
Curing the Curable: Managing Low-Risk Acute Lymphoblastic Leukemia in Resource Limited Countries


Abstract
:1. Introduction
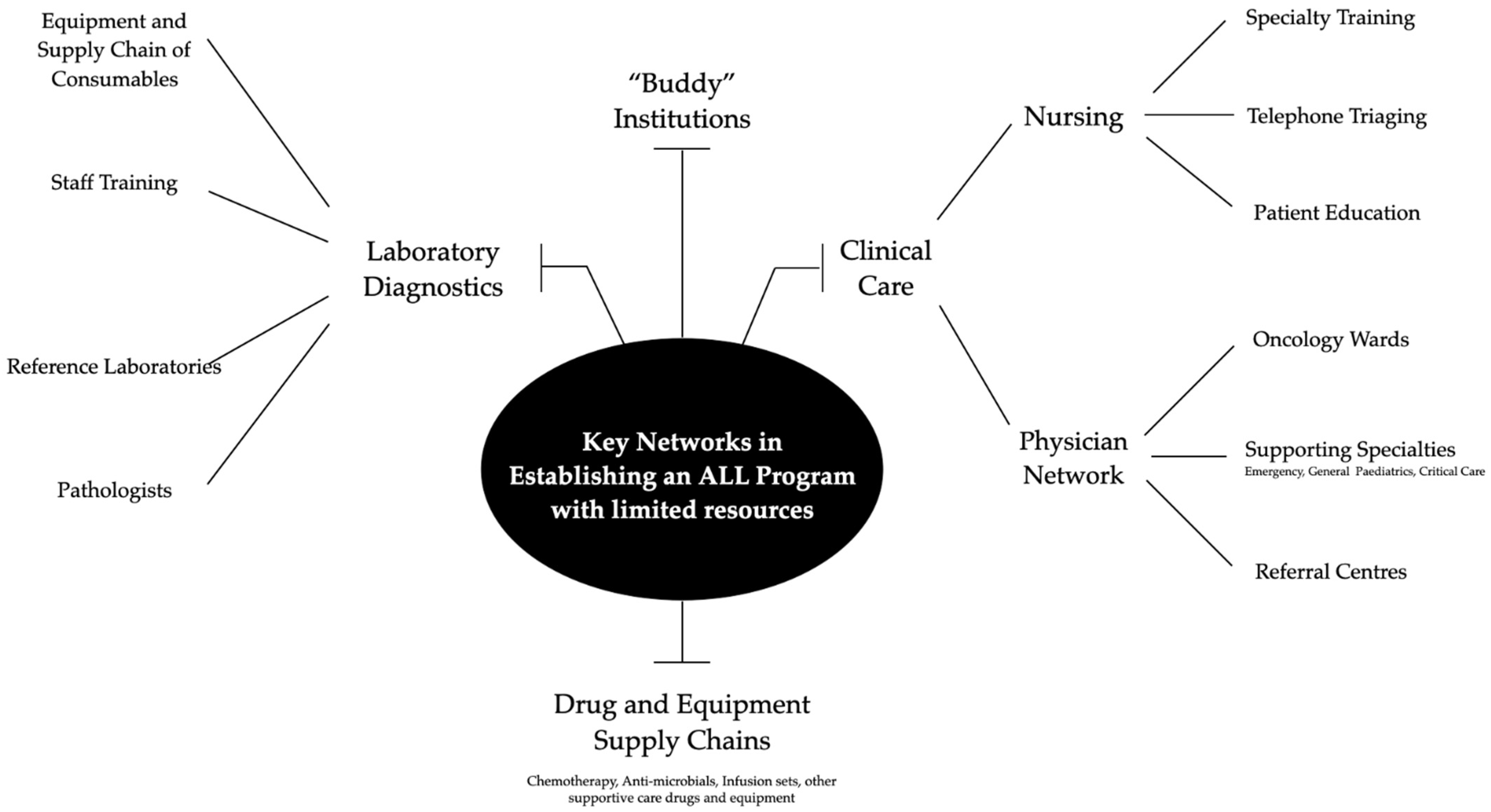
2. Causes of Failures in LMIC
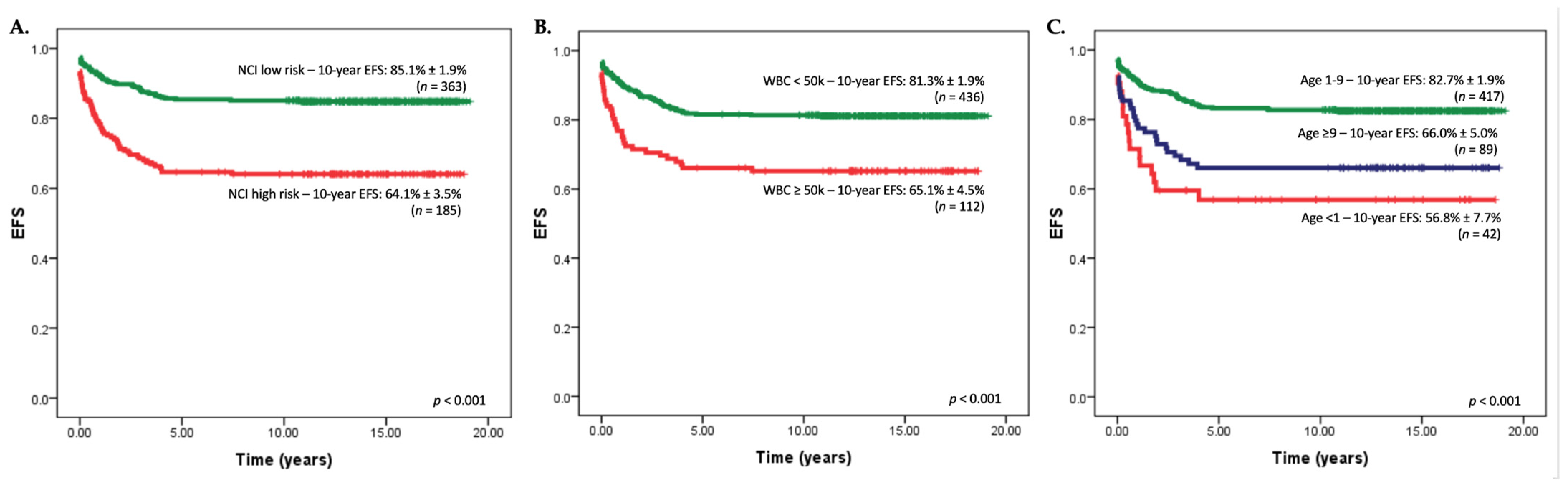
3. Identifying Low-Risk Groups in Resource Limited Settings
4. The Importance of the NCI Standard-Risk (SR) Criteria
5. Favorable ALL Genetics: Hyperdiploidy and ETV6-RUNX1
6. Democratization of Flow Cytometry
7. Specific Considerations for T-Lineage ALL
8. Delaying the First Intra-Thecal (IT) Chemotherapy
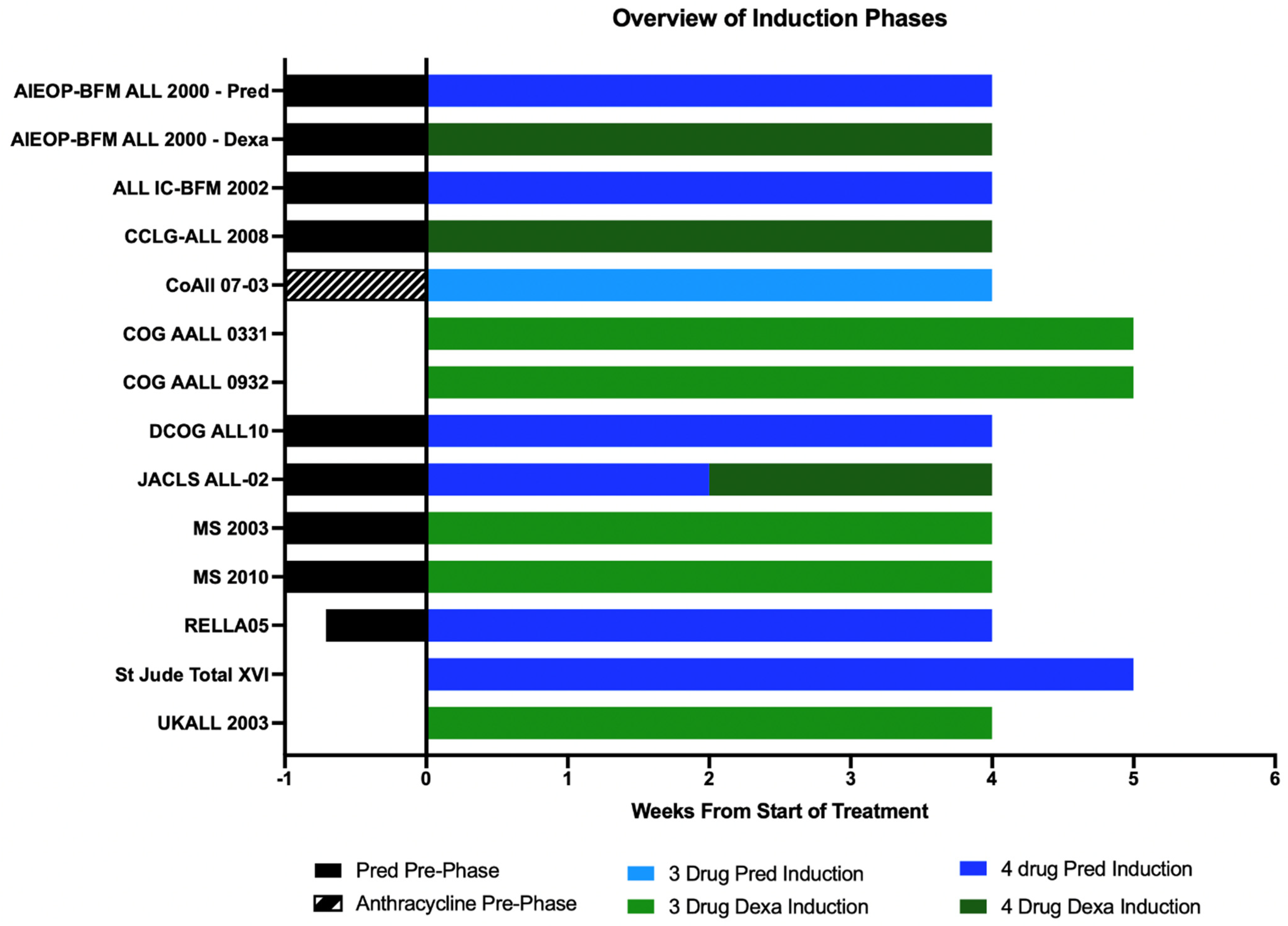
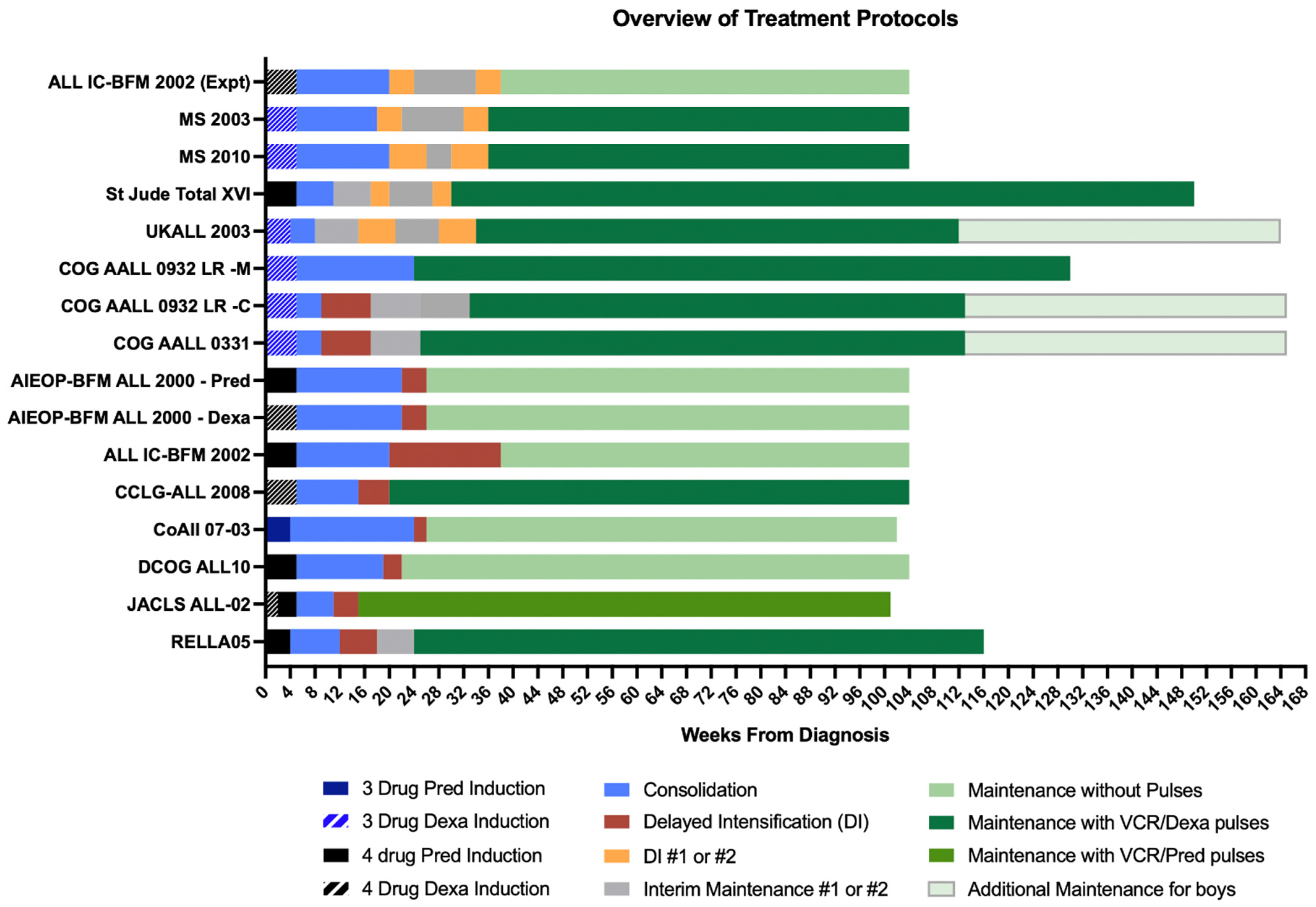
9. Prednisolone/Dexamethasone-Based and 3/4-Drug-Based Induction
10. L-Asp Doses in Induction and Delayed Intensification
11. Anthracycline-Free Regimens
12. Is High-Dose Methotrexate Really Necessary?
13. Delayed Intensification—Is More Necessarily Better?
14. The Malaysia Singapore Experience
15. Infections
16. Improving Supportive Care
17. Overview of Maintenance Therapy
18. Duration of Maintenance Phase
19. VCR/Steroid Pulses
20. TPMT and NUDT15 Variants on 6-MP Metabolism
21. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Pui, C.H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, A.E.J.; Tan, D.; Li, C.-K.; Hori, H.; Tse, E.; Pui, C.-H. Management of adult and paediatric acute lymphoblastic leukaemia in Asia: Resource-stratified guidelines from the Asian Oncology Summit 2013. Lancet Oncol. 2013, 14, e508–e523. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Galindo, C.; Friedrich, P.; Alcasabas, P.; Antillon, F.; Banavali, S.; Castillo, L.; Israels, T.; Jeha, S.; Harif, M.; Sullivan, M.J.; et al. Toward the Cure of All Children With Cancer Through Collaborative Efforts: Pediatric Oncology As a Global Challenge. J. Clin. Oncol. 2015, 33, 3065–3073. [Google Scholar] [CrossRef]
- Bhakta, N.; Force, L.M.; Allemani, C.; Atun, R.; Bray, F.; Coleman, M.P.; Steliarova-Foucher, E.; Frazier, A.L.; Robison, L.L.; Rodriguez-Galindo, C.; et al. Childhood cancer burden: A review of global estimates. Lancet Oncol. 2019, 20, e42–e53. [Google Scholar] [CrossRef] [Green Version]
- Atun, R.; Bhakta, N.; Denburg, A.; Frazier, A.L.; Friedrich, P.; Gupta, S.; Lam, C.G.; Ward, Z.J.; Yeh, J.M.; Allemani, C.; et al. Sustainable care for children with cancer: A Lancet Oncology Commission. Lancet Oncol. 2020, 21, e185–e224. [Google Scholar] [CrossRef]
- Locatelli, F.; Schrappe, M.; Bernardo, M.E.; Rutella, S. How I treat relapsed childhood acute lymphoblastic leukemia. Blood 2012, 120, 2807–2816. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.S.; Challinor, J.M.; Howard, S.C.; Israels, T. Improving Care for Children With Cancer in Low- and Middle-Income Countries—A SIOP PODC Initiative. Pediatr. Blood Cancer 2016, 63, 387–391. [Google Scholar] [CrossRef] [Green Version]
- Suarez, A.; Pina, M.; Nichols-Vinueza, D.X.; Lopera, J.; Rengifo, L.; Mesa, M.; Cardenas, M.; Morrissey, L.; Veintemilla, G.; Vizcaino, M.; et al. A strategy to improve treatment-related mortality and abandonment of therapy for childhood ALL in a developing country reveals the impact of treatment delays. Pediatr. Blood Cancer 2015, 62, 1395–1402. [Google Scholar] [CrossRef]
- Ladas, E.J.; Arora, B.; Howard, S.C.; Rogers, P.C.; Mosby, T.T.; Barr, R.D. A Framework for Adapted Nutritional Therapy for Children With Cancer in Low- and Middle-Income Countries: A Report From the SIOP PODC Nutrition Working Group. Pediatr. Blood Cancer 2016, 63, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Israels, T.; Renner, L.; Hendricks, M.; Hesseling, P.; Howard, S.; Molyneux, E.; Paediatric Oncology in Developing Countries. SIOP PODC: Recommendations for supportive care of children with cancer in a low-income setting. Pediatr. Blood Cancer 2013, 60, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Ariffin, H.; Navaratnam, P.; Kee, T.K.; Balan, G. Antibiotic resistance patterns in nosocomial gram-negative bacterial infections in units with heavy antibiotic usage. J. Trop. Pediatr. 2004, 50, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariffin, H.; Ab Rahman, S.; Leong, S.H.; Chiew, E.K.-H.; Lin, H.P.; Quah, T.C.; Yeoh, A.E.-J. Malaysia-Singapore (MASPORE) leukaemia study group: From common history to successful collaboration. Pediatr. Hematol. Oncol. J. 2020, 5, 11–16. [Google Scholar] [CrossRef]
