
IRF3 Knockout Results in Partial or Complete Rejection of Murine Mesothelioma

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tumor Cell Lines and Mice
2.2. Mouse Genotyping
2.3. In Vivo Tumor Growth Experiments
2.4. Local Radiotherapy (LRT)
2.5. Single Cell Preparation from Tumor Tissues
2.6. Flow Cytometry
2.7. Patients with Mesothelioma
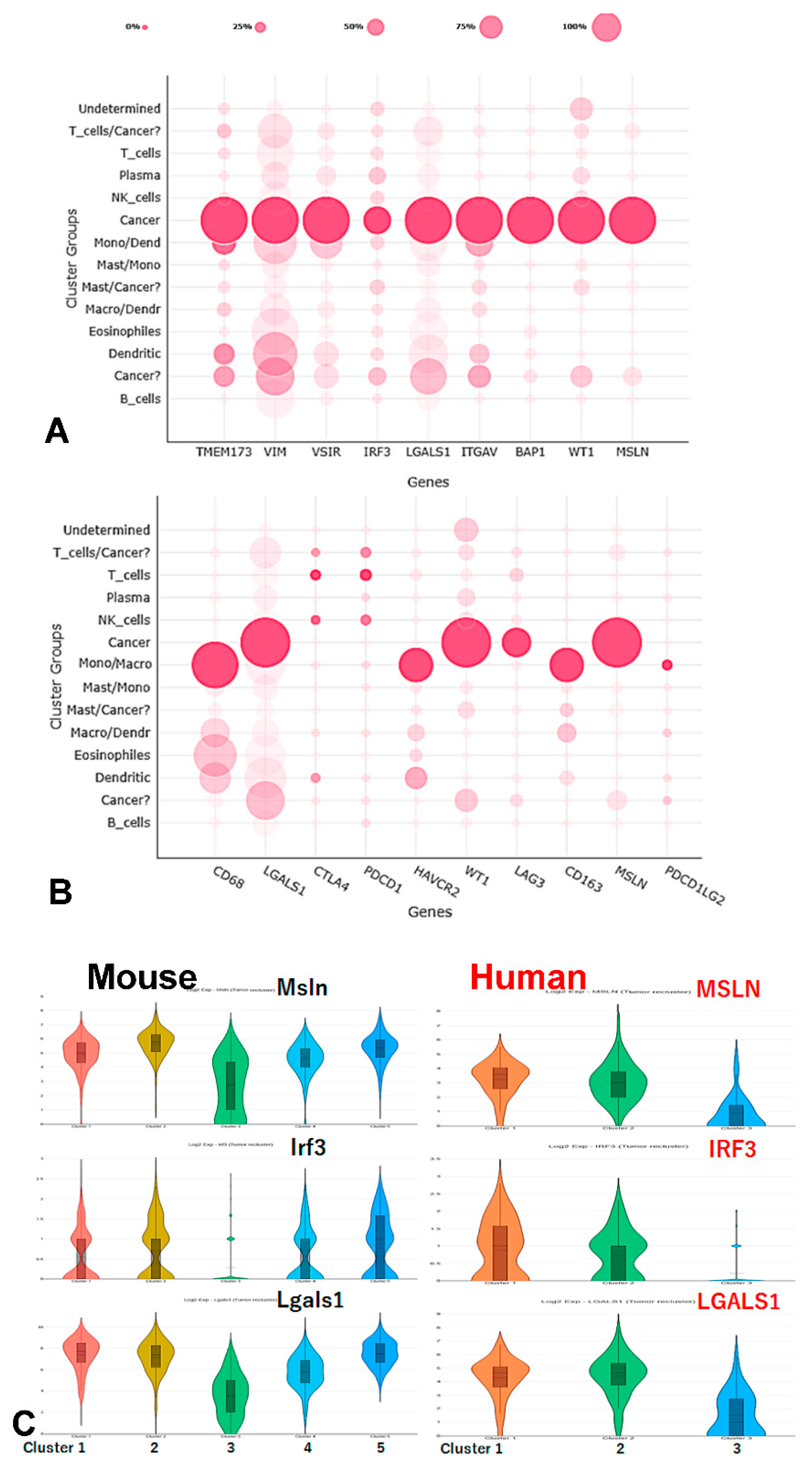
2.8. Single Cell RNA Sequencing (scRNA-Seq)
2.9. Analytical Tools
2.10. Statistical Analysis
3. Results
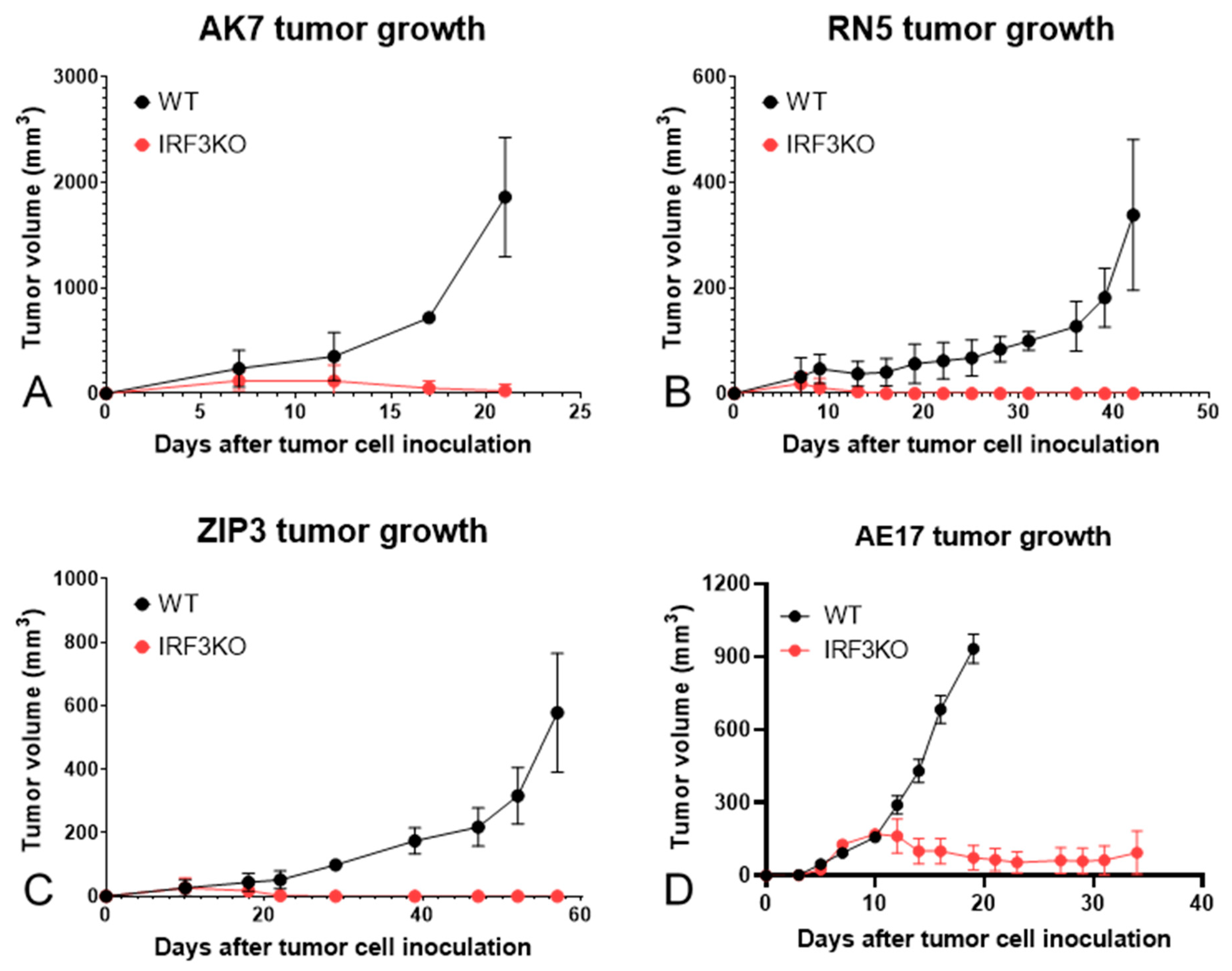
3.1. Tumor Growth Curves of Various Murine Mesothelioma Models
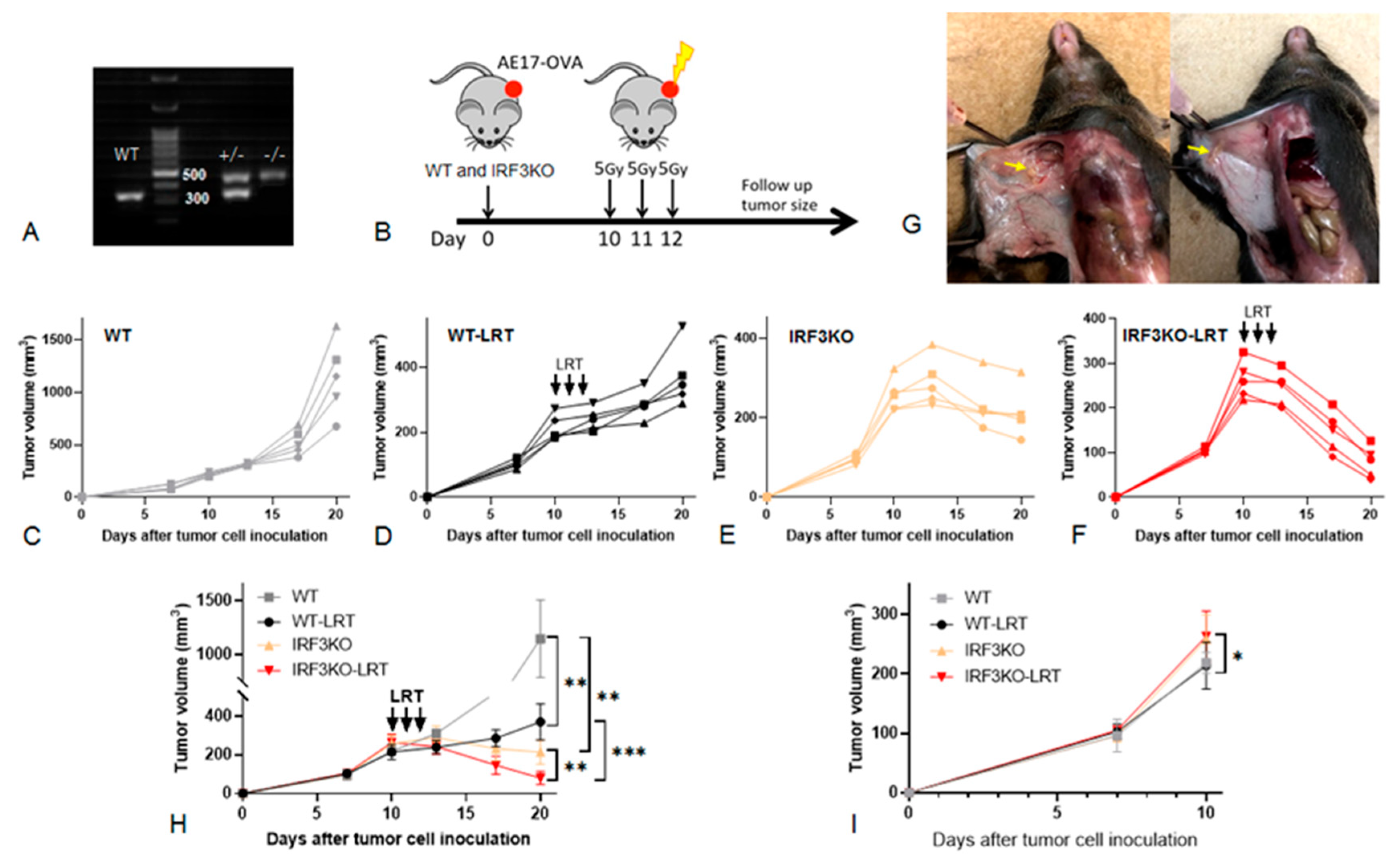
3.2. The Effect of Local Radiotherapy (LRT) on Tumor Growth in WT vs. IRF3KO Mice
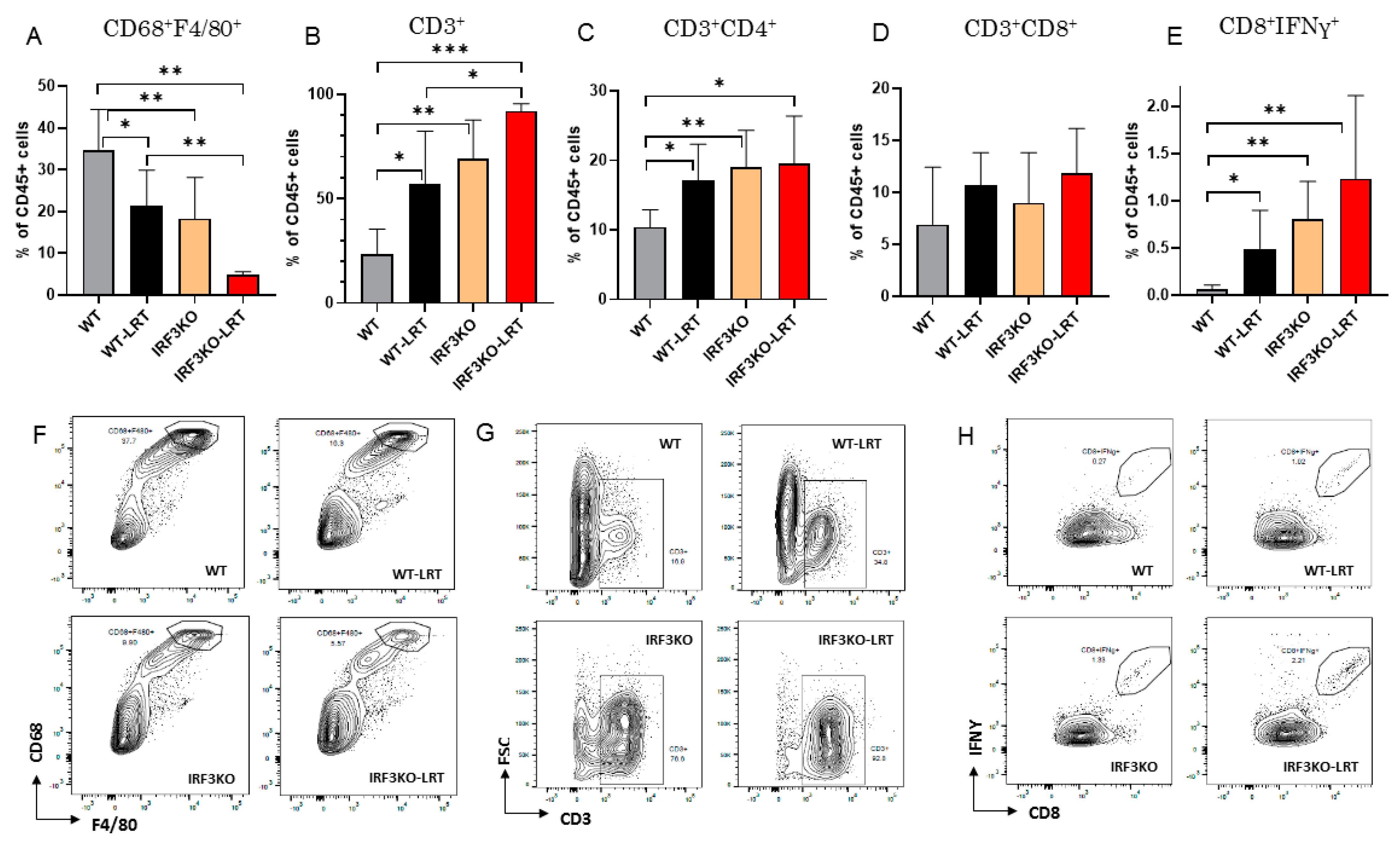
3.3. Immune Cell Infiltration in Tumor Microenvironment with LRT in WT vs. IRF3KO Mice
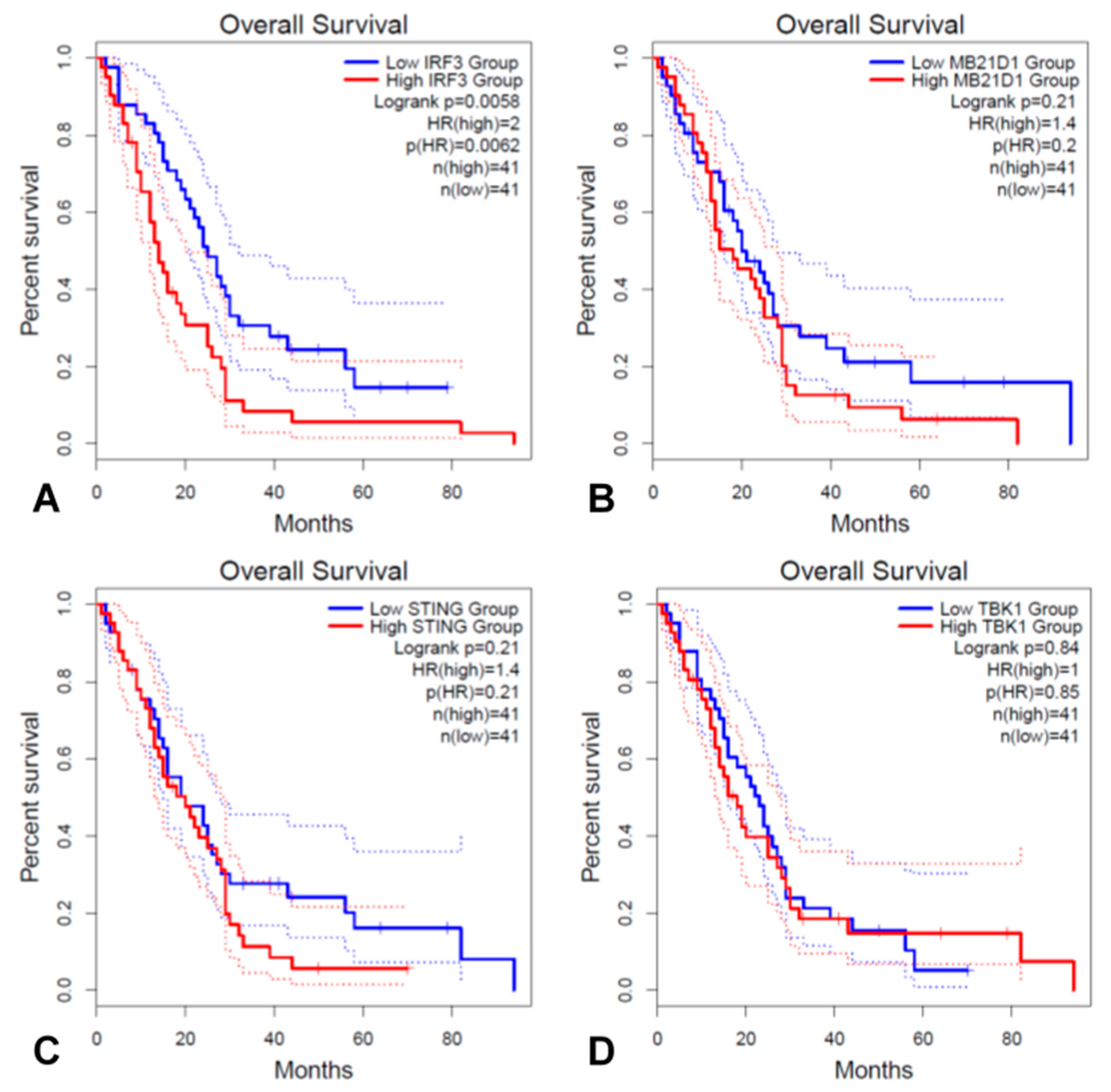
3.4. IRF3 and Key Genes in the cGAS/STING Signaling Pathway Associated with Prognosis in TCGA MESO Cohort
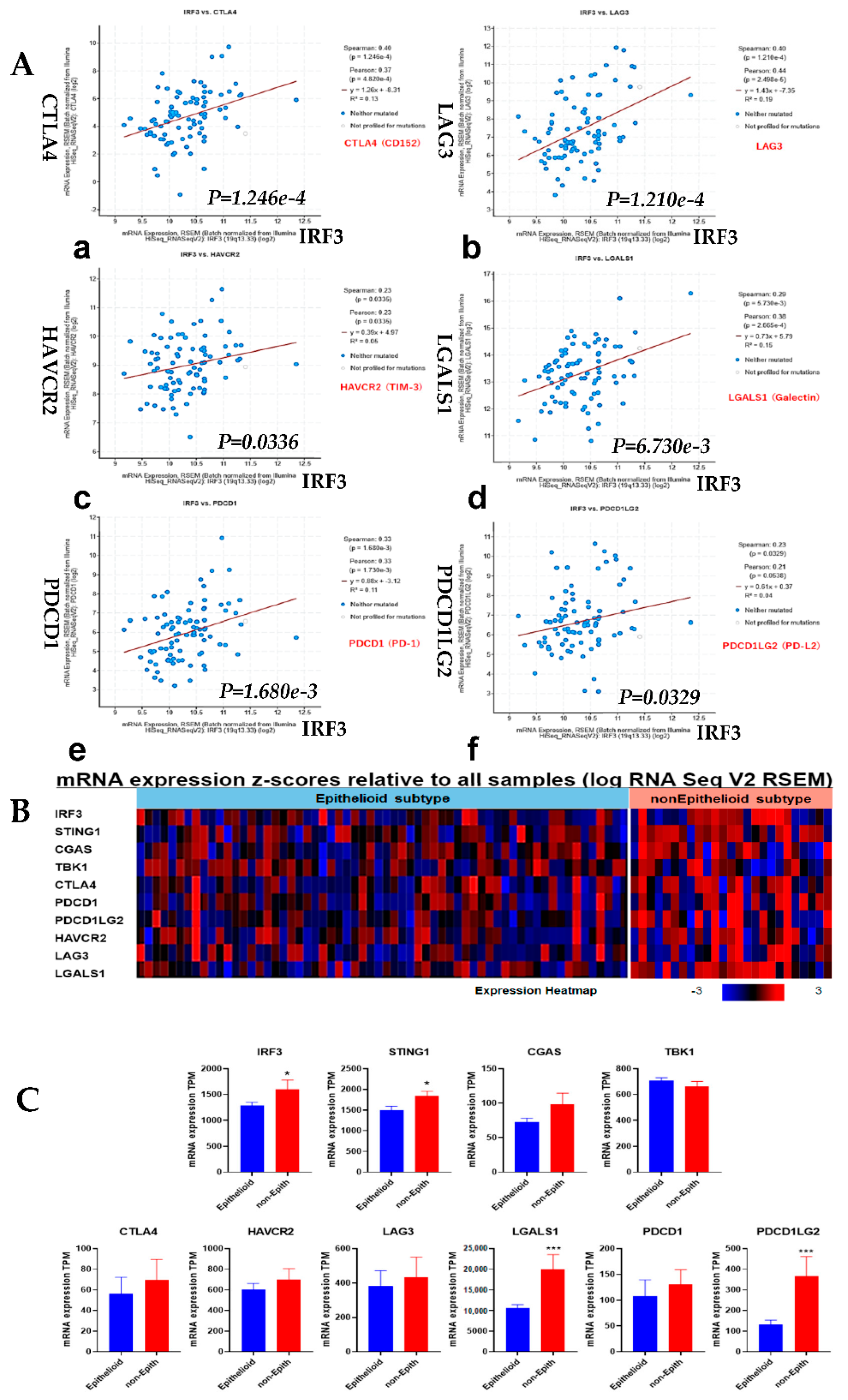
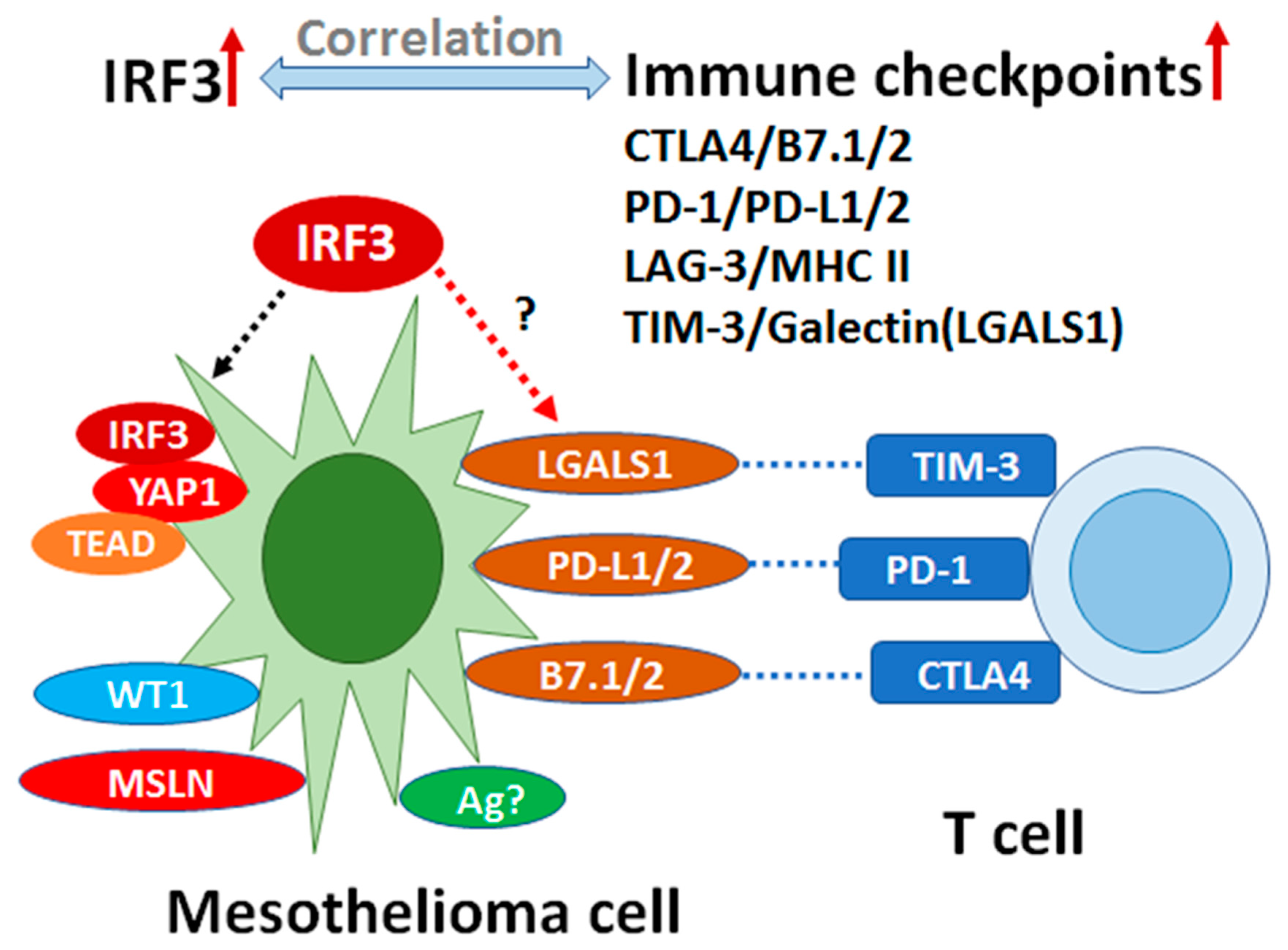
3.5. Correlation Analysis of IRF3 Expression with the Immune Checkpoints and Comparison of Epithelioid vs. Non-Epithelioid Subtype in TCGA MESO
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rusch, V.W.; Piantadosi, S.; Holmes, E.C. The role of extrapleural pneumonectomy in malignant pleural mesothelioma. A Lung Cancer Study Group trial. J. Thorac. Cardiovasc. Surg. 1991, 102, 1–9. [Google Scholar] [CrossRef]
- Pass, H.I.; Kranda, K.; Temeck, B.K.; Feuerstein, I.; Steinberg, S.M. Surgically debulked malignant pleural mesothelioma: Results and prognostic factors. Ann. Surg. Oncol. 1997, 4, 215–222. [Google Scholar] [CrossRef] [PubMed]
- de Perrot, M.; Wu, L.; Wu, M.; Cho, B.C.J. Radiotherapy for the treatment of malignant pleural mesothelioma. Lancet Oncol. 2017, 18, e532–e542. [Google Scholar] [CrossRef]
