168, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Hayden, A.; Lin, M.; Park, S.; Pudek, M.; Schneider, M.; Jordan, M.B.; Mattman, A.; Chen, L.Y.C. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017, 1, 2529–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaak, C.; Nyvlt, P.; Schuster, F.S.; Spies, C.; Heeren, P.; Schenk, T.; Balzer, F.; La Rosée, P.; Janka, G.; Brunkhorst, F.M.; et al. Hemophagocytic lymphohistiocytosis in critically ill patients: Diagnostic reliability of HLH-2004 criteria and HScore. Crit. Care 2020, 24, 244. [Google Scholar] [CrossRef]
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 Receptor Blockade Is Associated With Reduced Mortality in Sepsis Patients with Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial. Crit Care Med. 2016, 44, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Feature | Cut Off |
|---|---|
| Fever | ≥38.5 °C |
| Splenomegaly | |
| Cytopenias | ≥2 cell lines |
| —Hemoglobin * | <9 g/dL |
| —Platelets * | <100 × 109/L |
| —Neutrophils * | <1 × 109/L |
| Hyperferritinemia | >500 mcg/L |
| Hypofibrinogenemia or hypertriglyceridemia | <150 mg/dL >265 mg/dL |
| Elevated soluble CD25 | >2400 U/mL |
| Hemophagocytosis | Bone marrow, other tissues |
| Reduced or absent NK cytotoxicity | |
| Other features | |
| Elevated transaminase and bilirubin | |
| Elevated LDH | |
| Elevated d-dimers | |
| Cerebrospinal fluid pleocytosis and/or elevated protein |
| Laboratory Tests | Day 1 | Day 7 | Normal Ranges |
|---|---|---|---|
| Hb (g/dL) | 6.5 | 8.9 | 13.5–17.5 |
| PLT (10e9/L) | 22 | 12 | 130–400 |
| WBC (10e9/L) | 3.9 | 14.7 | 4.8–10.8 |
| Neutrophils (10e9/L) | 0.63 | 4.8 | 1.5–6.5 |
| Lymphocytes (10e9/L) | 3.3 | 9.4 | 1.2–3.4 |
| LDH (U/L) | 21759 | 17111 | 135–225 |
| AST (U/L) | 927 | 424 | 8–48 |
| ALT (U/L) | 101 | 43 | 9–59 |
| GGT (U/L) | 61 | 116 | 8–61 |
| ALP (U/L) | 95 | 137 | 40–129 |
| Total bilirubin (mg/dL) | 1.99 | 0.12–1.1 | |
| Unconjugated bilirubin (mg/dL) | 1.1 | 0–0.8 | |
| Creatinine (mg/dL) | 1.6 | 1.48 | 0.72–1.18 |
| PT ratio | 2.19 | 1.3 | 0.84–1.20 |
| aPTT ratio | 1.27 | 0.78 | 0.86–1.2 |
| CRP (mg/dL) | 4.28 | 1.33 | <0.5 |
| Procalcitonin (mcg/L) | 11.2 | 2.82 | <0.05 |
| Reticulocytes (10e12/L) | 0.022 | 0.097 | 0.02–0.10 |
| Haptoglobin (mg/dL) | <10 | <10 | 30–200 |
| Beta2-microglobulin (mg/L) | 15.2 | - | <0.2 |
| Laboratory Tests | Admission | Week 3 | Week 6 | Normal Ranges |
|---|---|---|---|---|
| Hb (g/dL) | 8.8 | 8.8 | 7.8 | 13.5–17.5 |
| PLT (10e9/L) | 147 | 50 | 19 | 130–400 |
| WBC (10e9/L) | 4.7 | 4.6 | 18.8 | 4.8–10.8 |
| Neutrophils (10e9/L) | 3.0 | 2.9 | 14.8 | 1.5–6.5 |
| Lymphocytes (10e9/L) | 0.3 | 0.26 | 2.1 | 1.2–3.4 |
| Reticulocytes (10e12/L) | 0.107 | - | - | 0.02–0.1 |
| LDH (U/L) | 742 | 734 | 1390 | 135–225 |
| Total bilirubin (mg/dL) | 1.2 | 2.13 | 1.99 | 0.12–1.1 |
| Unconjugated bilirubin (mg/dL) | 1.0 | 1.64 | 1.4 | 0–0.8 |
| Haptoglobin (mg/dL) | 45 | - | 24 | 30–200 |
| Beta2-microglobulin (mg/dL) | 5.0 | - | - | <0.2 |
| Creatinine (mg/dL) | 1.14 | 1.05 | 1.34 | 0.72–1.18 |
| Ferritin (mcg/L) | 2740 | 3707 | 5507 | 30–400 |
| PT ratio | 1.23 | 1.32 | 2.19 | 0.84–1.2 |
| aPTT ratio | 0.86 | 0.88 | 2.4 | 0.86–1.2 |
| Fibrinogen (mg/dL) | 353 | 162 | 53 | 165–350 |
| Triglycerides (mg/dL) | 293 | 286 | 396 | <150 |
| CRP (mg/dL) | 13.44 | 6.76 | 5.8 | <0.5 |
| Secondary HLH | Frequency | Notes | Ref |
|---|---|---|---|
| Infections | <5% to 50% | Virus (CMV, EBV, influenza, adenovirus, dengue, SARS-CoV-2, HIV); bacteria (E. coli, mycoplasma, mycobacterium, tick-borne bacteria); fungi (histoplasma, aspergillus); parasites (malaria, Leishmania) | [11,12,13,14,15,16,17,18,19,20,21,22,23] |
