
The Pathophysiology and Management of Hemorrhagic Shock in the Polytrauma Patient


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathophysiology of Hemorrhagic Sock
Oxygen Delivery/Utilization Imbalance
3. Trauma-Induced Coagulopathy
3.1. Specific Defects in Hemostasis Induced by Trauma
3.1.1. Upregulated Protein C Expression
3.1.2. Von Willebrand Factor
3.1.3. Hypofibrinogenemia
3.1.4. Platelet Dysfunction
3.2. Dysregulation of Fibrinolysis
3.2.1. Hyperfibrinolysis
3.2.2. Fibrinolysis Shutdown
4. Management of the Polytrauma Victim
4.1. Pre-Hospital Care
4.1.1. Physician-Staffed EMS Response
4.1.2. Prehospital Transfusion
4.1.3. Empiric Administration of Tranexamic Acid (TXA)
4.2. Hospital Management of the Polytrauma Patient
4.2.1. Initial Assessment
4.2.2. Damage Control Resuscitation (DCR)
- Resuscitation with whole blood
- Fixed Component Ratio-based DCR
- Goal-directed DCR
- Resuscitation with Concentrates
4.2.3. Secondary Assessment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Demetriades, D.; Murray, J.; Martin, M.; Velmahos, G.; Salim, A.; Alo, K.; Rhee, P. Pedestrians injured by automobiles: Relationship of age to injury type and severity. J. Am. Coll. Surg. 2004, 199, 382–387. [Google Scholar] [CrossRef]
- Moore, F.A.; Nelson, T.; McKinley, B.A.; Moore, E.E.; Nathens, A.B.; Rhee, P.; Puyana, J.C.; Beilman, G.J.; Cohn, S.M.; StO2 Study Group. Is there a role for aggressive use of fresh frozen plasma in massive transfusion of civilian trauma patients? Am. J. Surg. 2008, 196, 948–958; discussion 958–960. [Google Scholar] [CrossRef]
- Cannon, J.W. Hemorrhagic Shock. N. Engl. J. Med. 2018, 378, 370–379. [Google Scholar] [CrossRef]
- Tisherman, S.A.; Schmicker, R.H.; Brasel, K.J.; Bulger, E.M.; Kerby, J.D.; Minei, J.P.; Powell, J.L.; Reiff, D.A.; Rizoli, S.B.; Schreiber, M.A. Detailed description of all deaths in both the shock and traumatic brain injury hypertonic saline trials of the Resuscitation Outcomes Consortium. Ann. Surg. 2015, 261, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Hauser, C.J.; Boffard, K.; Dutton, R.; Bernard, G.R.; Croce, M.A.; Holcomb, J.B.; Leppaniemi, A.; Parr, M.; Vincent, J.L.; Tortella, B.J.; et al. Results of the CONTROL trial: Efficacy and safety of recombinant activated Factor VII in the management of refractory traumatic hemorrhage. J. Trauma 2010, 69, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Holcomb, J.B.; del Junco, D.J.; Fox, E.E.; Wade, C.E.; Cohen, M.J.; Schreiber, M.A.; Alarcon, L.H.; Bai, Y.; Brasel, K.J.; Bulger, E.M.; et al. The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study: Comparative effectiveness of a time-varying treatment with competing risks. JAMA Surg. 2013, 148, 127–136. [Google Scholar] [CrossRef]
- Fox, E.E.; Holcomb, J.B.; Wade, C.E.; Bulger, E.M.; Tilley, B.C.; Group, P.S. Earlier Endpoints are Required for Hemorrhagic Shock Trials Among Severely Injured Patients. Shock 2017, 47, 567–573. [Google Scholar] [CrossRef]
- Mullins, R.J.; Trunkey, D.D. Samuel, D. Gross: Pioneer academic trauma surgeon of 19th century America. J. Trauma 1990, 30, 528–538. [Google Scholar] [CrossRef]
- Maegele, M.; Lefering, R.; Yucel, N.; Tjardes, T.; Rixen, D.; Paffrath, T.; Simanski, C.; Neugebauer, E.; Bouillon, B.; AG Polytrauma of the German Trauma Society (DGU). Early coagulopathy in multiple injury: An analysis from the German Trauma Registry on 8724 patients. Injury 2007, 38, 298–304. [Google Scholar] [CrossRef]
- MacLeod, J.B.; Lynn, M.; McKenney, M.G.; Cohn, S.M.; Murtha, M. Early coagulopathy predicts mortality in trauma. J. Trauma 2003, 55, 39–44. [Google Scholar] [CrossRef]
- Niles, S.E.; McLaughlin, D.F.; Perkins, J.G.; Wade, C.E.; Li, Y.; Spinella, P.C.; Holcomb, J.B. Increased mortality associated with the early coagulopathy of trauma in combat casualties. J. Trauma 2008, 64, 1459–1463; discussion 1463–1465. [Google Scholar] [CrossRef] [Green Version]
- Brohi, K.; Cohen, M.J.; Ganter, M.T.; Matthay, M.A.; Mackersie, R.C.; Pittet, J.F. Acute traumatic coagulopathy: Initiated by hypoperfusion: Modulated through the protein C pathway? Ann. Surg. 2007, 245, 812–818. [Google Scholar] [CrossRef]
- Brohi, K.; Singh, J.; Heron, M.; Coats, T. Acute traumatic coagulopathy. J. Trauma 2003, 54, 1127–1130. [Google Scholar] [CrossRef] [Green Version]
- Kutcher, M.E.; Howard, B.M.; Sperry, J.L.; Hubbard, A.E.; Decker, A.L.; Cuschieri, J.; Minei, J.P.; Moore, E.E.; Brownstein, B.H.; Maier, R.V.; et al. Evolving beyond the vicious triad: Differential mediation of traumatic coagulopathy by injury, shock, and resuscitation. J. Trauma Acute Care Surg. 2015, 78, 516–523. [Google Scholar] [CrossRef]
- Ekeloef, N.P.; Eriksen, J.; Kancir, C.B. Evaluation of two methods to calculate p50 from a single blood sample. Acta Anaesthesiol. Scand. 2001, 45, 550–552. [Google Scholar] [CrossRef]
- Srinivasan, A.J.; Morkane, C.; Martin, D.S.; Welsby, I.J. Should modulation of p50 be a therapeutic target in the critically ill? Expert Rev. Hematol. 2017, 10, 449–458. [Google Scholar] [CrossRef]
- Leach, R.M.; Treacher, D.F. The pulmonary physician in critical care * 2: Oxygen delivery and consumption in the critically ill. Thorax 2002, 57, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Abdelsalam, M.; Cheifetz, I.M. Goal-directed therapy for severely hypoxic patients with acute respiratory distress syndrome: Permissive hypoxemia. Respir. Care 2010, 55, 1483–1490. [Google Scholar]
- Mercado, P.; Maizel, J.; Beyls, C.; Titeca-Beauport, D.; Joris, M.; Kontar, L.; Riviere, A.; Bonef, O.; Soupison, T.; Tribouilloy, C.; et al. Transthoracic echocardiography: An accurate and precise method for estimating cardiac output in the critically ill patient. Crit. Care 2017, 21, 136. [Google Scholar] [CrossRef] [Green Version]
- Jozwiak, M.; Monnet, X.; Teboul, J.L. Monitoring: From cardiac output monitoring to echocardiography. Curr. Opin. Crit. Care 2015, 21, 395–401. [Google Scholar] [CrossRef]
- Cecconi, M.; De Backer, D.; Antonelli, M.; Beale, R.; Bakker, J.; Hofer, C.; Jaeschke, R.; Mebazaa, A.; Pinsky, M.R.; Teboul, J.L.; et al. Consensus on circulatory shock and hemodynamic monitoring. Task force of the European Society of Intensive Care Medicine. Intensive Care Med. 2014, 40, 1795–1815. [Google Scholar] [CrossRef]
- Antonelli, M.; Levy, M.; Andrews, P.J.; Chastre, J.; Hudson, L.D.; Manthous, C.; Meduri, G.U.; Moreno, R.P.; Putensen, C.; Stewart, T.; et al. Hemodynamic monitoring in shock and implications for management. International Consensus Conference, Paris, France, 27–28 April 2006. Intensive Care Med. 2007, 33, 575–590. [Google Scholar] [CrossRef]
- Narang, N.; Thibodeau, J.T.; Levine, B.D.; Gore, M.O.; Ayers, C.R.; Lange, R.A.; Cigarroa, J.E.; Turer, A.T.; de Lemos, J.A.; McGuire, D.K. Inaccuracy of estimated resting oxygen uptake in the clinical setting. Circulation 2014, 129, 203–210. [Google Scholar] [CrossRef]