- Cho, B.J.; Feld, R.; Leighl, N.; Opitz, I.; Anraku, M.; Tsao, M.-S.; Hwang, D.M.; Hope, A.; de Perrot, M. A Feasibility Study Evaluating Surgery for Mesothelioma After Radiation Therapy: The “SMART” Approach for Resectable Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2014, 9, 397–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.C.J.; Donahoe, L.; A Bradbury, P.; Leighl, N.; Keshavjee, S.; Hope, A.; Pal, P.; Cabanero, M.; Czarnecka, K.; McRae, K.; et al. Surgery for malignant pleural mesothelioma after radiotherapy (SMART): Final results from a single-centre, phase 2 trial. Lancet Oncol. 2021, 22, 190–197. [Google Scholar] [CrossRef]
- Rusch, V.W.; Rosenzweig, K.; Venkatraman, E.; Leon, L.; Raben, A.; Harrison, L.; Bains, M.S.; Downey, R.J.; Ginsberg, R.J. A phase II trial of surgical resection and adjuvant high-dose hemithoracic radiation for malignant pleural mesothelioma. J. Thorac. Cardiovasc. Surg. 2001, 122, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weder, W.; Stahel, R.; Bernhard, J.; Bodis, S.; Vogt, P.; Ballabeni, P.; Lardinois, D.; Betticher, D.; Schmid, R.A.; Stupp, R.; et al. Multicenter trial of neo-adjuvant chemotherapy followed by extrapleural pneumonectomy in malignant pleural mesothelioma. Ann. Oncol. 2007, 18, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.M.; Pass, H.I.; Rusch, V.W.; Kindler, H.L.; Sugarbaker, D.J.; Rosenzweig, K.E.; Flores, R.; Friedberg, J.S.; Pisters, K.; Monberg, M.; et al. Multicenter phase II trial of neoadjuvant pemetrexed plus cisplatin followed by extrapleural pneumonectomy and radiation for malignant pleural mesothelioma. J. Clin. Oncol. 2009, 27, 3007–3013. [Google Scholar] [CrossRef] [Green Version]