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef]
- Pedrosa, F.; Coustan-Smith, E.; Zhou, Y.; Cheng, C.; Pedrosa, A.; Lins, M.M.; Pedrosa, M.; Lucena-Silva, N.; Ramos, A.M.L.; Vinhas, E.; et al. Reduced-dose intensity therapy for pediatric lymphoblastic leukemia: Long-term results of the Recife RELLA05 pilot study. Blood 2020, 135, 1458–1466. [Google Scholar] [CrossRef]
- Narula, G.; Prasad, M.; Jatia, S.; Subramanian, P.G.; Patkar, N.; Tembhare, P.; Shetty, D.; Khanna, N.; Laskar, S.; Shet, T.; et al. Clinicoepidemiological profiles, clinical practices, and the impact of holistic care interventions on outcomes of pediatric hematolymphoid malignancies—A 7-year audit of the pediatric hematolymphoid disease management group at Tata Memorial Hospital. Indian J. Cancer 2017, 54, 609–615. [Google Scholar] [CrossRef]
- Stary, J.; Zimmermann, M.; Campbell, M.; Castillo, L.; Dibar, E.; Donska, S.; Gonzalez, A.; Izraeli, S.; Janic, D.; Jazbec, J.; et al. Intensive chemotherapy for childhood acute lymphoblastic leukemia: Results of the randomized intercontinental trial ALL IC-BFM 2002. J. Clin. Oncol. 2014, 32, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Li, Z.G.; Chai, Y.H.; Yu, J.; Gao, J.; Zhu, X.F.; Jin, R.M.; Shi, X.D.; Zhang, L.P.; Gao, Y.J.; et al. Outcome of children with newly diagnosed acute lymphoblastic leukemia treated with CCLG-ALL 2008: The first nation-wide prospective multicenter study in China. Am. J. Hematol. 2018, 93, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Yeoh, A.E.; Ariffin, H.; Chai, E.L.; Kwok, C.S.; Chan, Y.H.; Ponnudurai, K.; Campana, D.; Tan, P.L.; Chan, M.Y.; Kham, S.K.; et al. Minimal residual disease-guided treatment deintensification for children with acute lymphoblastic leukemia: Results from the Malaysia-Singapore acute lymphoblastic leukemia 2003 study. J. Clin. Oncol. 2012, 30, 2384–2392. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, A.E.J.; Lu, Y.; Chin, W.H.N.; Chiew, E.K.H.; Lim, E.H.; Li, Z.; Kham, S.K.Y.; Chan, Y.H.; Abdullah, W.A.; Lin, H.P.; et al. Intensifying Treatment of Childhood B-Lymphoblastic Leukemia With IKZF1 Deletion Reduces Relapse and Improves Overall Survival: Results of Malaysia-Singapore ALL 2010 Study. J. Clin. Oncol. 2018, 36, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Bleckmann, K.; Zimmermann, M.; Biondi, A.; Moricke, A.; Locatelli, F.; Cario, G.; Rizzari, C.; Attarbaschi, A.; Valsecchi, M.G.; et al. Reduced-Intensity Delayed Intensification in Standard-Risk Pediatric Acute Lymphoblastic Leukemia Defined by Undetectable Minimal Residual Disease: Results of an International Randomized Trial (AIEOP-BFM ALL 2000). J. Clin. Oncol. 2018, 36, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Schramm, F.; Zur Stadt, U.; Zimmermann, M.; Jorch, N.; Pekrun, A.; Borkhardt, A.; Imschweiler, T.; Christiansen, H.; Faber, J.; Schmid, I.; et al. Results of CoALL 07-03 study childhood ALL based on combined risk assessment by in vivo and in vitro pharmacosensitivity. Blood Adv. 2019, 3, 3688–3699. [Google Scholar] [CrossRef]
- Schore, R.J.; Angiolillo, A.J.; Kairalla, J.A.; Devidas, M.; Rabin, K.R.; Zweidler-McKay, P.A.; Borowitz, M.J.; Wood, B.L.; Carroll, A.J.; Heerema, N.A.; et al. Outcomes with reduced intensity therapy in a low-risk subset of children with National Cancer Institute (NCI) standard-risk (SR) B-lymphoblastic leukemia (B-ALL): A report from Children’s Oncology Group (COG) AALL0932. J. Clin. Oncol. 2020, 38, 10509. [Google Scholar] [CrossRef]