- Lubarsky, D.A.; Smith, L.R.; Sladen, R.N.; Mault, J.R.; Reed, R.L., 2nd. Defining the relationship of oxygen delivery and consumption: Use of biologic system models. J. Surg. Res. 1995, 58, 503–508. [Google Scholar] [CrossRef]
- Kornblith, L.Z.; Moore, H.B.; Cohen, M.J. Trauma-induced coagulopathy: The past, present, and future. J. Thromb. Haemost. 2019, 17, 852–862. [Google Scholar] [CrossRef]
- Gonzalez Rodriguez, E.; Ostrowski, S.R.; Cardenas, J.C.; Baer, L.A.; Tomasek, J.S.; Henriksen, H.H.; Stensballe, J.; Cotton, B.A.; Holcomb, J.B.; Johansson, P.I.; et al. Syndecan-1: A Quantitative Marker for the Endotheliopathy of Trauma. J. Am. Coll. Surg. 2017, 225, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Di Battista, A.P.; Rizoli, S.B.; Lejnieks, B.; Min, A.; Shiu, M.Y.; Peng, H.T.; Baker, A.J.; Hutchison, M.G.; Churchill, N.; Inaba, K.; et al. Sympathoadrenal Activation is Associated with Acute Traumatic Coagulopathy and Endotheliopathy in Isolated Brain Injury. Shock 2016, 46, 96–103. [Google Scholar] [CrossRef]
- Johansson, P.; Stensballe, J.; Ostrowski, S. Shock induced endotheliopathy (SHINE) in acute critical illness—A unifying pathophysiologic mechanism. Crit. Care 2017, 21, 25. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, S.R.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E.; Johansson, P.I. Sympathoadrenal activation and endotheliopathy are drivers of hypocoagulability and hyperfibrinolysis in trauma: A prospective observational study of 404 severely injured patients. J. Trauma Acute Care Surg. 2017, 82, 293–301. [Google Scholar] [CrossRef]
- Pati, S.; Potter, D.R.; Baimukanova, G.; Farrel, D.H.; Holcomb, J.B.; Schreiber, M.A. Modulating the endotheliopathy of trauma: Factor concentrate versus fresh frozen plasma. J. Trauma Acute Care Surg. 2016, 80, 576–584; discussion 584–585. [Google Scholar] [CrossRef]
- Wu, F.; Chipman, A.; Pati, S.; Miyasawa, B.; Corash, L.; Kozar, R.A. Resuscitative Strategies to Modulate the Endotheliopathy of Trauma: From Cell to Patient. Shock 2020, 53, 575–584. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Lambris, J.D.; Ward, P.A. Innate immune responses to trauma. Nat. Immunol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Rahbar, E.; Cardenas, J.C.; Baimukanova, G.; Usadi, B.; Bruhn, R.; Pati, S.; Ostrowski, S.R.; Johansson, P.I.; Holcomb, J.B.; Wade, C.E. Endothelial glycocalyx shedding and vascular permeability in severely injured trauma patients. J. Transl. Med. 2015, 13, 117. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, S.R.; Johansson, P.I. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J. Trauma Acute Care Surg. 2012, 73, 60–66. [Google Scholar] [CrossRef]
- Schlimp, C.J.; Schochl, H. The role of fibrinogen in trauma-induced coagulopathy. Hamostaseologie 2014, 34, 29–39. [Google Scholar] [CrossRef]
- Fries, D.; Martini, W.Z. Role of fibrinogen in trauma-induced coagulopathy. Br. J. Anaesth. 2010, 105, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.I.; Sorensen, A.M.; Perner, A.; Welling, K.L.; Wanscher, M.; Larsen, C.F.; Ostrowski, S.R. Disseminated intravascular coagulation or acute coagulopathy of trauma shock early after trauma? An observational study. Crit. Care 2011, 15, R272. [Google Scholar] [CrossRef] [Green Version]
- Oshiro, A.; Yanagida, Y.; Gando, S.; Henzan, N.; Takahashi, I.; Makise, H. Hemostasis during the early stages of trauma: Comparison with disseminated intravascular coagulation. Crit. Care 2014, 18, R61. [Google Scholar] [CrossRef] [Green Version]
- Spahn, D.R.; Bouillon, B.; Cerny, V.; Duranteau, J.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Maegele, M.; Nardi, G.; Riddez, L.; et al. The European guideline on management of major bleeding and coagulopathy following trauma: Fifth edition. Crit. Care 2019, 23, 98. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Carbucicchio, A.; Benatti, M.; Cervellin, G. The mean platelet volume is decreased in patients with mild head trauma and brain injury. Blood Coagul. Fibrinolysis 2013, 24, 780–783. [Google Scholar] [CrossRef]
- Windelov, N.A.; Sorensen, A.M.; Perner, A.; Wanscher, M.; Larsen, C.F.; Ostrowski, S.R.; Johansson, P.I.; Rasmussen, L.S. Platelet aggregation following trauma: A prospective study. Blood Coagul. Fibrinolysis 2014, 25, 67–73. [Google Scholar] [CrossRef]
- Wohlauer, M.V.; Moore, E.E.; Thomas, S.; Sauaia, A.; Evans, E.; Harr, J.; Silliman, C.C.; Ploplis, V.; Castellino, F.J.; Walsh, M. Early platelet dysfunction: An unrecognized role in the acute coagulopathy of trauma. J. Am. Coll. Surg. 2012, 214, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Sillesen, M.; Johansson, P.I.; Rasmussen, L.S.; Jin, G.; Jepsen, C.H.; Imam, A.M.; Hwabejire, J.; Lu, J.; Duggan, M.; Velmahos, G.; et al. Platelet activation and dysfunction in a large-animal model of traumatic brain injury and hemorrhage. J. Trauma Acute Care Surg. 2013, 74, 1252–1259. [Google Scholar] [CrossRef]
- Solomon, C.; Traintinger, S.; Ziegler, B.; Hanke, A.; Rahe-Meyer, N.; Voelckel, W.; Schochl, H. Platelet function following trauma. A multiple electrode aggregometry study. Thromb. Haemost. 2011, 106, 322–330. [Google Scholar] [CrossRef]
- Di Scipio, R.G.; Hermodson, M.A.; Yates, S.G.; Davie, E.W. A comparison of human prothrombin, factor IX (Christmas factor), factor X (Stuart factor), and protein S. Biochemistry 1977, 16, 698–706. [Google Scholar] [CrossRef]
- Esmon, C.T.; Owen, W.G. The discovery of thrombomodulin. J. Thromb. Haemost. 2004, 2, 209–213. [Google Scholar] [CrossRef]
- Fuentes-Prior, P.; Iwanaga, Y.; Huber, R.; Pagila, R.; Rumennik, G.; Seto, M.; Morser, J.; Light, D.R.; Bode, W. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature 2000, 404, 518–525. [Google Scholar] [CrossRef]
- Sillen, M.; Declerck, P.J. Thrombin Activatable Fibrinolysis Inhibitor (TAFI): An Updated Narrative Review. Int. J. Mol. Sci. 2021, 22, 3670. [Google Scholar] [CrossRef]
- Nishimura, T.; Myles, T.; Piliponsky, A.M.; Kao, P.N.; Berry, G.J.; Leung, L.L. Thrombin-activatable procarboxypeptidase B regulates activated complement C5a in vivo. Blood 2007, 109, 1992–1997. [Google Scholar] [CrossRef] [Green Version]