| Cancers | 10 to 47% 3% | Hematologic cancers, particularly lymphoma Solid cancers, particularly thymoma, ovarian and prostate cancer | [12,16,19,20,24] |
| Autoimmune diseases | 10 to 40% | Systemic lupus erythematosus; rheumatoid arthritis; juvenile idiopathic arthritis; inflammatory bowel syndromes; Still’s disease; diabetes mellitus; sarcoidosis; psoriasis; Kawasaki and Kikuchi diseases; Steven-Johnson syndrome. | [12,19,20,23,25] |
| Transplants | Up to 17% Case reports to 6% | Stem cell transplants Solid organ transplants | [12,19,26] |
| Primary immunodeficiencies | Case series | Severe combined immunodeficiency; DiGeorge syndrome; Wiskott–Aldrich syndrome; chronic granulomatous disease | [12,19,27,28,29] |
| Drugs | Case reports | Several drugs including targeted anti-cancer immunosuppressants and anti-microbial drugs | [12,23,30,31] |
| Vaccinations | Case reports | Influenza and Bacillus Calmette Guerin vaccinations | [32,33] |
| Secondary AIHA | Frequency | Notes | Ref |
| Infections | Case reports to 20% | Parvovirus B19, HCV, HBV, HAV, HIV, Mycoplasma spp, tuberculosis, babesiosis, brucellosis, syphilis, EBV, respiratory syncytial virus | [34,35,36,37,38] |
| Cancers | <5% to 30% | Solid tumors Hematologic cancers | [34,36,39,40] |
| Autoimmune diseases | 1 to 14% | Systemic lupus erythematosus, systemic sclerosis, autoimmune thyroiditis, Sjogren syndrome, inflammatory bowel syndrome, autoimmune hepatitis/primary biliary cirrhosis | [41,42,43,44,45,46] |
| Primary immunodeficiencies | 2 to > 50% | Autoimmune lymphoproliferative syndrome, common variable immunodeficiency, IgA deficiency | [36,47,48] |
| Transplants | 2 to 15% | Stem cell transplants and solid organ transplants | [49,50,51,52,53] |
| Drugs and vaccines | Case reports | Antibiotics, anti-fungal, antipsychotics, anti-convulsive, anti-neoplastic, anti-diabetics, and novel anti-cancers (Fludarabine, Bruton tyrosine kinase inhibitor, phosphoinositide 3-kinase and checkpoint inhibitors) | [6,7,34,36,54] |
| AIHA | Membrane/ Enzyme Defects | CDA | PNH | TMA | Intravascular Devices | HLH | SIRS/Sepsis | |
|---|---|---|---|---|---|---|---|---|
| Hb | − to − − − | −/− − | − −/− − − | − −/− − − | − −/− − − | − | = to − − − | = to − |
| Reticulocytes | − to + + + | − to + + + | −/= | − to + + | + | + | − to + | − |
| Schistocytes | = | = | = | = | + + | + | = | = |
| LDH | +/+ + | + | + | + + + | + + | + + | + to + + + | = to + |
| Haptoglobin | − − − | − − − | − − | − − − | − | − − | = to + | +/+ + |
| Bilirubin | + | + + | + | + | + | + | = to+ | = |
| Ferritin | =/+ | + + | + + + | − to + | =/+ | =/+ | + + + | +/+ + |
| PLT | =/− − | =/− | = | =/− | − − | =/− | = to − − − | + |
| WBC | = | = | = | =/− | = | =/− | = to − − − | − to + + |
| Hemosiderinuria | =/+ | = | = | + to + + + | =/+ | =/+ | = | = |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fattizzo, B.; Ferraresi, M.; Giannotta, J.A.; Barcellini, W. Secondary Hemophagocytic Lymphohistiocytosis and Autoimmune Cytopenias: Case Description and Review of the Literature. J. Clin. Med. 2021, 10, 870. https://doi.org/10.3390/jcm10040870
Fattizzo B, Ferraresi M, Giannotta JA, Barcellini W. Secondary Hemophagocytic Lymphohistiocytosis and Autoimmune Cytopenias: Case Description and Review of the Literature. Journal of Clinical Medicine. 2021; 10(4):870. https://doi.org/10.3390/jcm10040870
Chicago/Turabian StyleFattizzo, Bruno, Marta Ferraresi, Juri Alessandro Giannotta, and Wilma Barcellini. 2021. "Secondary Hemophagocytic Lymphohistiocytosis and Autoimmune Cytopenias: Case Description and Review of the Literature" Journal of Clinical Medicine 10, no. 4: 870. https://doi.org/10.3390/jcm10040870