- Mattano, L.A., Jr.; Devidas, M.; Maloney, K.W.; Wang, C.; Friedmann, A.M.; Buckley, P.; Borowitz, M.J.; Carroll, A.J.; Gastier-Foster, J.M.; Heerema, N.A.; et al. Favorable Trisomies and ETV6-RUNX1 Predict Cure in Low-Risk B-Cell Acute Lymphoblastic Leukemia: Results From Children’s Oncology Group Trial AALL0331. J. Clin. Oncol. 2021, 39, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; de Groot-Kruseman, H.; Van der Velden, V.; Fiocco, M.; van den Berg, H.; de Bont, E.; Egeler, R.M.; Hoogerbrugge, P.; Kaspers, G.; Van der Schoot, E.; et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J. Clin. Oncol. 2016, 34, 2591–2601. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, D.; Imamura, T.; Yumura-Yagi, K.; Takahashi, Y.; Usami, I.; Suenobu, S.I.; Nishimura, S.; Suzuki, N.; Hashii, Y.; Deguchi, T.; et al. Risk-adjusted therapy for pediatric non-T cell ALL improves outcomes for standard risk patients: Results of JACLS ALL-02. Blood Cancer J. 2020, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391. [Google Scholar] [CrossRef]
- Vora, A.; Goulden, N.; Wade, R.; Mitchell, C.; Hancock, J.; Hough, R.; Rowntree, C.; Richards, S. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): A randomised controlled trial. Lancet Oncol. 2013, 14, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Attarbaschi, A.; Mann, G.; Dworzak, M.; Wiesbauer, P.; Schrappe, M.; Gadner, H. Mediastinal mass in childhood T-cell acute lymphoblastic leukemia: Significance and therapy response. Med. Pediatr. Oncol. 2002, 39, 558–565. [Google Scholar] [CrossRef]
- Pommert, L.; Tasian, S.K. Chemotherapy Drug Shortages in Pediatric Oncology: A Global Public Health Crisis Threatening Our Children. Hematologist 2021. [Google Scholar] [CrossRef]
- Cohen, P.; Friedrich, P.; Lam, C.; Jeha, S.; Metzger, M.L.; Qaddoumi, I.; Naidu, P.; Faughnan, L.; Rodriguez-Galindo, C.; Bhakta, N. Global Access to Essential Medicines for Childhood Cancer: A Cross-Sectional Survey. J. Glob. Oncol. 2018, 4, 1–11. [Google Scholar] [CrossRef]
- Howard, S.C.; Davidson, A.; Luna-Fineman, S.; Israels, T.; Chantada, G.; Lam, C.G.; Hunger, S.P.; Bailey, S.; Ribeiro, R.C.; Arora, R.S.; et al. A framework to develop adapted treatment regimens to manage pediatric cancer in low- and middle-income countries: The Pediatric Oncology in Developing Countries (PODC) Committee of the International Pediatric Oncology Society (SIOP). Pediatr. Blood Cancer 2017, 64 (Suppl. S5), e26879. [Google Scholar] [CrossRef] [Green Version]
- Loh, L.H.; Chen, S.P.; Quah, T.C.; Yeoh, A.E.; Ariffin, H. Real-time quantitative polymerase chain reaction (RO-PCR) using the LightCycler: A rapid, high-throughput method for detecting and quantifying fusion transcripts in childhood leukaemias for disease stratification and prognostication. Ann. Acad. Med. Singap. 2003, 32, S18–S21. [Google Scholar] [PubMed]
- Ibrahim, K.; Daud, S.S.; Seah, Y.L.; Yeoh, A.E.; Ariffin, H.; Malaysia-Singapore Leukemia Study, G. Rapid detection of prognostically important childhood acute lymphoblastic leukemia chimeric transcripts using multiplex SYBR green real-time reverse transcription PCR. Ann. Clin. Lab. Sci. 2008, 38, 338–343. [Google Scholar]
- Lee, S.H.R.; Li, Z.; Tai, S.T.; Oh, B.L.Z.; Yeoh, A.E.J. Genetic Alterations in Childhood Acute Lymphoblastic Leukemia: Interactions with Clinical Features and Treatment Response. Cancers 2021, 13, 4068. [Google Scholar] [CrossRef]
- Smith, M.; Arthur, D.; Camitta, B.; Carroll, A.J.; Crist, W.; Gaynon, P.; Gelber, R.; Heerema, N.; Korn, E.L.; Link, M.; et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J. Clin. Oncol. 1996, 14, 18–24. [Google Scholar] [CrossRef]
- Moricke, A.; Zimmermann, M.; Valsecchi, M.G.; Stanulla, M.; Biondi, A.; Mann, G.; Locatelli, F.; Cazzaniga, G.; Niggli, F.; Arico, M.; et al. Dexamethasone vs prednisone in induction treatment of pediatric ALL: Results of the randomized trial AIEOP-BFM ALL 2000. Blood 2016, 127, 2101–2112. [Google Scholar] [CrossRef]
- Maloney, K.W.; Devidas, M.; Wang, C.; Mattano, L.A.; Friedmann, A.M.; Buckley, P.; Borowitz, M.J.; Carroll, A.J.; Gastier-Foster, J.M.; Heerema, N.A.; et al. Outcome in Children With Standard-Risk B-Cell Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0331. J. Clin. Oncol. 2020, 38, 602–612. [Google Scholar] [CrossRef]