- Simurda, T.; Dobrotova, M.; Skornova, I.; Sokol, J.; Kubisz, P.; Stasko, J. Successful Use of a Highly Purified Plasma von Willebrand Factor Concentrate Containing Little FVIII for the Long-Term Prophylaxis of Severe (Type 3) von Willebrand’s Disease. Semin. Thromb. Hemost. 2017, 43, 639–641. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, M.S.; Tricomi, S.M. Plasma levels of the three endothelial-specific proteins von Willebrand factor, tissue factor pathway inhibitor, and thrombomodulin do not predict the development of acute respiratory distress syndrome. Intensive Care Med. 1999, 25, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Zeineddin, A.; Dong, J.F.; Wu, F.; Terse, P.; Kozar, R.A. Role of Von Willebrand Factor after Injury: It May Do More Than We Think. Shock 2021, 55, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Kermode, J.C.; Zheng, Q.; Milner, E.P. Marked temperature dependence of the platelet calcium signal induced by human von Willebrand factor. Blood 1999, 94, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; de Moerloose, P.; Neerman-Arbez, M. Clinical Features and Management of Congenital Fibrinogen Deficiencies. Semin. Thromb. Hemost. 2016, 42, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Mengoli, C.; Franchini, M.; Marano, G.; Pupella, S.; Vaglio, S.; Marietta, M.; Liumbruno, G.M. The use of fibrinogen concentrate for the management of trauma-related bleeding: A systematic review and meta-analysis. Blood Transfus. 2017, 15, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Rourke, C.; Curry, N.; Khan, S.; Taylor, R.; Raza, I.; Davenport, R.; Stanworth, S.; Brohi, K. Fibrinogen levels during trauma hemorrhage, response to replacement therapy, and association with patient outcomes. J. Thromb. Haemost. 2012, 10, 1342–1351. [Google Scholar] [CrossRef]
- Schochl, H.; Frietsch, T.; Pavelka, M.; Jambor, C. Hyperfibrinolysis after major trauma: Differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J. Trauma 2009, 67, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Schlimp, C.J.; Voelckel, W.; Inaba, K.; Maegele, M.; Ponschab, M.; Schochl, H. Estimation of plasma fibrinogen levels based on hemoglobin, base excess and Injury Severity Score upon emergency room admission. Crit. Care 2013, 17, R137. [Google Scholar] [CrossRef] [Green Version]
- Hagemo, J.S.; Stanworth, S.; Juffermans, N.P.; Brohi, K.; Cohen, M.; Johansson, P.I.; Roislien, J.; Eken, T.; Naess, P.A.; Gaarder, C. Prevalence, predictors and outcome of hypofibrinogenaemia in trauma: A multicentre observational study. Crit. Care 2014, 18, R52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foldesi, M.; Merkei, Z.; Ferenci, T.; Nardai, G. Fibrinogen level at hospital admission after multiple injury correlates with BMI and is negatively associated with the need for transfusion and early multiple organ failure. Injury 2021, 52 (Suppl. 1), S15–S20. [Google Scholar] [CrossRef]
- Agren, A.; Wikman, A.T.; Ostlund, A.; Edgren, G. TEG(R) Functional Fibrinogen Analysis May Overestimate Fibrinogen Levels. Anesth. Analg. 2014, 118, 933–935. [Google Scholar] [CrossRef] [Green Version]
- Castellano, G.; Woltman, A.M.; Nauta, A.J.; Roos, A.; Trouw, L.A.; Seelen, M.A.; Schena, F.P.; Daha, M.R.; van Kooten, C. Maturation of dendritic cells abrogates C1q production in vivo and in vitro. Blood 2004, 103, 3813–3820. [Google Scholar] [CrossRef] [Green Version]
- Davis, P.K.; Musunuru, H.; Walsh, M.; Cassady, R.; Yount, R.; Losiniecki, A.; Moore, E.E.; Wohlauer, M.V.; Howard, J.; Ploplis, V.A.; et al. Platelet dysfunction is an early marker for traumatic brain injury-induced coagulopathy. Neurocrit. Care 2013, 18, 201–208. [Google Scholar] [CrossRef]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Moore, H.B.; Moore, E.E.; Gonzalez, E.; Chapman, M.P.; Chin, T.L.; Silliman, C.C.; Banerjee, A.; Sauaia, A. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: The spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J. Trauma Acute Care Surg. 2014, 77, 811–817; discussion 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, H.B.; Moore, E.E.; Lawson, P.J.; Gonzalez, E.; Fragoso, M.; Morton, A.P.; Gamboni, F.; Chapman, M.P.; Sauaia, A.; Banerjee, A.; et al. Fibrinolysis shutdown phenotype masks changes in rodent coagulation in tissue injury versus hemorrhagic shock. Surgery 2015, 158, 386–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liras, I.N.; Cotton, B.A.; Cardenas, J.C.; Harting, M.T. Prevalence and impact of admission hyperfibrinolysis in severely injured pediatric trauma patients. Surgery 2015, 158, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Cotton, B.A.; Harvin, J.A.; Kostousouv, V.; Minei, K.M.; Radwan, Z.A.; Schochl, H.; Wade, C.E.; Holcomb, J.B.; Matijevic, N. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J. Trauma Acute Care Surg 2012, 73, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Blackbourne, L.H.; Baer, D.G.; Cestero, R.F.; Inaba, K.; Rasmussen, T.E. Exsanguination shock: The next frontier in prevention of battlefield mortality. J. Trauma 2011, 71, S1–S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brohi, K.; Cohen, M.J.; Ganter, M.T.; Schultz, M.J.; Levi, M.; Mackersie, R.C.; Pittet, J.F. Acute coagulopathy of trauma: Hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J. Trauma 2008, 64, 1211–1217. [Google Scholar] [CrossRef] [Green Version]
- Morton, A.P.; Moore, E.E.; Wohlauer, M.V.; Lo, K.; Silliman, C.C.; Burlew, C.C.; Banerjee, A. Revisiting early postinjury mortality: Are they bleeding because they are dying or dying because they are bleeding? J. Surg. Res. 2013, 179, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Schochl, H.; Cadamuro, J.; Seidl, S.; Franz, A.; Solomon, C.; Schlimp, C.J.; Ziegler, B. Hyperfibrinolysis is common in out-of-hospital cardiac arrest: Results from a prospective observational thromboelastometry study. Resuscitation 2013, 84, 454–459. [Google Scholar] [CrossRef]
- Chapman, M.P.; Moore, E.E.; Moore, H.B.; Gonzalez, E.; Gamboni, F.; Chandler, J.G.; Mitra, S.; Ghasabyan, A.; Chin, T.L.; Sauaia, A.; et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J. Trauma Acute Care Surg. 2016, 80, 16. [Google Scholar] [CrossRef] [Green Version]
- Brohi, K.; Cohen, M.J.; Davenport, R.A. Acute coagulopathy of trauma: Mechanism, identification and effect. Curr. Opin. Crit. Care 2007, 13, 680–685. [Google Scholar] [CrossRef]
- Madurska, M.J.; Sachse, K.A.; Jansen, J.O.; Rasmussen, T.E.; Morrison, J.J. Fibrinolysis in trauma: A review. Eur. J. Trauma Emerg. Surg. 2018, 44, 35–44. [Google Scholar] [CrossRef]
- Meizoso, J.P.; Dudaryk, R.; Mulder, M.B.; Ray, J.J.; Karcutskie, C.A.; Eidelson, S.A.; Namias, N.; Schulman, C.I.; Proctor, K.G. Increased risk of fibrinolysis shutdown among severely injured trauma patients receiving tranexamic acid. J. Trauma Acute Care Surg. 2018, 84, 426–432. [Google Scholar] [CrossRef]
- Moore, H.B.; Moore, E.E.; Neal, M.D.; Sheppard, F.R.; Kornblith, L.Z.; Draxler, D.F.; Walsh, M.; Medcalf, R.L.; Cohen, M.J.; Cotton, B.A.; et al. Fibrinolysis Shutdown in Trauma: Historical Review and Clinical Implications. Anesth. Analg. 2019, 129, 762–773. [Google Scholar] [CrossRef]