- Van Schil, P.E.; Baas, P.; Gaafar, R.; Maat, A.P.; Van de Pol, M.; Hasan, B.; Klomp, H.M.; Abdelrahman, A.M.; Welch, J.; van Meerbeeck, J.P. European Organisation for Research and Treatment of Cancer (EORTC) Lung Cancer Group. Trimodality therapy for malignant pleural mesothelioma: Results from an EORTC phase II multicentre trial. Eur. Respir. J. 2010, 36, 1362–1369. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, S.; Okada, M.; Tanaka, F.; Yamanaka, T.; Soejima, T.; Kamikonya, N.; Tsujimura, T.; Fukuoka, K.; Yokoi, K.; Nakano, T. Trimodality strategy for treating malignant pleural mesothelioma: Results of a feasibility study of induction pemetrexed plus cisplatin followed by extrapleural pneumonectomy and postoperative hemithoracic radiation (Japan Mesothelioma Interest Group 0601 Trial). Int. J. Clin. Oncol. 2015, 21, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Quispel-Janssen, J.; van der Noort, V.; de Vries, J.F.; Zimmerman, M.; Lalezari, F.; Thunnissen, E.; Monkhorst, K.; Schouten, R.; Schunselaar, L.; Disselhorst, M.; et al. Programmed Death 1 Blockade With Nivolumab in Patients With Recurrent Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2018, 13, 1569–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherpereel, A.; Mazieres, J.; Greillier, L.; Lantuejoul, S.; Dô, P.; Bylicki, O.; Monnet, I.; Corre, R.; Audigier-Valette, C.; Locatelli-Sanchez, M.; et al. Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): A multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol. 2019, 20, 239–253. [Google Scholar] [CrossRef]
- Popat, S.; Curioni-Fontecedro, A.; Dafni, U.; Shah, R.; O’Brien, M.; Pope, A.; Fisher, P.; Spicer, J.; Roy, A.; Gilligan, D.; et al. A multicentre randomised phase III trial comparing pembrolizumab versus single-agent chemotherapy for advanced pre-treated malignant pleural mesothelioma: The European Thoracic Oncology Platform (ETOP 9-15) PROMISE-meso trial. Ann. Oncol. 