
Effects of Recombinant Human Growth Hormone Treatment, Depending on the Therapy Start in Different Nutritional Phases in Paediatric Patients with Prader–Willi Syndrome: A Polish Multicentre Study

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Material and Methods
3. Data Analysis
4. Results
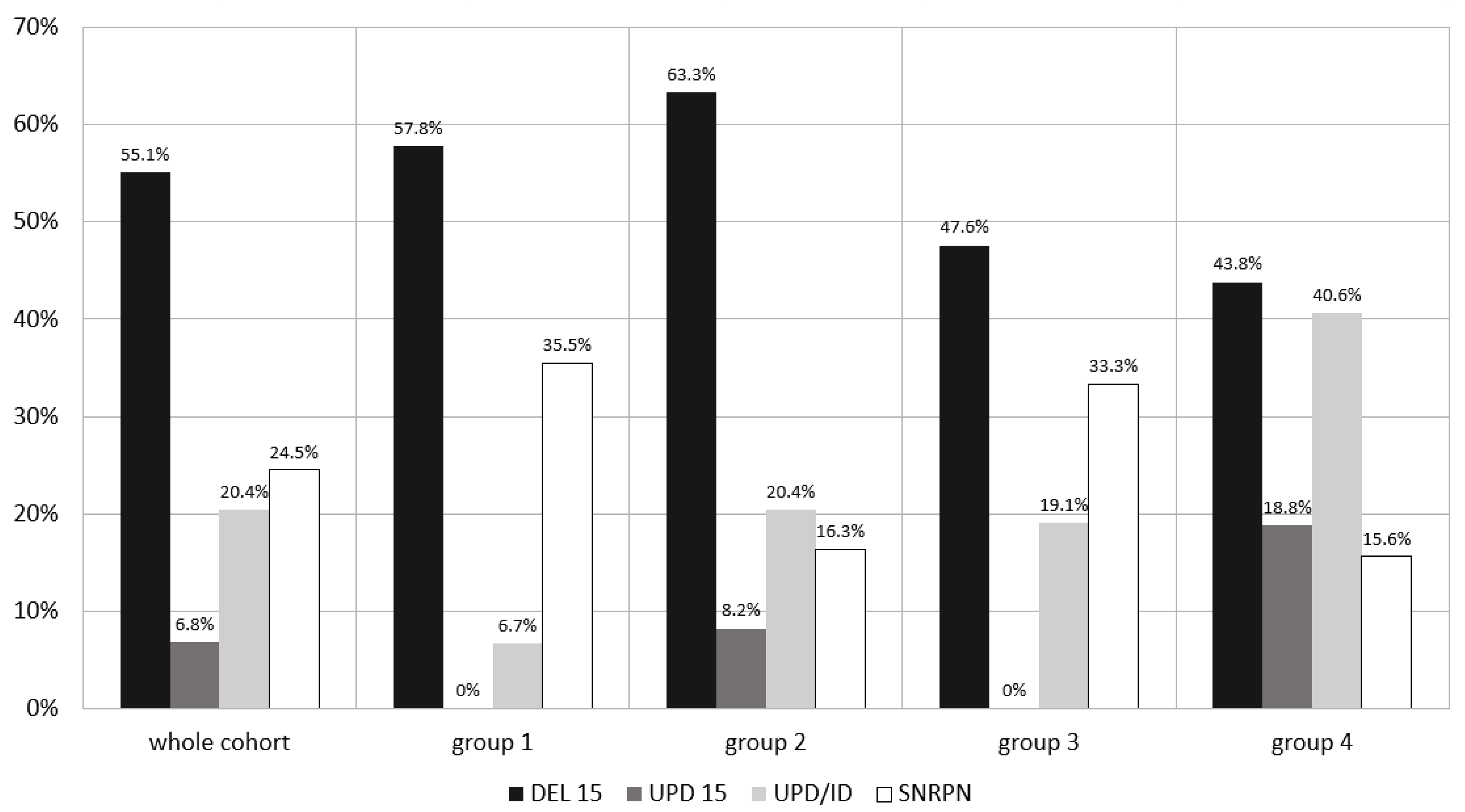
4.1. Molecular Type of Diagnosis and Patients’ Clinical Characteristics
4.2. Age of rhGH Start and rhGH Dosage
4.3. Effects of the Therapy in the Whole Cohort of Patients and Comparison between the Separate Groups
4.4. Effects of the Therapy—Comparison of Four Time Points of Follow-Up
4.5. Metabolic Parameters Evaluation
4.6. Bone Maturation Assessment
4.7. Adrenarche and Central Puberty Evaluation
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sävendahl, L.; Polak, M.; Backeljauw, P.; Blair, J.; Miller, B.S.; Rohrer, T.R.; Pietropoli, A.; Ostrow, V.; Ross, J. Treatment of children with GH in the United States and Europe: Long-term follow-up from NordiNet IOS and ANSWER Program. J. Clin. Endocrinol. Metab. 2019, 104, 4730–4742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wit, J.M.; Ranke, M.B.; Albertsson-Wikland, K.; Carrascosa, A.; Rosenfeld, R.G.; van Buuren, S.; Kristrom, B.; Schoenau, E.; Audi, L.; Hokken-Koelega, A.C.S.; et al. Personalized approach to growth hormone treatment: Clinical use of growth prediction models. Horm. Res. Paediatr. 2013, 79, 257–270. [Google Scholar] [CrossRef]
- Goldstone, A.P.; Holland, A.J.; Hauffa, B.P.; Hokken-Koelega, A.C.; Tauber, M. On behalf of speakers and contributors at the Second Expert Meeting of the Comprehensive Care of Patients with PWS. Recommendations for the diagnosis and management of Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4183–4197. [Google Scholar] [CrossRef] [PubMed]
- Deal, C.L.; Tony, M.; Hoybye, C.; Allen, D.B.; Tauber, M.; Christiansen, J.S. Growth hormone in Prader-Willi syndrome clinical care guidelines workshop participants. Growth Hormone Research Society workshop summary. Consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2013, 98, E1072–E1087. [Google Scholar] [CrossRef]
- De Lind van Wijngaarden, R.F.A.; Siemensma, E.P.C.; Festen, D.A.M.; Otten, B.J.; van Mil, E.G.; Rotteveel, J.; Odink, R.J.H.; Bindels-de Heus, G.C.B.K.; van Leeuwen, M.; Haring, D.A.J.P.; et al. Efficacy and safety of long-term continuous growth hormone treatment in children with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2009, 94, 4205–4215. [Google Scholar] [CrossRef] [PubMed]