- Gando, S. Microvascular thrombosis and multiple organ dysfunction syndrome. Crit. Care Med. 2010, 38, S35–S42. [Google Scholar] [CrossRef]
- Pfeiler, S.; Massberg, S.; Engelmann, B. Biological basis and pathological relevance of microvascular thrombosis. Thromb. Res. 2014, 133 (Suppl. 1), S35–S37. [Google Scholar] [CrossRef]
- Tsikouris, J.P.; Suarez, J.A.; Meyerrose, G.E. Plasminogen activator inhibitor-1: Physiologic role, regulation, and the influence of common pharmacologic agents. J. Clin. Pharmacol. 2002, 42, 1187–1199. [Google Scholar] [CrossRef]
- Carroll, S.L.; Dye, D.W.; Smedley, W.A.; Stephens, S.W.; Reiff, D.A.; Kerby, J.D.; Holcomb, J.B.; Jansen, J.O. Early and prehospital trauma deaths: Who might benefit from advanced resuscitative care? J. Trauma Acute Care Surg. 2020, 88, 776–782. [Google Scholar] [CrossRef]
- Geeraedts, L.M., Jr.; Kaasjager, H.A.; van Vugt, A.B.; Frolke, J.P. Exsanguination in trauma: A review of diagnostics and treatment options. Injury 2009, 40, 11–20. [Google Scholar] [CrossRef]
- Clarke, J.R.; Trooskin, S.Z.; Doshi, P.J.; Greenwald, L.; Mode, C.J. Time to laparotomy for intra-abdominal bleeding from trauma does affect survival for delays up to 90 minutes. J. Trauma 2002, 52, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Berkeveld, E.; Popal, Z.; Schober, P.; Zuidema, W.P.; Bloemers, F.W.; Giannakopoulos, G.F. Prehospital time and mortality in polytrauma patients: A retrospective analysis. BMC Emerg. Med. 2021, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Elkbuli, A.; Dowd, B.; Sanchez, C.; Shaikh, S.; Sutherland, M.; McKenney, M. Emergency Medical Service Transport Time and Trauma Outcomes at an Urban Level 1 Trauma Center: Evaluation of Prehospital Emergency Medical Service Response. Am. Surg. 2021, 3134820988827. [Google Scholar] [CrossRef]
- Chen, X.; Gestring, M.L.; Rosengart, M.R.; Billiar, T.R.; Peitzman, A.B.; Sperry, J.L.; Brown, J.B. Speed is not everything: Identifying patients who may benefit from helicopter transport despite faster ground transport. J. Trauma Acute Care Surg. 2018, 84, 549–557. [Google Scholar] [CrossRef]
- Choi, J.; Carlos, G.; Nassar, A.K.; Knowlton, L.M.; Spain, D.A. The impact of trauma systems on patient outcomes. Curr. Probl. Surg. 2021, 58, 100840. [Google Scholar] [CrossRef]
- Schroder, H.; Beckers, S.K.; Ogrodzki, K.; Borgs, C.; Ziemann, S.; Follmann, A.; Rossaint, R.; Felzen, M. Tele-EMS physicians improve life-threatening conditions during prehospital emergency missions. Sci. Rep. 2021, 11, 14366. [Google Scholar] [CrossRef]
- Zhu, C.S.; Cobb, D.; Jonas, R.B.; Pokorny, D.; Rani, M.; Cotner-Pouncy, T.; Oliver, J.; Cap, A.; Cestero, R.; Nicholson, S.E.; et al. Shock index and pulse pressure as triggers for massive transfusion. J. Trauma Acute Care Surg. 2019, 87, S159–S164. [Google Scholar] [CrossRef]
- Sperry, J.L.; Guyette, F.X.; Brown, J.B.; Yazer, M.H.; Triulzi, D.J.; Early-Young, B.J.; Adams, P.W.; Daley, B.J.; Miller, R.S.; Harbrecht, B.G.; et al. Prehospital Plasma during Air Medical Transport in Trauma Patients at Risk for Hemorrhagic Shock. N. Engl. J. Med. 2018, 379, 315–326. [Google Scholar] [CrossRef]
- Pusateri, A.E.; Moore, E.E.; Moore, H.B.; Le, T.D.; Guyette, F.X.; Chapman, M.P.; Sauaia, A.; Ghasabyan, A.; Chandler, J.; McVaney, K.; et al. Association of Prehospital Plasma Transfusion With Survival in Trauma Patients With Hemorrhagic Shock When Transport Times Are Longer Than 20 Minutes: A Post Hoc Analysis of the PAMPer and COMBAT Clinical Trials. JAMA Surg. 2020, 155, e195085. [Google Scholar] [CrossRef]
- Braverman, M.A.; Smith, A.; Pokorny, D.; Axtman, B.; Shahan, C.P.; Barry, L.; Corral, H.; Jonas, R.B.; Shiels, M.; Schaefer, R.; et al. Prehospital whole blood reduces early mortality in patients with hemorrhagic shock. Transfusion 2021, 61 (Suppl. 1), S15–S21. [Google Scholar] [CrossRef] [PubMed]
- Shakur, H.; Roberts, I.; Bautista, R.; Caballero, J.; Coats, T.; Dewan, Y.; El-Sayed, H.; Gogichaishvili, T.; Gupta, S.; Herrera, J.; et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): A randomised, placebo-controlled trial. Lancet 2010, 376, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.M.; Cohen, M.J.; Cotton, B.A.; Schreiber, M.A.; Moore, E.E. Tranexamic acid in trauma: How should we use it? J. Trauma Acute Care Surg. 2013, 74, 1575–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binz, S.; McCollester, J.; Thomas, S.; Miller, J.; Pohlman, T.; Waxman, D.; Shariff, F.; Tracy, R.; Walsh, M. CRASH-2 Study of Tranexamic Acid to Treat Bleeding in Trauma Patients: A Controversy Fueled by Science and Social Media. J. Blood Transfus. 2015, 2015, 874920. [Google Scholar] [CrossRef] [Green Version]
- Imach, S.; Wafaisade, A.; Lefering, R.; Bohmer, A.; Schieren, M.; Suarez, V.; Frohlich, M.; TraumaRegister, D.G.U. The impact of prehospital tranexamic acid on mortality and transfusion requirements: Match-pair analysis from the nationwide German TraumaRegister DGU(R). Crit. Care 2021, 25, 277. [Google Scholar] [CrossRef]
- Wafaisade, A.; Lefering, R.; Bouillon, B.; Bohmer, A.B.; Gassler, M.; Ruppert, M.; TraumaRegister, D.G.U. Prehospital administration of tranexamic acid in trauma patients. Crit. Care 2016, 20, 143. [Google Scholar] [CrossRef] [Green Version]
- Moore, H.B.; Moore, E.E.; Huebner, B.R.; Stettler, G.R.; Nunns, G.R.; Einersen, P.M.; Silliman, C.C.; Sauaia, A. Tranexamic acid is associated with increased mortality in patients with physiological fibrinolysis. J. Surg. Res. 2017, 220, 438–443. [Google Scholar] [CrossRef]
- Diebel, M.E.; Martin, J.V.; Liberati, D.M.; Diebel, L.N. The temporal response and mechanism of action of tranexamic acid in endothelial glycocalyx degradation. J. Trauma Acute Care Surg. 2018, 84, 75–80. [Google Scholar] [CrossRef]
- Duque, P.; Gonzalez-Zarco, L.; Martinez, R.; Gago, S.; Varela, J.A. Tranexamic acid use in severely injured patients, is it always appropriate? Rev. Esp. Anestesiol. Reanim. (Engl. Ed.) 2021, 68, 301–303. [Google Scholar] [CrossRef]
- Jenkins, D.H.; Rappold, J.F.; Badloe, J.F.; Berseus, O.; Blackbourne, L.; Brohi, K.H.; Butler, F.K.; Cap, A.P.; Cohen, M.J.; Davenport, R.; et al. Trauma hemostasis and oxygenation research position paper on remote damage control resuscitation: Definitions, current practice, and knowledge gaps. Shock 2014, 41 (Suppl. 1), 3–12. [Google Scholar] [CrossRef] [Green Version]
- Kheirbek, T.; Martin, T.J.; Cao, J.; Hall, B.M.; Lueckel, S.; Adams, C.A. Prehospital shock index outperforms hypotension alone in predicting significant injury in trauma patients. Trauma Surg. Acute Care Open 2021, 6, e000712. [Google Scholar] [CrossRef]
- Schroll, R.; Swift, D.; Tatum, D.; Couch, S.; Heaney, J.B.; Llado-Farrulla, M.; Zucker, S.; Gill, F.; Brown, G.; Buffin, N.; et al. Accuracy of shock index versus ABC score to predict need for massive transfusion in trauma patients. Injury 2018, 49, 15–19. [Google Scholar] [CrossRef]