- Parihar, M.; Singh, M.K.; Islam, R.; Saha, D.; Mishra, D.K.; Saha, V.; Krishnan, S. A triple-probe FISH screening strategy for risk-stratified therapy of acute lymphoblastic leukaemia in low-resource settings. Pediatr. Blood Cancer 2018, 65, e27366. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Rana, S.; Sreedharanunni, S.; Gautam, A.; Sachdeva, M.U.S.; Naseem, S.; Varma, N.; Jain, R.; Bansal, D.; Trehan, A. An Evaluation of a Fluorescence In Situ Hybridization Strategy Using Air-dried Blood and Bone-marrow Smears in the Risk Stratification of Pediatric B-Lineage Acute Lymphoblastic Leukemia in Resource-limited Settings. J. Pediatr. Hematol. Oncol. 2021, 43, e481–e485. [Google Scholar] [CrossRef]
- O’Connor, D.; Moorman, A.V.; Wade, R.; Hancock, J.; Tan, R.M.; Bartram, J.; Moppett, J.; Schwab, C.; Patrick, K.; Harrison, C.J.; et al. Use of Minimal Residual Disease Assessment to Redefine Induction Failure in Pediatric Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 660–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coustan-Smith, E.; Ribeiro, R.C.; Stow, P.; Zhou, Y.; Pui, C.H.; Rivera, G.K.; Pedrosa, F.; Campana, D. A simplified flow cytometric assay identifies children with acute lymphoblastic leukemia who have a superior clinical outcome. Blood 2006, 108, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Vinhas, E.; Lucena-Silva, N.; Pedrosa, F. Implementation of a simplified flow cytometric assays for minimal residual disease monitoring in childhood acute lymphoblastic leukemia. Cytometry B Clin. Cytom. 2018, 94, 94–99. [Google Scholar] [CrossRef]
- Sidhom, I.; Shaaban, K.; Youssef, S.H.; Ali, N.; Gohar, S.; Rashed, W.M.; Mehanna, M.; Salem, S.; Soliman, S.; Yassin, D.; et al. Reduced-intensity therapy for pediatric lymphoblastic leukemia: Impact of residual disease early in remission induction. Blood 2021, 137, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.S.; Dunsmore, K.P.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children’s Oncology Group AALL0434 Methotrexate Randomization. J. Clin. Oncol. 2018, 36, 2926–2934. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Campana, D.; Pei, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manabe, A.; Tsuchida, M.; Hanada, R.; Ikuta, K.; Toyoda, Y.; Okimoto, Y.; Ishimoto, K.; Okawa, H.; Ohara, A.; Kaneko, T.; et al. Delay of the diagnostic lumbar puncture and intrathecal chemotherapy in children with acute lymphoblastic leukemia who undergo routine corticosteroid testing: Tokyo Children’s Cancer Study Group study L89-12. J. Clin. Oncol. 2001, 19, 3182–3187. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.C.; Liang, D.C.; Hou, J.Y.; Jaing, T.H.; Lin, D.T.; Yang, C.P.; Peng, C.T.; Hung, I.J.; Lin, K.H.; Hsiao, C.C.; et al. Treatment of childhood acute lymphoblastic leukemia with delayed first intrathecal therapy and omission of prophylactic cranial irradiation: Results of the TPOG-ALL-2002 study. Cancer 2018, 124, 4538–4547. [Google Scholar] [CrossRef]
- Teuffel, O.; Kuster, S.P.; Hunger, S.P.; Conter, V.; Hitzler, J.; Ethier, M.C.; Shah, P.S.; Beyene, J.; Sung, L. Dexamethasone versus prednisone for induction therapy in childhood acute lymphoblastic leukemia: A systematic review and meta-analysis. Leukemia 2011, 25, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, S.; Manabe, A.; Ohara, A.; Kumagai, M.; Saito, T.; Okimoto, Y.; Kamijo, T.; Isoyama, K.; Kajiwara, M.; Sotomatsu, M.; et al. No advantage of dexamethasone over prednisolone for the outcome of standard- and intermediate-risk childhood acute lymphoblastic leukemia in the Tokyo Children’s Cancer Study Group L95-14 protocol. J. Clin. Oncol. 2005, 23, 6489–6498. [Google Scholar] [CrossRef]
- Hurwitz, C.A.; Silverman, L.B.; Schorin, M.A.; Clavell, L.A.; Dalton, V.K.; Glick, K.M.; Gelber, R.D.; Sallan, S.E. Substituting dexamethasone for prednisone complicates remission induction in children with acute lymphoblastic leukemia. Cancer 2000, 88, 1964–1969. [Google Scholar] [CrossRef]
- Mitchell, C.D.; Richards, S.M.; Kinsey, S.E.; Lilleyman, J.; Vora, A.; Eden, T.O.; Medical Research Council Childhood Leukaemia Working Party. Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: Results of the UK Medical Research Council ALL97 randomized trial. Br. J. Haematol. 2005, 129, 734–745. [Google Scholar] [CrossRef]