2020, 31, 1734–1745. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Murakami, J.; Wu, L.; Chan, M.-L.; Yun, Z.; Cho, B.C.J.; De Perrot, M. Foxp3+ Regulatory T Cell Depletion after Nonablative Oligofractionated Irradiation Boosts the Abscopal Effects in Murine Malignant Mesothelioma. J. Immunol. 2020, 205, 2519–2531. [Google Scholar] [CrossRef]
- Murakami, J.; Wu, L.; Kohno, M.; Chan, M.-L.; Zhao, Y.; Yun, Z.; Cho, B.C.J.; de Perrot, M. Triple-modality therapy maximizes antitumor immune responses in a mouse model of mesothelioma. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Menis, J.; Pasello, G.; Remon, J. Immunotherapy in malignant pleural mesothelioma: A review of literature data. Transl. Lung Cancer Res. 2021, 10, 2988–3000. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2012, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, Y.; Shu, H.-B.; Wang, Y.-Y. MITA/STING: A central and multifaceted mediator in innate immune response. Cytokine Growth Factor Rev. 2014, 25, 631–639. [Google Scholar] [CrossRef]
- An, X.; Zhu, Y.; Zheng, T.; Wang, G.; Zhang, M.; Li, J.; Ji, H.; Li, S.; Yang, S.; Xu, D.; et al. An Analysis of the Expression and Association with Immune Cell Infiltration of the cGAS/STING Pathway in Pan-Cancer. Mol. Ther. Nucleic Acids 2018, 14, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Leng, X.; Pan, Z.; Xu, L.; Zhang, H. Overexpression of IRF3 Predicts Poor Prognosis in Clear Cell Renal Cell Carcinoma. Int. J. Gen. Med. 2021, 14, 5675–5692. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Li, J.; Yao, M.; Fang, C. Potential for treatment benefit of STING agonists plus immune checkpoint inhibitors in oral squamous cell carcinoma. BMC Oral Heal. 2021, 21, 1–8. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 1–22. [Google Scholar] [CrossRef]