- Liu, Y.C.; Liu, J.H.; Fang, Z.A.; Shan, G.L.; Xu, J.; Qi, Z.W.; Zhu, H.D.; Wang, Z.; Yu, X.Z. Modified shock index and mortality rate of emergency patients. World J. Emerg. Med. 2012, 3, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Campbell, R.; Ardagh, M.W.; Than, M. Validation of the pulse rate over pressure evaluation index as a detector of early occult hemorrhage: A prospective observational study. J. Trauma Acute Care Surg. 2012, 73, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A.; Sumislawski, J.J.; Zarzaur, B.L.; Dutton, W.P.; Croce, M.A.; Fabian, T.C. The new metric to define large-volume hemorrhage: Results of a prospective study of the critical administration threshold. J. Trauma Acute Care Surg. 2015, 78, 224–229; discussion 229–230. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.W.; Al Khan, S.; Wang, A.Y.; Dawe, P.; Young, P.Y.; Greene, A.; Hudoba, M.; Vu, E. Systematic reviews of scores and predictors to trigger activation of massive transfusion protocols. J. Trauma Acute Care Surg. 2019, 87, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Uhlich, R.; Black, J.; Jansen, J.O.; Kerby, J.; Holcomb, J.B. A new definition for massive transfusion in the modern era of whole blood resuscitation. Transfusion 2021, 61 (Suppl. 1), S252–S263. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, L.S.L.; Bulger, E.M.; Ciesielski, W.A.; Carlbom, D.J.; Fisk, D.M.; Sheehan, K.L.; Asplund, K.M.; Schenkman, K.A. Muscle Oxygenation as an Early Predictor of Shock Severity in Trauma Patients. Shock 2017, 47, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkman, K.A.; Carlbom, D.J.; Bulger, E.M.; Ciesielski, W.A.; Fisk, D.M.; Sheehan, K.L.; Asplund, K.M.; Shaver, J.M.; Arakaki, L.S.L. Muscle oxygenation as an indicator of shock severity in patients with suspected severe sepsis or septic shock. PLoS ONE 2017, 12, e0182351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkman, K.A.; Hawkins, D.S.; Ciesielski, W.A.; Delaney, M.; Arakaki, L.S. Non-invasive assessment of muscle oxygenation may aid in optimising transfusion threshold decisions in ambulatory paediatric patients. Transfus. Med. 2017, 27, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Arakaki, L.S.; Schenkman, K.A.; Ciesielski, W.A.; Shaver, J.M. Muscle oxygenation measurement in humans by noninvasive optical spectroscopy and Locally Weighted Regression. Anal. Chim. Acta 2013, 785, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, E.E.; Moore, H.B.; Kornblith, L.Z.; Neal, M.D.; Hoffman, M.; Mutch, N.J.; Schochl, H.; Hunt, B.J.; Sauaia, A. Trauma-induced coagulopathy. Nat. Rev. Dis. Prim. 2021, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, J.B.; Jenkins, D.; Rhee, P.; Johannigman, J.; Mahoney, P.; Mehta, S.; Cox, E.D.; Gehrke, M.J.; Beilman, G.J.; Schreiber, M.; et al. Damage control resuscitation: Directly addressing the early coagulopathy of trauma. J. Trauma 2007, 62, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Van, P.Y.; Holcomb, J.B.; Schreiber, M.A. Novel concep.pts for damage control resuscitation in trauma. Curr. Opin. Crit. Care 2017, 23, 498–502. [Google Scholar] [CrossRef]
- Moore, F.D. Should blood be whole or in parts? N. Engl. J. Med. 1969, 280, 327–328. [Google Scholar] [CrossRef]
- Yazer, M.H.; Spinella, P.C.; Anto, V.; Dunbar, N.M. Survey of group A plasma and low-titer group O whole blood use in trauma resuscitation at adult civilian level 1 trauma centers in the US. Transfusion 2021, 61, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Troughton, M.; Young, P.P. Conservation of Rh negative Low Titer O Whole Blood (LTOWB) and the need for a national conversation to define its use in trauma transfusion protocols. Transfusion 2021, 61, 1966–1971. [Google Scholar] [CrossRef] [PubMed]
- Malkin, M.; Nevo, A.; Brundage, S.I.; Schreiber, M. Effectiveness and safety of whole blood compared to balanced blood components in resuscitation of hemorrhaging trauma patients—A systematic review. Injury 2021, 52, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Shea, S.M.; Staudt, A.M.; Thomas, K.A.; Schuerer, D.; Mielke, J.E.; Folkerts, D.; Lowder, E.; Martin, C.; Bochicchio, G.V.; Spinella, P.C. The use of low-titer group O whole blood is independently associated with improved survival compared to component therapy in adults with severe traumatic hemorrhage. Transfusion 2020, 60 (Suppl. 3), S2–S9. [Google Scholar] [CrossRef]
- Clements, T.; McCoy, C.; Assen, S.; Cardenas, J.; Wade, C.; Meyer, D.; Cotton, B.A. The prehospital use of younger age whole blood is associated with an improved arrival coagulation profile. J. Trauma Acute Care Surg. 2021, 90, 607–614. [Google Scholar] [CrossRef]
- Fadeyi, E.A.; Saha, A.K.; Naal, T.; Martin, H.; Fenu, E.; Simmons, J.H.; Jones, M.R.; Pomper, G.J. A comparison between leukocyte reduced low titer whole blood vs non-leukocyte reduced low titer whole blood for massive transfusion activation. Transfusion 2020, 60, 2834–2840. [Google Scholar] [CrossRef] [PubMed]
- Salamea-Molina, J.C.; Himmler, A.N.; Valencia-Angel, L.I.; Ordonez, C.A.; Parra, M.W.; Caicedo, Y.; Guzman-Rodriguez, M.; Orlas, C.; Granados, M.; Macia, C.; et al. Whole blood for blood loss: Hemostatic resuscitation in damage control. Colomb. Med. (Cali) 2020, 51, e4044511. [Google Scholar] [CrossRef]
- Yazer, M.H.; Triulzi, D.J.; Sperry, J.L.; Seheult, J.N. Rate of RhD-alloimmunization after the transfusion of multiple RhD-positive primary red blood cell-containing products. Transfusion 2021, 61 (Suppl. 1), S150–S158. [Google Scholar] [CrossRef]
- Shackelford, S.A.; Gurney, J.M.; Taylor, A.L.; Keenan, S.; Corley, J.B.; Cunningham, C.W.; Drew, B.G.; Jensen, S.D.; Kotwal, R.S.; Montgomery, H.R.; et al. Joint Trauma System, Defense Committee on Trauma, and Armed Services Blood Program consensus statement on whole blood. Transfusion 2021, 61 (Suppl. 1), S333–S335. [Google Scholar] [CrossRef]
- Williams, J.; Merutka, N.; Meyer, D.; Bai, Y.; Prater, S.; Cabrera, R.; Holcomb, J.B.; Wade, C.E.; Love, J.D.; Cotton, B.A. Safety profile and impact of low-titer group O whole blood for emergency use in trauma. J. Trauma Acute Care Surg. 2020, 88, 87–93. [Google Scholar] [CrossRef]
- Yazer, M.H.; Freeman, A.; Harrold, I.M.; Anto, V.; Neal, M.D.; Triulzi, D.J.; Sperry, J.L.; Seheult, J.N. Injured recipients of low-titer group O whole blood have similar clinical outcomes compared to recipients of conventional component therapy: A single-center, retrospective study. Transfusion 2021, 61, 1710–1720. [Google Scholar] [CrossRef]