- Carrel, A.L.; Myers, S.E.; Whitman, B.Y.; Eickhoff, J.; Allen, D.B. Long-term growth hormone therapy changes the natural history of body composition and motor function in children with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 1131–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, N.E.; Kuppens, R.J.; Siemensma, E.P.C.; Tummers-de Lind van Wijngaarden, R.F.A.; Festen, D.A.M.; Bindels-de Heus, G.C.B.; Bocca, G.; Haring, D.A.J.P.; Hoorweg-Nijman, J.J.G.; Houdijk, E.C.A.M.; et al. Eight years of growth hormone treatment in children with Prader-Willi syndrome: Maintaining the positive effects. J. Clin. Endocrinol. Metab. 2013, 98, 4013–4022. [Google Scholar] [CrossRef] [Green Version]
- Bakker, N.E.; Siemensma, E.P.C.; van Rijn, M.; Festen, D.; Hokken-Koelega, A.C.S. Beneficial effect of growth hormone treatment on health-related quality of life in children with Prader-Willi syndrome: A randomized controlled trial and longitudinal study. Horm. Res. Paediatr. 2015, 84, 4. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, F.; Hallberg, M. Growth hormone and cognitive function. Nat. Rev. Endocrinol. 2013, 9, 357–365. [Google Scholar] [CrossRef]
- Donze, S.H.; Damen, L.; Mahabier, E.F.; Hokken-Koelega, A.C.S. Cognitive functioning in children with Prader–Willi syndrome during 8 years of growth hormone treatment. Eur. J. Endocrinol. 2020, 182, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Deal, C.L.; Rogol, A.D. Growth hormone treatments and cognitive functioning in children with Prader–Willi syndrome. Eur. J. Endocrinol. 2020, 182, C21–C25. [Google Scholar] [CrossRef]
- Donze, S.H.; Damen, L.; Mahabier, E.F.; Hokken-Koelega, A.C.S. Improved mental and motor development during 3 years of GH treatment in very young children with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 3714–3719. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Zheng, Z.; Yang, Y.; Bai, X.; Yang, H.; Zhu, H.; Pan, H.; Chen, S. Effects of growth hormone on cognitive, motor, and behavioral development in Prader-Willi syndrome children: A meta-analysis of randomized controlled trials. Endocrine 2021, 71, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [Green Version]
- Duis, J.; van Wattum, P.J.; Scheimann, A.; Salehi, P.; Brokamp, E.; Fairbrother, L.; Childers, A.; Shelton, A.R.; Bingham, N.C.; Shoemaker, A.H.; et al. A multidisciplinary approach to the clinical management of Prader–Willi syndrome. Mol. Genet. Genom. Med. 2019, 7, e514. [Google Scholar] [CrossRef] [PubMed]