- Oh, B.L.Z.; Lee, S.H.R.; Foo, K.M.; Chiew, K.H.; Seeto, Z.Z.L.; Chen, Z.W.; Neoh, C.C.C.; Liew, G.S.M.; Eng, J.J.; Lam, J.C.M.; et al. Successful toxicity reduction during delayed intensification in the non-high-risk arm of Malaysia-Singapore Acute Lymphoblastic Leukaemia 2010 study. Eur. J. Cancer 2021, 142, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Merryman, R.; Stevenson, K.E.; Gostic, W.J., 2nd; Neuberg, D.; O’Brien, J.; Sallan, S.E.; Silverman, L.B. Asparaginase-associated myelosuppression and effects on dosing of other chemotherapeutic agents in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2012, 59, 925–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakta, N.; Liu, Q.; Ness, K.K.; Baassiri, M.; Eissa, H.; Yeo, F.; Chemaitilly, W.; Ehrhardt, M.J.; Bass, J.; Bishop, M.W.; et al. The cumulative burden of surviving childhood cancer: An initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet 2017, 390, 2569–2582. [Google Scholar] [CrossRef]
- Suh, E.; Stratton, K.L.; Leisenring, W.M.; Nathan, P.C.; Ford, J.S.; Freyer, D.R.; McNeer, J.L.; Stock, W.; Stovall, M.; Krull, K.R.; et al. Late mortality and chronic health conditions in long-term survivors of early-adolescent and young adult cancers: A retrospective cohort analysis from the Childhood Cancer Survivor Study. Lancet Oncol. 2020, 21, 421–435. [Google Scholar] [CrossRef]
- Tubergen, D.G.; Gilchrist, G.S.; O’Brien, R.T.; Coccia, P.F.; Sather, H.N.; Waskerwitz, M.J.; Hammond, G.D. Improved outcome with delayed intensification for children with acute lymphoblastic leukemia and intermediate presenting features: A Childrens Cancer Group phase III trial. J. Clin. Oncol. 1993, 11, 527–537. [Google Scholar] [CrossRef]
- Khera, S.; Kapoor, R.; Pramanik, S.K. Solitary serum methotrexate level 36 hours post high-dose methotrexate: A safe, efficacious, and cost-effective strategy to monitor methotrexate toxicities in childhood leukemia in resource-limited centers. Pediatr. Blood Cancer 2020, 67, e28387. [Google Scholar] [CrossRef]
- Dhingra, H.; Kalra, M.; Mahajan, A. Safe administration of high-dose methotrexate with minimal drug level monitoring: Experience from a center in north India. Pediatr. Blood Cancer 2020, 67, e28394. [Google Scholar] [CrossRef]
- Larsen, E.C.; Devidas, M.; Chen, S.; Salzer, W.L.; Raetz, E.A.; Loh, M.L.; Mattano, L.A., Jr.; Cole, C.; Eicher, A.; Haugan, M.; et al. Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group Study AALL0232. J. Clin. Oncol. 2016, 34, 2380–2388. [Google Scholar] [CrossRef]
- Matloub, Y.; Bostrom, B.C.; Hunger, S.P.; Stork, L.C.; Angiolillo, A.; Sather, H.; La, M.; Gastier-Foster, J.M.; Heerema, N.A.; Sailer, S.; et al. Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Blood 2011, 118, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Pei, D.; Wolf, J.; Howard, S.C.; Hayden, R.T.; Go, M.; Varechtchouk, O.; Hahn, T.; Buaboonnam, J.; Metzger, M.L.; et al. Infection-related complications during treatment for childhood acute lymphoblastic leukemia. Ann. Oncol. 2017, 28, 386–392. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.; Bate, J.; Wade, R.; Clack, R.; Dhir, S.; Hough, R.; Vora, A.; Goulden, N.; Samarasinghe, S. Infection-related mortality in children with acute lymphoblastic leukemia: An analysis of infectious deaths on UKALL2003. Blood 2014, 124, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Vora, A. Childhood Acute Lymphoblastic Leukemia, Chapter: Developing World Perspective; Springer: Cham, Switzerland, 2017; pp. 323–336. [Google Scholar] [CrossRef]
- Schmiegelow, K. Maintenance therapy of childhood acute lymphoblastic leukemia: Do all roads lead to Rome? Pediatr. Blood Cancer 2020, 67, e28418. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duration and intensity of maintenance chemotherapy in acute lymphoblastic leukaemia: Overview of 42 trials involving 12,000 randomised children. Lancet 1996, 347, 1783–1788. [CrossRef]