- Seheult, J.N.; Anto, V.; Alarcon, L.H.; Sperry, J.L.; Triulzi, D.J.; Yazer, M.H. Clinical outcomes among low-titer group O whole blood recipients compared to recipients of conventional components in civilian trauma resuscitation. Transfusion 2018, 58, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, M.; Franchini, M.; Mengoli, C.; Marano, G.; Pati, I.; Masiello, F.; Veropalumbo, E.; Pupella, S.; Vaglio, S.; Agostini, V.; et al. The use of whole blood in traumatic bleeding: A systematic review. Intern. Emerg. Med. 2021, 16, 209–220. [Google Scholar] [CrossRef]
- Holcomb, J.B.; Tilley, B.C.; Baraniuk, S.; Fox, E.E.; Wade, C.E.; Podbielski, J.M.; del Junco, D.J.; Brasel, K.J.; Bulger, E.M.; Callcut, R.A.; et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs. a 1:1:2 ratio and mortality in patients with severe trauma: The PROPPR randomized clinical trial. JAMA 2015, 313, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Kemp Bohan, P.M.; McCarthy, P.M.; Wall, M.E.; Adams, A.M.; Chick, R.C.; Forcum, J.E.; Radowsky, J.S.; How, R.A.; Sams, V.G. Safety and efficacy of low-titer O whole blood resuscitation in a civilian level I trauma center. J. Trauma Acute Care Surg. 2021, 91, S162–S168. [Google Scholar] [CrossRef]
- Levy, J.H.; Neal, M.D.; Herman, J.H. Bacterial contamination of platelets for transfusion: Strategies for prevention. Crit. Care 2018, 22, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devine, D.V.; Serrano, K. The platelet storage lesion. Clin. Lab. Med. 2010, 30, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Becker, G.A.; Tuccelli, M.; Kunicki, T.; Chalos, M.K.; Aster, R.H. Studies of platelet concentrates stored at 22 C nad 4 C. Transfusion 1973, 13, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Reddoch, K.M.; Pidcoke, H.F.; Montgomery, R.K.; Fedyk, C.G.; Aden, J.K.; Ramasubramanian, A.K.; Cap, A.P. Hemostatic function of apheresis platelets stored at 4 °C and 22 °C. Shock 2014, 41 (Suppl. 1), 54–61. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.; Gardner, F.H. Effect of storage temperature on maintenance of platelet viability--deleterious effect of refrigerated storage. N. Engl. J. Med. 1969, 280, 1094–1098. [Google Scholar] [CrossRef]
- Milford, E.M.; Reade, M.C. Comprehensive review of platelet storage methods for use in the treatment of active hemorrhage. Transfusion 2016, 56 (Suppl. 2), S140–S148. [Google Scholar] [CrossRef] [Green Version]
- Pidcoke, H.F.; Spinella, P.C.; Ramasubramanian, A.K.; Strandenes, G.; Hervig, T.; Ness, P.M.; Cap, A.P. Refrigerated platelets for the treatment of acute bleeding: A review of the literature and reexamination of current standards. Shock 2014, 41 (Suppl. 1), 51–53. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xiong, Y.; Wang, R.; Tang, F.; Wang, X. Blood banking-induced alteration of red blood cell oxygen release ability. Blood Transfus. 2016, 14, 238–244. [Google Scholar] [CrossRef]
- Tinmouth, A.; Fergusson, D.; Yee, I.C.; Hebert, P.C.; ABLE Investigators and the Canadian Critical Care Trials Group. Clinical consequences of red cell storage in the critically ill. Transfusion 2006, 46, 2014–2027. [Google Scholar] [CrossRef]
- Fabron, A., Jr.; Lopes, L.B.; Bordin, J.O. Transfusion-related acute lung injury. J. Bras. Pneumol. 2007, 33, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, R.L. Red blood cell storage duration and trauma. Transfus Med. Rev. 2015, 29, 120–126. [Google Scholar] [CrossRef]
- Stan, A.; Zsigmond, E. The restoration in vivo of 2,3-diphosphoglycerate (2,3-DPG) in stored red cells, after transfusion. The levels of red cells 2,3-DPG. Rom. J. Intern. Med. 2009, 47, 173–177. [Google Scholar]
- Sowers, N.; Froese, P.C.; Erdogan, M.; Green, R.S. Impact of the age of stored blood on trauma patient mortality: A systematic review. Can. J. Surg. 2015, 58, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.R.; Patel, R.P.; Marques, M.B.; Donnelly, J.P.; Griffin, R.L.; Pittet, J.F.; Kerby, J.D.; Stephens, S.W.; DeSantis, S.M.; Hess, J.R.; et al. Older Blood Is Associated With Increased Mortality and Adverse Events in Massively Transfused Trauma Patients: Secondary Analysis of the PROPPR Trial. Ann. Emerg. Med. 2019, 73, 650–661. [Google Scholar] [CrossRef]
- Remy, K.E.; Sun, J.; Wang, D.; Welsh, J.; Solomon, S.B.; Klein, H.G.; Natanson, C.; Cortes-Puch, I. Transfusion of recently donated (fresh) red blood cells (RBCs) does not improve survival in comparison with current practice, while safety of the oldest stored units is yet to be established: A meta-analysis. Vox Sang. 2016, 111, 43–54. [Google Scholar] [CrossRef]
- Milford, E.M.; Reade, M.C. Resuscitation Fluid Choices to Preserve the Endothelial Glycocalyx. Crit. Care 2019, 23, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardigan, R.; Green, L. Thawed and liquid plasma--what do we know? Vox Sang. 2015, 109, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, W.P.; Bhakta, V.; Mastronardi, C.; Ramirez-Arcos, S.; Howe, D.; Jenkins, C. Changes in coagulation factor activity and content of di(2-ethylhexyl)phthalate in frozen plasma units during refrigerated storage for up to five days after thawing. Transfusion 2012, 52, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Alhumaidan, H.; Cheves, T.; Holme, S.; Sweeney, J. Stability of coagulation factors in plasma prepared after a 24-hour room temperature hold. Transfusion 2010, 50, 1934–1942. [Google Scholar] [CrossRef]
- Chhibber, V.; Greene, M.; Vauthrin, M.; Bailey, J.; Weinstein, R. Is group A thawed plasma suitable as the first option for emergency release transfusion? (CME). Transfusion 2014, 54, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- Cooling, L. Going from A to B: The safety of incompatible group A plasma for emergency release in trauma and massive transfusion patients. Transfusion 2014, 54, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehr, C.R.; Gupta, R.; von Recklinghausen, F.M.; Szczepiorkowski, Z.M.; Dunbar, N.M. Balancing risk and benefit: Maintenance of a thawed Group A plasma inventory for trauma patients requiring massive transfusion. J. Trauma Acute Care Surg 2013, 74, 1425–1431. [Google Scholar] [CrossRef]
- Meledeo, M.A.; Peltier, G.C.; McIntosh, C.S.; Bynum, J.A.; Corley, J.B.; Cap, A.P. Coagulation function of never frozen liquid plasma stored for 40 days. Transfusion 2021, 61 (Suppl. 1), S111–S118. [Google Scholar] [CrossRef] [PubMed]
- Mok, G.; Hoang, R.; Khan, M.W.; Pannell, D.; Peng, H.; Tien, H.; Nathens, A.; Callum, J.; Karkouti, K.; Beckett, A.; et al. Freeze-dried plasma for major trauma—Systematic review and meta-analysis. J. Trauma Acute Care Surg. 2021, 90, 589–602. [Google Scholar] [CrossRef]
- Pusateri, A.E.; Given, M.B.; Schreiber, M.A.; Spinella, P.C.; Pati, S.; Kozar, R.A.; Khan, A.; Dacorta, J.A.; Kupferer, K.R.; Prat, N.; et al. Dried plasma: State of the science and recent developments. Transfusion 2016, 56 (Suppl. 2), S128–S139. [Google Scholar] [CrossRef]