- Emerick, J.E.; Vogt, K.S. Endocrine manifestations and management of Prader-Willi syndrome. Int. J. Pediatr. Endocrinol. 2013, 2013, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.L. Approach to the child with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2012, 97, 3837–3844. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.; Franco, R.R. Prader-Willi syndrome: Endocrine manifestations and management. Arch. Endocrinol. Metab. 2020, 64, 3. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.P.; Butler, G.E. Prader-Willi syndrome: Clinical features and management. Paediatr. Child. Health 2009, 19, 473–478. [Google Scholar] [CrossRef]
- Goldstone, A.P. Prader-Willi syndrome: Advances in genetics, pathophysiology and treatment. Trends Endocrinol. Metab. 2004, 15, 12–20. [Google Scholar] [CrossRef]
- Butler, M.G.; Duis, J. Chromosome 15 imprinting disorders: Genetic laboratory methodology and approaches. Front. Pediatr. 2020, 8, 154. [Google Scholar] [CrossRef]
- Irizarry, K.A.; Miller, M.; Freemark, M.; Haqq, A.M. Prader Willi Syndrome: Genetics, metabolomics, hormonal function, and new approaches to therapy. Adv. Pediatr. 2016, 63, 47–77. [Google Scholar] [CrossRef] [Green Version]
- Kanber, D.; Giltay, J.; Wieczorek, D.; Zogel, C.; Hochstenbach, R.; Caliebe, A.; Kuechler, A.; Horsthemke, B.; Buiting, K. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader–Willi syndrome. Eur. J. Hum. Genet. 2009, 17, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Tennese, A.A.; Wevrick, R. Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in Magel2-null mice. Endocrinology 2011, 152, 967–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.J.; Gerasimidis, K.; Edwards, C.A.; Shaikh, M.G. Mechanisms of obesity in Prader-Willi syndrome. J. Ped. Obes. 2018, 13, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Crinò, A.; Fintini, D.; Bocchini, S.; Grugni, G. Obesity management in Prader–Willi syndrome: Current perspectives. Diabetes, Metabolic Syndrome and Obesity. Targets Ther. 2018, 11, 579–593. [Google Scholar]
- Gantz, M.G.; Andrews, S.M.; Wheeler, A.C. Food and non-food-related behavior across settings in children with Prader–Willi syndrome. Genes 2020, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Kimonis, V.; Dykens, E.; Gold, J.A.; Miller, J.; Tamura, R.; Driscoll, D.J. Prader–Willi syndrome and early-onset morbid obesity NIH rare disease consortium: A review of natural history study. Am. J. Med. Genet. A 2018, 176, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.; Barone, B.; Morra, S.; Celentano, G.; La Rocca, R.; Capece, M.; Morgera, V.; Turco, C.; Caputo, V.F.; Spena, G.; et al. Hypogonadism in patients with Prader Willi syndrome: A narrative review. Int. J. Mol. Sci. 2021, 22, 1993. [Google Scholar] [CrossRef]