- Teachey, D.T.; Hunger, S.P.; Loh, M.L. Optimizing therapy in the modern age: Differences in length of maintenance therapy in acute lymphoblastic leukemia. Blood 2021, 137, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.C.; Yang, C.P.; Lin, D.T.; Hung, I.J.; Lin, K.H.; Chen, J.S.; Hsiao, C.C.; Chang, T.T.; Peng, C.T.; Lin, M.T.; et al. Long-term results of Taiwan Pediatric Oncology Group studies 1997 and 2002 for childhood acute lymphoblastic leukemia. Leukemia 2010, 24, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Ishimaru, S.; Seki, M.; Yoshida, K.; Shiraishi, Y.; Chiba, K.; Kakiuchi, N.; Sato, Y.; Ueno, H.; Tanaka, H.; et al. Long-term outcome of 6-month maintenance chemotherapy for acute lymphoblastic leukemia in children. Leukemia 2017, 31, 580–584. [Google Scholar] [CrossRef]
- Toyoda, Y.; Manabe, A.; Tsuchida, M.; Hanada, R.; Ikuta, K.; Okimoto, Y.; Ohara, A.; Ohkawa, Y.; Mori, T.; Ishimoto, K.; et al. Six months of maintenance chemotherapy after intensified treatment for acute lymphoblastic leukemia of childhood. J. Clin. Oncol. 2000, 18, 1508–1516. [Google Scholar] [CrossRef]
- Moricke, A.; Zimmermann, M.; Reiter, A.; Henze, G.; Schrauder, A.; Gadner, H.; Ludwig, W.D.; Ritter, J.; Harbott, J.; Mann, G.; et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 2010, 24, 265–284. [Google Scholar] [CrossRef] [Green Version]
- Brandalise, S.R.; Pinheiro, V.R.; Aguiar, S.S.; Matsuda, E.I.; Otubo, R.; Yunes, J.A.; Pereira, W.V.; Carvalho, E.G.; Cristofani, L.M.; Souza, M.S.; et al. Benefits of the intermittent use of 6-mercaptopurine and methotrexate in maintenance treatment for low-risk acute lymphoblastic leukemia in children: Randomized trial from the Brazilian Childhood Cooperative Group-protocol ALL-99. J. Clin. Oncol. 2010, 28, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Eden, T.; Pieters, R.; Richards, S.; Childhood Acute Lymphoblastic Leukaemia Collaborative Group. Systematic review of the addition of vincristine plus steroid pulses in maintenance treatment for childhood acute lymphoblastic leukaemia—An individual patient data meta-analysis involving 5659 children. Br. J. Haematol 2010, 149, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Conter, V.; Valsecchi, M.G.; Silvestri, D.; Campbell, M.; Dibar, E.; Magyarosy, E.; Gadner, H.; Stary, J.; Benoit, Y.; Zimmermann, M.; et al. Pulses of vincristine and dexamethasone in addition to intensive chemotherapy for children with intermediate-risk acute lymphoblastic leukaemia: A multicentre randomised trial. Lancet 2007, 369, 123–131. [Google Scholar] [CrossRef]
- Yang, W.; Cai, J.; Shen, S.; Gao, J.; Yu, J.; Hu, S.; Jiang, H.; Fang, Y.; Liang, C.; Ju, X.; et al. Pulse therapy with vincristine and dexamethasone for childhood acute lymphoblastic leukaemia (CCCG-ALL-2015): An open-label, multicentre, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2021, 29, 1322–1332. [Google Scholar] [CrossRef]
- De Moerloose, B.; Suciu, S.; Bertrand, Y.; Mazingue, F.; Robert, A.; Uyttebroeck, A.; Yakouben, K.; Ferster, A.; Margueritte, G.; Lutz, P.; et al. Improved outcome with pulses of vincristine and corticosteroids in continuation therapy of children with average risk acute lymphoblastic leukemia (ALL) and lymphoblastic non-Hodgkin lymphoma (NHL): Report of the EORTC randomized phase 3 trial 58,951. Blood 2010, 116, 36–44. [Google Scholar] [CrossRef]
- Angiolillo, A.L.; Schore, R.J.; Kairalla, J.A.; Devidas, M.; Rabin, K.R.; Zweidler-McKay, P.; Borowitz, M.J.; Wood, B.; Carroll, A.J.; Heerema, N.A.; et al. Excellent Outcomes With Reduced Frequency of Vincristine and Dexamethasone Pulses in Standard-Risk B-Lymphoblastic Leukemia: Results From Children’s Oncology Group AALL0932. J. Clin. Oncol. 2021, 39, 1437–1447. [Google Scholar] [CrossRef]
- Jeha, S.; Choi, J.; Roberts, K.G.; Pei, D.; Coustan-Smith, E.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; Gruber, T.A.; Raimondi, S.C.; et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease-directed therapy. Blood Cancer Discov. 2021, 2, 326–337. [Google Scholar] [CrossRef]