- Le Naour, J.; Zitvogel, L.; Galluzzi, L.; Vacchelli, E.; Kroemer, G. Trial watch: STING agonists in cancer therapy. OncoImmunology 2020, 9, 1777624. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Pecze, L.; Felley-Bosco, E.; Worthmüller-Rodriguez, J.; Wu, L.; Vrugt, B.; De Perrot, M.; Schwaller, B. Establishment of immortalized murine mesothelial cells and a novel mesothelioma cell line. Vitr. Cell. Dev. Biol. Anim. 2015, 51, 714–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehrauer, H.; Wu, L.; Blum, W.; Pecze, L.; Henzi, T.; Serre-Beinier, V.; Aquino, C.; Vrugt, B.; de Perrot, M.; Schwaller, B.; et al. How asbestos drives the tissue towards tumors: YAP activation, macrophage and mesothelial precursor recruitment, RNA editing, and somatic mutations. Oncogene 2018, 37, 2645–2659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Maza, L.; Wu, M.; Wu, L.; Yun, H.; Zhao, Y.; Cattral, M.; McCart, A.; Cho, B.J.; de Perrot, M. In Situ Vaccination after Accelerated Hypofractionated Radiation and Surgery in a Mesothelioma Mouse Model. Clin Cancer Res. 2017, 23, 5502–5513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.Y.C.; Lheureux, S.; Karakasis, K.; Burnier, J.V.; Bruce, J.P.; Clouthier, D.L.; Danesh, A.; Quevedo, R.; Dowar, M.; Hanna, Y.; et al. Landscape of genomic alterations in high-grade serous ovarian cancer from exceptional long- and short-term survivors. Genome Med. 2018, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Cindy Yang, S.Y.; Lien, S.C.; Wang, B.X.; Clouthier, D.L.; Hanna, Y.; Cirlan, I.; Zhu, K.; Bruce, J.P.; El Ghamrasni, S.; Iafolla, M.A.; et al. Pan-cancer analysis of longitudinal metastatic tumors reveals genomic alterations and immune landscape dynamics associated with pembrolizumab sensitivity. Nat. Commun. 2021, 12, 5137. [Google Scholar] [CrossRef] [PubMed]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.-D.; Carroll, T.; Funabiki, H. The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 2019, 178, 302–315.e23. [Google Scholar] [CrossRef]