- Gross-Tsur, V.; Hirsch, H.J.; Benarroch, F.; Eldar-Geva, T. The FSH-inhibin axis in Prader-Willi syndrome: Heterogeneity of gonadal dysfunction. Reprod. Biol. Endocrinol. 2012, 10, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, H.J.; Eldar-Geva, T.; Bennaroch, F.; Pollak, Y.; Gross-Tsur, V. Sexual dichotomy of gonadal function in Prader-Willi syndrome from early infancy through the fourth decade. Hum. Reprod. 2015, 30, 2587–2596. [Google Scholar] [CrossRef] [Green Version]
- Lecka-Ambroziak, A.; Wysocka-Mincewicz, M.; Marszałek-Dziuba, K.; Rudzka-Kocjan, A.; Szalecki, M. Premature Adrenarche in Children with Prader-Willi syndrome treated with recombinant human growth hormone seems to not influence the course of central puberty and the efficacy and safety of the therapy. Life 2020, 10, 237. [Google Scholar] [CrossRef]
- Butler, J.V.; Whittington, J.E.; Holland, A.J.; McAllister, C.J.; Goldstone, A.P. The transition between the phenotypes of Prader-Willi syndrome during infancy and early childhood. Dev. Med. Child. Neurol. 2010, 52, e88–e93. [Google Scholar] [CrossRef]
- Miller, J.L.; Lynn, C.H.; Driscoll, D.C.; Goldstone, A.P.; Gold, J.A.; Kimonis, V.; Dykens, E.; Butler, M.G.; Shuster, J.J.; Driscoll, D.J. Nutritional phases in Prader-Willi syndrome. Am. J. Med. Genet. Part A 2011, 155, 1040–1049. [Google Scholar] [CrossRef] [Green Version]
- Lecka-Ambroziak, A.; Wysocka-Mincewicz, M.; Doleżal-Ołtarzewska, K.; Zygmunt-Górska, A.; Żak, T.; Noczyńska, A.; Birkholz-Walerzak, D.; Stawerska, R.; Hilczer, M.; Obara-Moszyńska, M.; et al. Correlation of genotype and perinatal period, time of diagnosis and anthropometric data before commencement of recombinant human growth hormone treatment in Polish patients with Prader–Willi Syndrome. Diagnostics 2021, 11, 798. [Google Scholar] [CrossRef]
- Magill, L.; Laemmer, C.; Woelfle, J.; Fimmers, R.; Gohlke, B. Early start of growth hormone is associated with positive effects on auxology and metabolism in Prader-Willi-syndrome. Orphanet J. Rare Dis. 2020, 15, 283. [Google Scholar] [CrossRef]
- Corripio, R.; Tubau, C.; Calvo, L.; Brun, C.; Capdevila, N.; Larramona, H.; Gabau, E. Safety and effectiveness of growth hormone therapy in infants with Prader-Willi syndrome younger than 2 years: A prospective study. J. Pediatr. Endocrinol. Metab. 2019, 32, 879–884. [Google Scholar] [CrossRef]