- Relling, M.V.; Hancock, M.L.; Rivera, G.K.; Sandlund, J.T.; Ribeiro, R.C.; Krynetski, E.Y.; Pui, C.H.; Evans, W.E. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J. Natl. Cancer Inst. 1999, 91, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.E.; Hon, Y.Y.; Bomgaars, L.; Coutre, S.; Holdsworth, M.; Janco, R.; Kalwinsky, D.; Keller, F.; Khatib, Z.; Margolin, J.; et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J. Clin. Oncol. 2001, 19, 2293–2301. [Google Scholar] [CrossRef]
- Cai, J.-P.; Ishibashi, T.; Takagi, Y.; Hayakawa, H.; Sekiguchi, M. Mouse MTH2 protein which prevents mutations caused by 8-oxoguanine nucleotides. Biochem. Biophys. Res. Commun. 2003, 305, 1073–1077. [Google Scholar] [CrossRef]
- Takagi, Y.; Setoyama, D.; Ito, R.; Kamiya, H.; Yamagata, Y.; Sekiguchi, M. Human MTH3 (NUDT18) protein hydrolyzes oxidized forms of guanosine and deoxyguanosine diphosphates: Comparison with MTH1 and MTH2. J. Biol. Chem. 2012, 287, 21541–21549. [Google Scholar] [CrossRef] [Green Version]
- Saiz-Rodriguez, M.; Ochoa, D.; Belmonte, C.; Roman, M.; Martinez-Ingelmo, C.; Ortega-Ruiz, L.; Sarmiento-Iglesias, C.; Herrador, C.; Abad-Santos, F. Influence of thiopurine S-methyltransferase polymorphisms in mercaptopurine pharmacokinetics in healthy volunteers. Basic Clin. Pharmacol. Toxicol. 2019, 124, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Landier, W.; Yang, W.; Liu, C.; Hageman, L.; Cheng, C.; Pei, D.; Chen, Y.; Crews, K.R.; Kornegay, N.; et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J. Clin. Oncol. 2015, 33, 1235–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relling, M.V.; Schwab, M.; Whirl-Carrillo, M.; Suarez-Kurtz, G.; Pui, C.H.; Stein, C.M.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018 Update. Clin. Pharmacol. Ther. 2019, 105, 1095–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Low-Income Countries (LIC) Setting | Middle-Income Countries (MIC) Setting | High-Income Countries (HIC) Setting | |
|---|---|---|---|
| Low-Risk features | NCI SR CNS I No mediastinal mass | NCI SR CNS I Flow T vs. B DNA index Cytogenetics/FISH OFT screening | NCI SR CNS I Flow T vs. B Cytogenetics/FISH OFT screening/RNA-Seq |
| Early response PB | D8 blast < 1 × 109/L | Day 8 blast < 1 × 109/L | Day 8 blast or PB flow MRD |
| Early response BM | Day 15 M1/2, EOI M3 | Day 15 M1/2, EOI M3 Flow-MRD-lite | Flow MRD or PCR MRD at EOI, EOC |
| Protocol | One protocol | SR/HR | SR/IR/HR |
| B-ALL SR | VCR-Dexa | 3-drug Dexa-based | 3-drug Dexa-based |
| T-ALL and B-HR | VCR-Dexa | 3-drug Dexa-based | 4-drug Pred-based |
| Central Nervous System (CNS) directed Rx | Cranial RT/IT MTX | IT MTX/Cranial RT | IT MTX/HDMTX |
| Delayed intensification | VCR-Dex-Doxo | VCR-Dex-L-asp + CTX-araC-MP | Protocol II |
| Maintenance Therapy | 4-weekly VCR/Dex pulse | 8-weekly VCR/Dex pulse | 12-weekly VCR/Dex pulse |
| TPMT and NUDT15 | In sensitive patients | In sensitive patients or routinely | Routine |
| Clinical Trial Examples | LMIC Examples: RELLA05 [14] ICiCLe ALL 14 [15] | ALL IC-BFM 2002 [16] CCLG-ALL 2008 [17] MS 2003 [18]/ 2010 [19] | AIEOP-BFM ALL 2000 [20] CoALL 07-03 [21] COG AALL 0932 [22]/0331 [23] DCOG ALL10 [24] JACLS ALL-02 [25] St Jude Total XVI [26] UKALL 2003 [27] |
| B-ALL | T-ALL | Age Criteria | Total White Cell Count at Diagnosis | Central Nervous System (CNS) Status | Specific Cytogenetic Inclusion Criteria | Prednisolone Response | Treatment Responses | Cumulative Cyclophosphamide Dosing (mg/m2) | Cumulative Anthracycline Dosing (mg/m2) | Cumulative L-Asparaginase × 1000 units/m2 Dosing ^^ | % of Study Population Treated in the Lowest-Risk Arms | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D8 | D15 | End of Induction | Later Timepoints | ||||||||||||
| AIEOP-BFM ALL 2000 [20,36] | Y | Y | 1–17 | Any | Any | Y | - | - | PCR MRD NEG | Day 78 PCR MRD NEG | 3000 | 240 | 80 | 28 | |
| ALL IC-BFM 2002 [16] | Y | Y | 1–6 | <20,000/μL | Any | Y | - | M1/M2 | - | - | 3000 | 180 | Standard: 80 Expt: 120 | 31 | |
| CCLG-ALL 2008 [17] | Y | - | 1–10 | <50,000/μL | No CNS 3 | Y | - | M1/M2 | PCR MRD NEG or Flow MRD < 0.01% | Week 12 PCR MRD NEG or Flow MRD < 0.01% | 2000 | 125 | 80 | 39 | |
| CoAll 07-03 [21] LR-R | Y | - | <10 | <25,000/μL | Any | - | - | MRD < 0.01% | ⍬ MRD < 0.01% | - | 900 | 120 | 125 | 13 | |