- Corrales, L.; Gajewski, T.F. Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer. Clin. Cancer Res. 2015, 21, 4774–4779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W., Jr.; Gajewski, T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duguay, D.; Mercier, F.; Stagg, J.; Martineau, D.; Bramson, J.; Servant, M.; Lin, R.; Galipeau, J.; Hiscott, J. In vivo interferon regulatory factor 3 tumor suppressor activity in B16 melanoma tumors. Cancer Res. 2002, 62, 5148–5152. [Google Scholar] [PubMed]
- Kim, T.Y.; Lee, K.-H.; Chang, S.; Chung, C.; Lee, H.-W.; Yim, J. Oncogenic Potential of a Dominant Negative Mutant of Interferon Regulatory Factor 3. J. Biol. Chem. 2003, 278, 15272–15278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, H.; Chiba, S.; Hangai, S.; Kometani, K.; Inoue, A.; Kimura, Y.; Abe, T.; Kiyonari, H.; Nishio, J.; Taguchi-Atarashi, N.; et al. Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 5253–5258. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Getahun, A.; Knowles, H.M.; Mogan, J.; Akerlund, L.J.; Packard, T.A.; Perraud, A.L.; Cambier, J.C. STING/MPYS mediates host defense against Listeria monocytogenes infection by regulating Ly6C(hi) monocyte migration. J. Immunol. 2013, 190, 2835–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Cheng, W.-L.; Jiang, X.; Wang, P.-X.; Fang, C.; Zhu, X.-Y.; Huang, Z.; She, Z.-G.; Li, H. Ablation of Interferon Regulatory Factor 3 Protects Against Atherosclerosis in Apolipoprotein E–Deficient Mice. Hypertension 2017, 69, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, L.; Gong, C.-Y.; Shi, H.-S.; Zeng, Y.-H.; Wang, X.-Z.; Zhao, Y.-W.; Wei, Y.-Q. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [Green Version]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.; Gallagher, W.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Cao, Y.; Markelc, B.; Kaeppler, J.; Vermeer, J.A.; Muschel, R.J. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Investig. 2019, 129, 4224–4238. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wu, M.O.; De La Maza, L.; Yun, Z.; Yu, J.; Zhao, Y.; Cho, J.; De Perrot, M. Targeting the inhibitory receptor CTLA-4 on T cells increased abscopal effects in murine mesothelioma model. Oncotarget 2015, 6, 12468–12480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daguenet, E.; Louati, S.; Wozny, A.-S.; Vial, N.; Gras, M.; Guy, J.-B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magné, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Heipertz, E.L.; Harper, J.; Goswami, D.G.; Lopez, C.A.; Nellikappallil, J.; Zamora, R.; Vodovotz, Y.; Walker, W.E. IRF3 Signaling within the Mouse Stroma Influences Sepsis Pathogenesis. J. Immunol. 2020, 206, 398–409. [Google Scholar] [CrossRef]
- Oweida, A.; Hararah, M.; Phan, A.V.; Binder, D.C.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef] [Green Version]
- Dixon, K.O.; Tabaka, M.; Schramm, M.A.; Xiao, S.; Tang, R.; Dionne, D.; Anderson, A.C.; Rozenblatt-Rosen, O.; Regev, A.; Kuchroo, V.K. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nat. Cell Biol. 2021, 595, 101–106. [Google Scholar]
- Rodríguez, E.; Schetters, S.T.T.; Van Kooyk, Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef]
- Mereiter, S.; Balmaña, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef]