- Bakker, N.E.; Lindberg, A.; Heissler, J.; Wollmann, H.A.; Camacho-Hübner, C.; Hokken-Koelega, A.C.; on behalf of the KIGS Steering Committee. Growth hormone treatment in children with Prader-Willi syndrome: Three years of longitudinal data in prepubertal children and adult height data from the KIGS database. J. Clin. Endocrinol. Metab. 2017, 102, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Angulo, M.; Abuzzahab, M.J.; Pietropoli, A.; Ostrow, V.; Kelepouris, N.; Tauber, M. Outcomes in children treated with growth hormone for Prader-Willi syndrome: Data from the ANSWER Program® and NordiNet® International Outcome Study. Int. J. Pediatr. Endocrinol. 2020, 20. [Google Scholar] [CrossRef]
- Passone, C.D.G.B.; Franco, R.R.; Ito, S.S.; Trindade, E.; Polak, M.; Damiani, D.; Bernardo, W.M. Growth hormone treatment in Prader-Willi syndrome patients: Systematic review and meta-analysis. BMJ Paediatr. Open 2020, 4. [Google Scholar] [CrossRef] [PubMed]
- Bridges, N. What is the value of growth hormone therapy in Prader Willi syndrome? Arch. Dis. Child 2014, 99, 166–170. [Google Scholar] [CrossRef]
- Lionti, T.; Reid, S.M.; White, S.M.; Rowell, M.M. A population-based profile of 160 Australians with Prader-Willi syndrome: Trends in diagnosis, birth prevalence and birth characteristics. Am. J. Med. Genet. A 2015, 167A, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Diene, G.; Mimoun, E.; Feigerlova, E.; Caula, S.; Molinas, C.; Grandjean, H.; Tauber, M. French Reference Centre for PWS. Endocrine disorders in children with Prader-Willi Syndrome—Data from 142 children of the French database. Horm. Res. Paediatr. 2010, 74, 121–128. [Google Scholar] [CrossRef]
- Grugni, G.; Marzullo, P. Diagnosis and treatment of GH deficiency in Prader-Willi syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 785–794. [Google Scholar] [CrossRef]
- Feigerlová, E.; Diene, G.; Oliver, I.; Gennero, I.; Salles, J.-P.; Arnaud, C.; Tauber, M. Elevated insulin-like growth factor-I values in children with Prader-Willi Syndrome compared with growth hormone (GH) deficiency children over two years of GH treatment. J. Clin. Endocrinol. Metab. 2010, 95, 4600–4608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, N.E.; van Doorn, J.; Renes, J.S.; Donker, G.H.; Hokken-Koelega, A.C.S. IGF-1 levels, complex formation, and IGF bioactivity in growth hormone-treated children with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2015, 100, 3041–3049. [Google Scholar] [CrossRef] [Green Version]