| COG AALL 0331 [23,37] LRS | Y | - | 1.01–9.99 | <50,000/μL | CNS 1 only | Triple trisomies of chromosomes 4, 10, and 17 Or ETV6-RUNX1 | - | M1 * | M1 * | ⍬ MRD < 0.01% | - | 1000 | 75 | 80 | 35 |
| COG AALL 0932 [22] LR-C | Y | - | 1.01–9.99 | <50,000/μL | CNS 1 only | Triple trisomies of chromosomes 4 and 10 Or ETV6-RUNX1 | - | PB MRD < 0.01% | - | ⍬ MRD < 0.01% | - | 1000 | 75 | 80 | 3.5 |
| COG AALL 0932 [22] LR-M | Y | - | 1.01–9.99 | <50,000/μL | CNS 1 only | Triple trisomies of chromosomes 4 and 10 Or ETV6-RUNX1 | - | PB MRD < 0.01% | - | ⍬ MRD < 0.01% | - | 0 | 0 | 40 | 3.5 |
| DCOG 10 [24] | Y | Y | 1–18 | Any | CNS 1 only | Y | - | - | PCR MRD NEG | Day 80 PCR MRD NEG | 2000 | 120 | 80 | 26 | |
| JACLS-ALL-02 [25] | Y | - | 1–9 | <10,000/μL | No CNS 3 | Y | - | M1/M2 | M1 | - | 1500 | ^ 90 | 72 | 40 | |
| MS 2003 [18] | Y | Y | 0–18 | Any | CNS 1 only | Y | - | - | PCR MRD NEG | Week 8 PCR MRD NEG | 3000 | 120 | 140 | 31 | |
| MS 2010 [19] | Y | - | 1–10 | Any | CNS 1 only | Y | - | - | PCR MRD NEG | Week 8 PCR MRD NEG | 3000 | 0 | 167.5 | 40 | |
| RELLA05 [14] | Y | - | 1–9 | <50,000/μL | CNS1/CNS2 Traumatic LP | DNA index of ≥1.16 or ETV6-RUNX1 | - | - | ** MRD <0.01% | - | - | 0 | 50 | 160 | 22 |
| St Jude Total XVI [26] | Y | - | 1–10 | <50,000/μL | Any | DNA index of >/=1.16 or ETV6-RUNX1 | - | - | MRD < 1% | - | D46 MRD <0.01% | 1000 | 110 | 208 | 43 |
| UKALL 2003 [27] | Y | Y | 1–10 | <50,000/μL | Any | - | - | M1/M2 | + MRD < 0.01% | - | 2000 | 150 | 64 | 33 | |
| Cumulative Int./High Dose MTX Dose (g/m2) | Number of Int./High MTX Doses | Dose of Int./High Dose MTX (g/m2) | Duration of 6MP (weeks) | Daily 6MP Dose (mg/m2) | Total 6MP Dose (mg/m2) | Number of Intrathecal Chemotherapy Injections | Other Drugs | |
|---|---|---|---|---|---|---|---|---|
| AIEOP-BFM ALL 2000 [20,36] | 20 | 4 | 5 | 8 | 25 | 1400 | 4 | - |
| ALL IC-BFM 2002 [16] | 8 | 4 | 2 | 8 | 25 | 1400 | 4 | - |
| CCLG-ALL 2008 [17] | 8 | 4 | 2 | 8 | 25 | 1400 | 4 | - |
| CoAll 07-03 [21] | 3 | 3 | 1 | 2 | 100 | 1400 | 3 | Teniposide 165 mg/m2 + Thioguanine (100 mg/m2/day) for 1 week + L-asp 45,000 units/m2 + PEG-Asp 5000 units/m2 + Cytarabine 12,300 mg/m2 |
| COG AALL 0932 [22] (LR-M) | 6 | 6 | 1 | 19 | 50 | 6650 | 6 | Dexamethasone 84 mg/m2 + Vincristine 6 mg/m2 |
| DCOG 10 [24] | 20 | 4 | 5 | 8 | 25 | 1400 | 4 | - |
| JACLS-ALL–02 [25] Arm A | 6 | 2 | 3 | 1 | 50 | 350 | 4 | Cyclophosphamide 1.5 g/m2 + Cytarabine 750 mg/m2 |
| JACLS-ALL–02 [25] Arm B | 6 | 2 | 3 | - | - | - | 4 | Dexamethasone 50 mg/m2 + Cyclophosphamide 1 g/m2 + Cytarabine 500 mg/m2 |
| MS 2003 [18] | 8 | 4 | 2 | 8 | 25 | 1400 | 4 | - |
| MS 2010 [19] | 10 | 4 | 2.5 | 8 | 25 | 1400 | 4 | Interspersed Cyclophosphamide blocks |
| RELLA05 [14] | 10 | 4 | 2.5 | 8 | 50 | 2800 | 4 | - |
| St Jude Total XVI [26] | 10 | 4 | 2.5 | 8 | 50 | 2800 | 4 | - |
| Total Dose of Dose Escalating MTX (g/m2) | No. of Dose Escalating MTX | Oral MTX (mg/m2) | Duration of 6MP (weeks) | Daily 6MP Dose (mg/m2) | Total 6MP Dose (mg/m2) | Number of IT Chemotherapy Injections | Other Drugs | |
| COG AALL 0932 [22] (LR-C) | 1 | 5 | - | 4 | 75 | 2100 | 4 | Vincristine 9 mg/m2 |
| COG AALL 0331 [23,37] | 1 | 5 | - | 4 | 75 | 2100 | 4 | Vincristine 9 mg/m2 (L-asp intensification arm: 4 additional doses of PEG-Asp (10,000 units/m2) |
| UKALL 2003 [27] | - | - | 140 | 4 | 75 | 2100 | 4 | Vincristine 4.5 mg/m2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, B.L.Z.; Lee, S.H.R.; Yeoh, A.E.J. Curing the Curable: Managing Low-Risk Acute Lymphoblastic Leukemia in Resource Limited Countries. J. Clin. Med. 2021, 10, 4728. https://doi.org/10.3390/jcm10204728
Oh BLZ, Lee SHR, Yeoh AEJ. Curing the Curable: Managing Low-Risk Acute Lymphoblastic Leukemia in Resource Limited Countries. Journal of Clinical Medicine. 2021; 10(20):4728. https://doi.org/10.3390/jcm10204728
Chicago/Turabian StyleOh, Bernice L. Z., Shawn H. R. Lee, and Allen E. J. Yeoh. 2021. "Curing the Curable: Managing Low-Risk Acute Lymphoblastic Leukemia in Resource Limited Countries" Journal of Clinical Medicine 10, no. 20: 4728. https://doi.org/10.3390/jcm10204728