- Sailliol, A.; Martinaud, C.; Cap, A.P.; Civadier, C.; Clavier, B.; Deshayes, A.V.; Mendes, A.C.; Pouget, T.; Demazeau, N.; Chueca, M.; et al. The evolving role of lyophilized plasma in remote damage control resuscitation in the French Armed Forces Health Service. Transfusion 2013, 53 (Suppl. 1), 65S–71S. [Google Scholar] [CrossRef]
- Shlaifer, A.; Siman-Tov, M.; Radomislensky, I.; Peleg, K.; Shina, A.; Baruch, E.N.; Glassberg, E.; Yitzhak, A.; ITG*. Prehospital administration of freeze-dried plasma, is it the solution for trauma casualties? J. Trauma Acute Care Surg. 2017, 83, 675–682. [Google Scholar] [CrossRef]
- Winearls, J.; Campbell, D.; Hurn, C.; Furyk, J.; Ryan, G.; Trout, M.; Walsham, J.; Holley, A.; Shuttleworth, M.; Dyer, W.; et al. Fibrinogen in traumatic haemorrhage: A narrative review. Injury 2017, 48, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Winearls, J.; Wullschleger, M.; Wake, E.; Hurn, C.; Furyk, J.; Ryan, G.; Trout, M.; Walsham, J.; Holley, A.; Cohen, J.; et al. Fibrinogen Early In Severe Trauma studY (FEISTY): Study protocol for a randomised controlled trial. Trials 2017, 18, 241. [Google Scholar] [CrossRef] [Green Version]
- Hayes, T. Dysfibrinogenemia and Thrombosis. Arch. Pathol. Lab. Med. 2002, 126, 1387–1390. [Google Scholar] [CrossRef]
- Meyer, M.A.; Ostrowski, S.R.; Sorensen, A.M.; Meyer, A.S.; Holcomb, J.B.; Wade, C.E.; Johansson, P.I.; Stensballe, J. Fibrinogen in trauma, an evaluation of thrombelastography and rotational thromboelastometry fibrinogen assays. J. Surg. Res. 2015, 194, 581–590. [Google Scholar] [CrossRef]
- Holcomb, J.B.; Fox, E.E.; Zhang, X.; White, N.; Wade, C.E.; Cotton, B.A.; Del Junco, D.J.; Bulger, E.M.; Cohen, M.J.; Schreiber, M.A.; et al. Cryoprecipitate Use in the Prospective Observational Multicenter Major Trauma Transfusion study (PROMMTT). J. Trauma Acute Care Surg. 2013, 75, S31–S39. [Google Scholar] [CrossRef] [Green Version]
- Barry, M.; Trivedi, A.; Miyazawa, B.Y.; Vivona, L.R.; Khakoo, M.; Zhang, H.; Pathipati, P.; Bagri, A.; Gatmaitan, M.G.; Kozar, R.; et al. Cryoprecipitate attenuates the endotheliopathy of trauma in mice subjected to hemorrhagic shock and trauma. J. Trauma Acute Care Surg. 2021, 90, 1022–1031. [Google Scholar] [CrossRef]
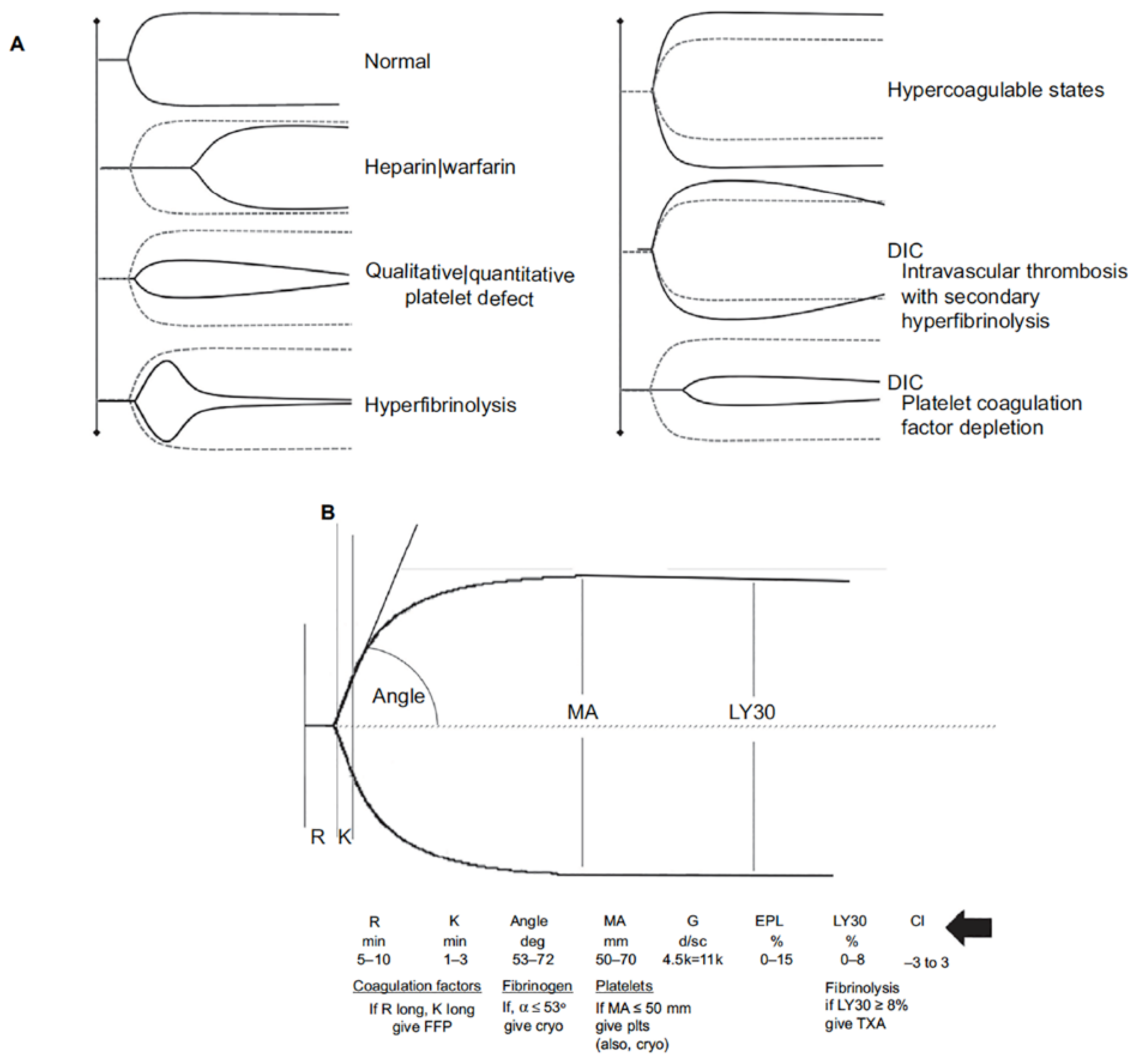
- Bugaev, N.; Como, J.J.; Golani, G.; Freeman, J.J.; Sawhney, J.S.; Vatsaas, C.J.; Yorkgitis, B.K.; Kreiner, L.A.; Garcia, N.M.; Aziz, H.A.; et al. Thromboelastography and rotational thromboelastometry in bleeding patients with coagulopathy: Practice management guideline from the Eastern Association for the Surgery of Trauma. J. Trauma Acute Care Surg. 2020, 89, 999–1017. [Google Scholar] [CrossRef]
- Einersen, P.M.; Moore, E.E.; Chapman, M.P.; Moore, H.B.; Gonzalez, E.; Silliman, C.C.; Banerjee, A.; Sauaia, A. Rapid thrombelastography thresholds for goal-directed resuscitation of patients at risk for massive transfusion. J. Trauma Acute Care Surg. 2017, 82, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Sharp, G.; Young, C.J. Point-of-care viscoelastic assay devices (rotational thromboelastometry and thromboelastography): A primer for surgeons. ANZ J. Surg. 2019, 89, 291–295. [Google Scholar] [CrossRef]
- Brill, J.B.; Cotton, B.A.; Brenner, M.; Duchesne, J.; Ferrada, P.; Horer, T.; Kauvar, D.; Khan, M.; Roberts, D.; Ordonez, C.; et al. The Role of TEG and ROTEM in Damage Control Resuscitation. Shock 2021. [Google Scholar] [CrossRef] [PubMed]
- Gall, L.S.; Vulliamy, P.; Gillespie, S.; Jones, T.F.; Pierre, R.S.J.; Breukers, S.E.; Gaarder, C.; Juffermans, N.P.; Maegele, M.; Stensballe, J.; et al. The S100A10 Pathway Mediates an Occult Hyperfibrinolytic Subtype in Trauma Patients. Ann. Surg. 2019, 269, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Curry, N.S.; Davenport, R.; Pavord, S.; Mallett, S.V.; Kitchen, D.; Klein, A.A.; Maybury, H.; Collins, P.W.; Laffan, M. The use of viscoelastic haemostatic assays in the management of major bleeding: A British Society for Haematology Guideline. Br. J. Haematol. 2018, 182, 789–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roullet, S.; de Maistre, E.; Ickx, B.; Blais, N.; Susen, S.; Faraoni, D.; Garrigue, D.; Bonhomme, F.; Godier, A.; Lasne, D.; et al. Position of the French Working Group on Perioperative Haemostasis (GIHP) on viscoelastic tests: What role for which indication in bleeding situations? Anaesth. Crit. Care Pain Med. 2019, 38, 539–548. [Google Scholar] [CrossRef]
- CRASH-2 Collaborators; Roberts, I.; Shakur, H.; Afolabi, A.; Brohi, K.; Coats, T.; Dewan, Y.; Gando, S.; Guyatt, G.; Hunt, B.J.; et al. The importance of early treatment with tranexamic acid in bleeding trauma patients: An exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 2011, 377, 1096–1101.e1–2. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.P.; Kutcher, M.E.; Rosengart, M.R.; Sperry, J.L.; Peitzman, A.B.; Brown, J.B.; Neal, M.D. Tranexamic acid administration is associated with an increased risk of posttraumatic venous thromboembolism. J. Trauma Acute Care Surg. 2019, 86, 20–27. [Google Scholar] [CrossRef]
- Moore, E.E.; Moore, H.B.; Gonzalez, E.; Chapman, M.P.; Hansen, K.C.; Sauaia, A.; Silliman, C.C.; Banerjee, A. Postinjury fibrinolysis shutdown: Rationale for selective tranexamic acid. J. Trauma Acute Care Surg. 2015, 78, S65–S69. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.D.; Moore, H.B.; Vigneshwar, N.; Dhara, S.; Chandler, J.; Chapman, M.P.; Sauaia, A.; Moore, E.E.; Yaffe, M.B. Plasmin thrombelastography rapidly identifies trauma patients at risk for massive transfusion, mortality, and hyperfibrinolysis: A diagnostic tool to resolve an international debate on tranexamic acid? J. Trauma Acute Care Surg. 2020, 89, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Jehan, F.; Bulger, E.M.; O’Keeffe, T.; Holcomb, J.B.; Wade, C.E.; Schreiber, M.A.; Joseph, B.; Group, P.S. Severely injured trauma patients with admission hyperfibrinolysis: Is there a role of tranexamic acid? Findings from the PROPPR trial. J. Trauma Acute Care Surg. 2018, 85, 851–857. [Google Scholar] [CrossRef]
- Selby, R. “TEG talk”: Expanding clinical roles for thromboelastography and rotational thromboelastometry. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 67–75. [Google Scholar] [CrossRef]
- British Committee for Standards in Haematology Writing Group; Stainsby, D.; MacLennan, S.; Thomas, D.; Isaac, J.; Hamilton, P.J. Guidelines on the management of massive blood loss. Br. J. Haematol. 2006, 135, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Hiippala, S.T.; Myllyla, G.J.; Vahtera, E.M. Hemostatic factors and replacement of major blood loss with plasma-poor red cell concentrates. Anesth. Analg. 1995, 81, 360–365. [Google Scholar] [PubMed]