- Bakker, N.E.; Siemensma, E.P.C.; Koopman, C.; Hokken-Koelega, A.C.S. Dietary energy intake, body composition and resting energy expenditure in prepubertal children with Prader-Willi syndrome before and during growth hormone treatment: A randomized controlled trial. Horm. Res. Paediatr. 2015, 83, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Miller, J.L.; Forster, J.L. Prader-Willi Syndrome—Clinical genetics, diagnosis and treatment approaches: An update. Curr. Pediatr. Rev. 2019, 15, 207–244. [Google Scholar] [CrossRef] [PubMed]
- Scheermeyer, E.; Harris, M.; Hughes, I.; Crock, P.A.; Ambler, G.; Verge, C.F.; Bergman, P.; Werther, G.; Craig, M.E.; Choong, C.S.; et al. Low dose growth hormone treatment in infants and toddlers with Prader-Willi syndrome is comparable to higher dosage regimens. Growth Horm. IGF Res. 2017, 34, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Carrel, A.L.; Myers, S.E.; Whitman, B.Y.; Allen, D.B. Benefits of long-term GH therapy in Prader-Willi syndrome: A 4-year study. J. Clin. Endocrinol. Metab. 2002, 87, 1581–1585. [Google Scholar] [CrossRef]
- Crinò, A.; Grugni, G. Update on diabetes mellitus and glucose metabolism alterations in Prader-Willi syndrome. Curr. Diabetes Rep. 2020, 20, 7. [Google Scholar] [CrossRef]
- Brambilla, P.; Crinò, A.; Bedogni, G.; Bosio, L.; Cappa, M.; Corrias, A.; Delvecchio, M.; Di Candia, S.; Gargantini, L.; Grechi, E. Metabolic syndrome in children with Prader-Willi syndrome: The effect of obesity. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 269–276. [Google Scholar] [CrossRef]
- Fintini, D.; Grugni, G.; Bocchini, S.; Brufani, C.; Di Candia, S.; Corrias, A.; Delvecchio, M.; Salvatoni, A.; Ragusa, L.; Greggio, N.; et al. Disorders of glucose metabolism in Prader-Willi syndrome: Results of a multicenter Italian cohort study. Nutr. Metab. Cardiovasc. Dis. 2016, 26. [Google Scholar] [CrossRef]
- Unanue, N.; Bazaes, R.; Iñiguez, G.; Cortes, F.; Ávila, A.; Mericq, V. Adrenarche in Prader-Willi syndrome appears not related to insulin sensitivity and serum adiponectin. Horm. Res. Paediatr. 2006, 67, 152–158. [Google Scholar] [CrossRef]
- Siemensma, E.; Wijngaarden, R.F.A.D.L.V.; Otten, B.J.; De Jong, F.H.; Hokken-Koelega, A.C.S. Pubarche and serum dehydroepiandrosterone sulphate levels in children with Prader-Willi syndrome. Clin. Endocrinol. 2011, 75, 83–89. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PWS Group | Whole Cohort | Group 1 | Group 2 | Group 3 | Group 4 |
|---|---|---|---|---|---|
| Number of patients | n = 147 | n = 45 (30.61) | n = 49 (33.33) | n = 21 (14.29) | n = 32 (21.77) |