- Jiao, S.; Guan, J.; Chen, M.; Wang, W.; Li, C.; Wang, Y.; Cheng, Y.; Zhou, Z. Targeting IRF3 as a YAP agonist therapy against gastric cancer. J. Exp. Med. 2018, 215, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Orozco, C.A.; Bosch, N.M.; Guerrero, P.E.; Vinaixa, J.; Dalotto-Moreno, T.; Iglesias, M.; Moreno, M.; Djurec, M.; Poirier, F.; Gabius, H.-J.; et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor–stroma crosstalk. Proc. Natl. Acad. Sci. 2018, 115, E3769–E3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagnoni, A.J.; Giribaldi, M.L.; Blidner, A.G.; Cutine, A.M.; Gatto, S.G.; Morales, R.M.; Salatino, M.; Abba, M.C.; Croci, D.O.; Mariño, K.V.; et al. Galectin-1 fosters an immunosuppressive microenvironment in colorectal cancer by reprogramming CD8+ regulatory T cells. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, D.K.; Aguilera, T.; Cao, H.; Kwok, S.; Kong, C.; Bloomstein, J.; Wang, Z.; Rangan, V.S.; Jiang, D.; Von Eyben, R.; et al. Galectin-1–driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. J. Clin. Investig. 2019, 129, 5553–5567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, M.; Wang, X.; Sun, J.; Lin, W.; Chen, L.; Liu, S.; Wu, X.; Shi, L.; Xu, P.; Cai, X.; et al. IRF3 prevents colorectal tumorigenesis via inhibiting the nuclear translocation of β-catenin. Nat. Commun. 2020, 11, 5762. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aoki, M.; Wu, L.; Murakami, J.; Zhao, Y.; Yun, H.; de Perrot, M. IRF3 Knockout Results in Partial or Complete Rejection of Murine Mesothelioma. J. Clin. Med. 2021, 10, 5196. https://doi.org/10.3390/jcm10215196
Aoki M, Wu L, Murakami J, Zhao Y, Yun H, de Perrot M. IRF3 Knockout Results in Partial or Complete Rejection of Murine Mesothelioma. Journal of Clinical Medicine. 2021; 10(21):5196. https://doi.org/10.3390/jcm10215196
Chicago/Turabian StyleAoki, Masaya, Licun Wu, Junichi Murakami, Yidan Zhao, Hana Yun, and Marc de Perrot. 2021. "IRF3 Knockout Results in Partial or Complete Rejection of Murine Mesothelioma" Journal of Clinical Medicine 10, no. 21: 5196. https://doi.org/10.3390/jcm10215196