- Inaba, K.; Branco, B.C.; Rhee, P.; Blackbourne, L.H.; Holcomb, J.B.; Teixeira, P.G.; Shulman, I.; Nelson, J.; Demetriades, D. Impact of plasma transfusion in trauma patients who do not require massive transfusion. J. Am. Coll. Surg. 2010, 210, 957–965. [Google Scholar] [CrossRef] [PubMed]
- McQuilten, Z.K.; Wood, E.M.; Bailey, M.; Cameron, P.A.; Cooper, D.J. Fibrinogen is an independent predictor of mortality in major trauma patients: A five-year statewide cohort study. Injury 2017, 48, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Ishikura, H.; Kushimoto, S.; Kiyomi, F.; Kato, H.; Sasaki, J.; Ogura, H.; Matsuoka, T.; Uejima, T.; Morimura, N.; et al. Fibrinogen level on admission is a predictor for massive transfusion in patients with severe blunt trauma: Analyses of a retrospective multicentre observational study. Injury 2017, 48, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Innerhofer, P.; Fries, D.; Mittermayr, M.; Innerhofer, N.; von Langen, D.; Hell, T.; Gruber, G.; Schmid, S.; Friesenecker, B.; Lorenz, I.H.; et al. Reversal of trauma-induced coagulopathy using first-line coagulation factor concentrates or fresh frozen plasma (RETIC): A single-centre, parallel-group, open-label, randomised trial. Lancet Haematol. 2017. [Google Scholar] [CrossRef]
- Nascimento, B.; Callum, J.; Tien, H.; Peng, H.; Rizoli, S.; Karanicolas, P.; Alam, A.; Xiong, W.; Selby, R.; Garzon, A.M.; et al. Fibrinogen in the initial resuscitation of severe trauma (FiiRST): A randomized feasibility trial. Br. J. Anaesth. 2016, 117, 775–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schochl, H.; Nienaber, U.; Maegele, M.; Hochleitner, G.; Primavesi, F.; Steitz, B.; Arndt, C.; Hanke, A.; Voelckel, W.; Solomon, C. Transfusion in trauma: Thromboelastometry-guided coagulation factor concentrate-based therapy versus standard fresh frozen plasma-based therapy. Crit. Care 2011, 15, R83. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Yamaguchi, A.; Sawano, M.; Matsuda, M.; Anan, M.; Inokuchi, K.; Sugiyama, S. Pre-emptive administration of fibrinogen concentrate contributes to improved prognosis in patients with severe trauma. Trauma Surg. Acute Care Open 2016, 1, e000037. [Google Scholar] [CrossRef] [Green Version]
- Simurda, T.; Stanciakova, L.; Stasko, J.; Dobrotova, M.; Kubisz, P. Yes or no for secondary prophylaxis in afibrinogenemia? Blood Coagul. Fibrinolysis 2015, 26, 978–980. [Google Scholar] [CrossRef]
- Su, Y.; Chen, Y.; Zhang, W.; Liu, L.; Cao, X.; Wu, J. Platelet factor 4 and beta-thromboglobulin mRNAs in circulating microparticles of trauma patients as diagnostic markers for deep vein thrombosis. J. Thromb. Thrombolysis 2020, 50, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Neisser-Svae, A.; Hegener, O.; Gorlinger, K. Differences in the biochemical composition of three plasma derived human fibrinogen concentrates. Thromb. Res. 2021, 205, 44–46. [Google Scholar] [CrossRef]
- Schlimp, C.J.; Cadamuro, J.; Solomon, C.; Redl, H.; Schochl, H. The effect of fibrinogen concentrate and factor XIII on thromboelastometry in 33% diluted blood with albumin, gelatine, hydroxyethyl starch or saline in vitro. Blood Transfus. 2013, 11, 510–517. [Google Scholar] [CrossRef]
- Nagashima, F.; Inoue, S.; Koami, H.; Miike, T.; Sakamoto, Y.; Kai, K. High-dose Factor XIII administration induces effective hemostasis for trauma-associated coagulopathy (TAC) both in vitro and in rat hemorrhagic shock in vivo models. J. Trauma Acute Care Surg. 2018, 85, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Perner, A.; Haase, N.; Wiis, J.; White, J.O.; Delaney, A. Central venous oxygen saturation for the diagnosis of low cardiac output in septic shock patients. Acta Anaesthesiol. Scand. 2010, 54, 98–102. [Google Scholar] [CrossRef]
- Reinhart, K.; Bloos, F. The value of venous oximetry. Curr. Opin. Crit. Care 2005, 11, 259–263. [Google Scholar] [CrossRef]
- Reinhart, K.; Kuhn, H.J.; Hartog, C.; Bredle, D.L. Continuous central venous and pulmonary artery oxygen saturation monitoring in the critically ill. Intensive Care Med. 2004, 30, 1572–1578. [Google Scholar] [CrossRef]
- Edwards, J.D.; Mayall, R.M. Importance of the sampling site for measurement of mixed venous oxygen saturation in shock. Crit. Care Med. 1998, 26, 1356–1360. [Google Scholar] [CrossRef]
- Martin, C.; Auffray, J.P.; Badetti, C.; Perrin, G.; Papazian, L.; Gouin, F. Monitoring of central venous oxygen saturation versus mixed venous oxygen saturation in critically ill patients. Intensive Care Med. 1992, 18, 101–104. [Google Scholar] [CrossRef]
- Pope, J.V.; Jones, A.E.; Gaieski, D.F.; Arnold, R.C.; Trzeciak, S.; Shapiro, N.I.; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO(2)) as a predictor of mortality in patients with sepsis. Ann. Emerg. Med. 2010, 55, 40–46 e41. [Google Scholar] [CrossRef] [Green Version]
- Mallat, J.; Lazkani, A.; Lemyze, M.; Pepy, F.; Meddour, M.; Gasan, G.; Temime, J.; Vangrunderbeeck, N.; Tronchon, L.; Thevenin, D. Repeatability of blood gas parameters, PCO2 gap, and PCO2 gap to arterial-to-venous oxygen content difference in critically ill adult patients. Medicine (Baltimore) 2015, 94, e415. [Google Scholar] [CrossRef]
- Mallat, J.; Lemyze, M.; Tronchon, L.; Vallet, B.; Thevenin, D. Use of venous-to-arterial carbon dioxide tension difference to guide resuscitation therapy in septic shock. World J. Crit. Care Med. 2016, 5, 47–56. [Google Scholar] [CrossRef]
- Vallet, B.; Pinsky, M.R.; Cecconi, M. Resuscitation of patients with septic shock: Please “mind the gap”! Intensive Care Med. 2013, 39, 1653–1655. [Google Scholar] [CrossRef] [Green Version]
- Pohlman, T.H.; Walsh, M.; Aversa, J.; Hutchison, E.M.; Olsen, K.P.; Lawrence Reed, R. Damage control resuscitation. Blood Rev. 2015, 29, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Gutterman, D.D.; Chabowski, D.S.; Kadlec, A.O.; Durand, M.J.; Freed, J.K.; Ait-Aissa, K.; Beyer, A.M. The Human Microcirculation: Regulation of Flow and Beyond. Circ. Res. 2016, 118, 157–172. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fecher, A.; Stimpson, A.; Ferrigno, L.; Pohlman, T.H. The Pathophysiology and Management of Hemorrhagic Shock in the Polytrauma Patient. J. Clin. Med. 2021, 10, 4793. https://doi.org/10.3390/jcm10204793
Fecher A, Stimpson A, Ferrigno L, Pohlman TH. The Pathophysiology and Management of Hemorrhagic Shock in the Polytrauma Patient. Journal of Clinical Medicine. 2021; 10(20):4793. https://doi.org/10.3390/jcm10204793
Chicago/Turabian StyleFecher, Alison, Anthony Stimpson, Lisa Ferrigno, and Timothy H. Pohlman. 2021. "The Pathophysiology and Management of Hemorrhagic Shock in the Polytrauma Patient" Journal of Clinical Medicine 10, no. 20: 4793. https://doi.org/10.3390/jcm10204793