| F/M | 69/78 (46.94/53.06) | 25/20 (55.56/44.44) | 25/24 (51.02/49.98) | 10/11 (47.62/52.38) | 9/23 (28.13/71.87) |
| Age of diagnosis (years) a | 1.67 ± 2.39 0.53 (0.02–12.49) | 0.29 ± 0.29 0.20 (0.04–1.22) | 0.79 ± 0.83 0.44 (0.02–2.85) | 2.98 ± 2.12 3.06 (0.15–7.31) | 4.24 ± 3.40 3.54 (0.02–12.49) |
| DEL 15 | 81 (55.10) | 26 (57.77) | 31 (63.26) | 10 (47.62) | 14 (43.75) |
| UPD 15 | 10 (6.80) | none | 4 (8.16) | none | 6 (18.75) |
| UPD/ID | 30 (20.41) | 3 (6.67) | 10 (20.41) | 4 (19.05) | 13 (40.62) |
| SNRPN | 36 (24.49) | 16 (35.56) | 8 (16.33) | 7 (33.33) | 5 (15.63) |
| Cryptorchidism | 70 (89.74) | 19 (95.00) | 23 (95.83) | 8 (72.73) | 20 (86.96) |
| PA | 22 (14.97) | 3 (6.67) | 8 (16.33) | 7 (33.33) | 4 (12.50) |
| T/E/E-P | 26 (25.49) b | none | 6 (12.25) | 5 (23.81) | 15 (46.88) |
| Secondary hypothyroidism | 51 (34.69) | 14 (31.11) | 14 (28.57) | 12 (57.14) | 11 (34.38) |
| Scoliosis | 34 (23.13) | 3 (6.67) | 15 (30.61) | 4 (19.05) | 12 (37.50) |
| Adenoid hypertrophy | 28 (19.05) | 2 (4.44) | 12 (24.49) | 6 (28.57) | 8 (25.00) |
| Severe behaviour disorders | 17 (11.57) | none | 3 (6.12) | 2 (9.52) | 12 (37.50) |
| Epilepsy | 12 (8.16) | 2 (4.44) | 3 (6.12) | 3 (14.29) | 4 (12.50) |
| rhGH Therapy | Baseline | rhGH1 | rhGH2 | rhGH3 |
|---|---|---|---|---|
| Number of patients (%) | n = 147 | n = 140 (95) | n = 126 (86) | n = 99 (67) |
| Age of patients (years) | 4.50 ± 3.73 3.03 (0.58–17.43) | 5.73 ± 3.89 4.17 (0.89–19.96) | 8.26 ± 4.27 7.12 (1.85–17.52) | 11.06 ± 4.71 10.67 (2.72–20.67) |
| Time of rhGH therapy (years) | NA | 1.21 ± 0.81 1.00 (0.13–3.76) | 3.77 ± 2.17 3.27 (0.60-10.60) | 6.50 ± 2.92 6.17 (1.23–13.67) |
| Height SDS | −2.11 ± 1.50 * −2.16 (−5.45-2.15) | −1.04 ± 1.37 * −1.3 (−3.87–2.39) | −0.6 ± 1.36 * −0.59 (−3.61–2.84) | −0.40 ± 1.46 * −0.64 (−3.88–3.55) |
| GV (cm/year) | NA | 10.41 ± 3.23 * 9.94 (3.83–26.07) | 7.49 ± 2.42 * 7.41 (0.73–13.48) | 5.79 ± 2.71 * 5.78 (0.41–3.55) |
| BMI SDS | 0.41 ± 1.55 0.57 (−3.92–4.24) | 0.42 ± 1.38 0.44 (−2.99–4.10) | 0.6 ± 1.29 * 0.80 (−2.56–4.28) | 0.88 ± 1.30 * 1.12 (−2.60–3.18) |
| Number of patients | n = 142 | n = 140 | n = 119 | n = 92 |
| Age of patients at IGF1 measurement (years) | 4.50 ± 3.76 2.98 (0.58–17.43) | 5.85 ± 3.88 4.18 (0.85–18.74) | 8.56 ± 4.21 8.17 (1.84–19.10) | 11.32 ± 4.45 11.36 (3.3–19.88) |
| Time of rhGH therapy at IGF1 measurement (years) | NA | 1.27 ± 1.00 1.00 (0.17–4.90) | 3.76 ± 2.26 3.21 (0.79–9.84) | 6.43 ± 2.58 6.12 (2.08–12.73) |
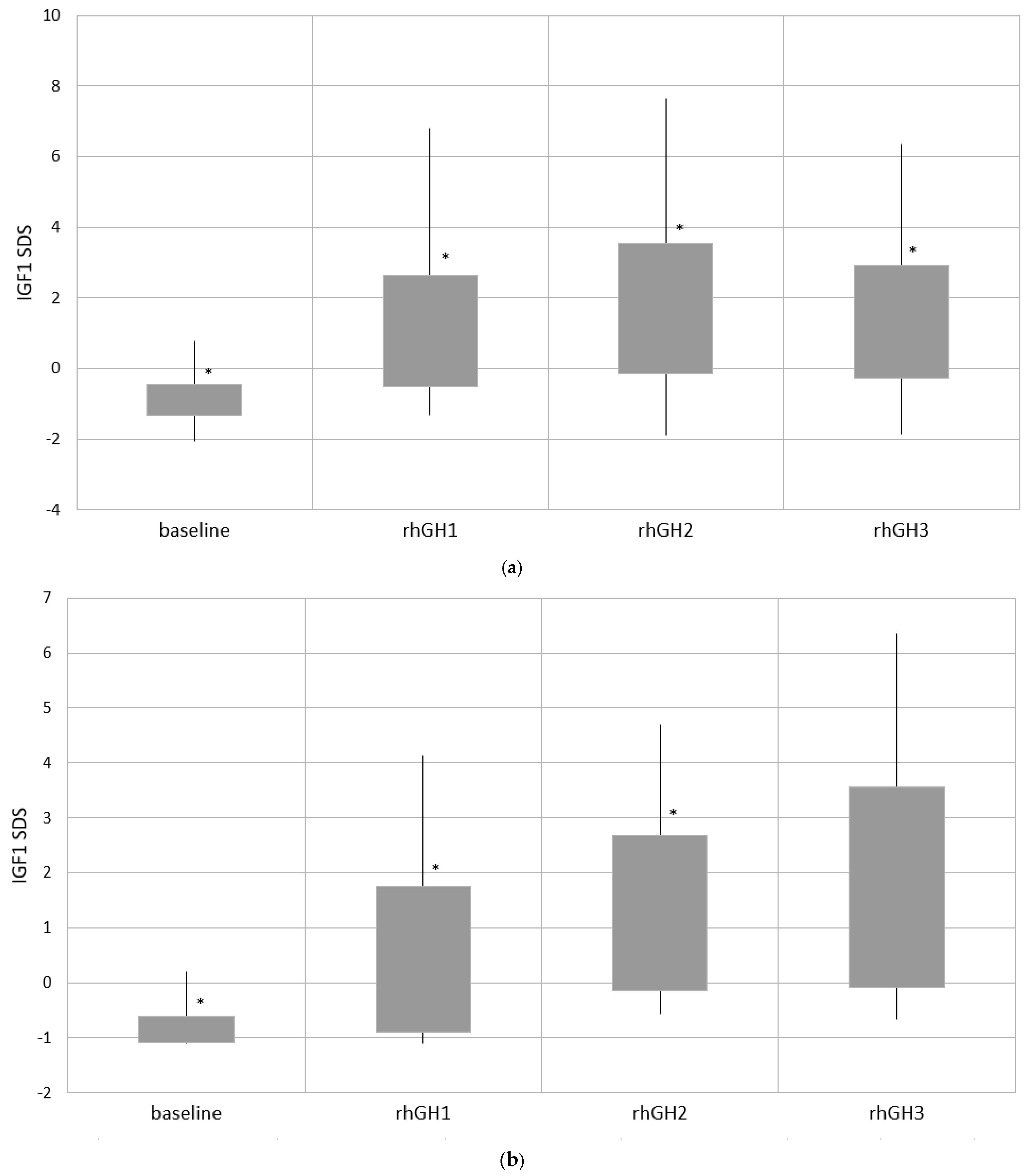
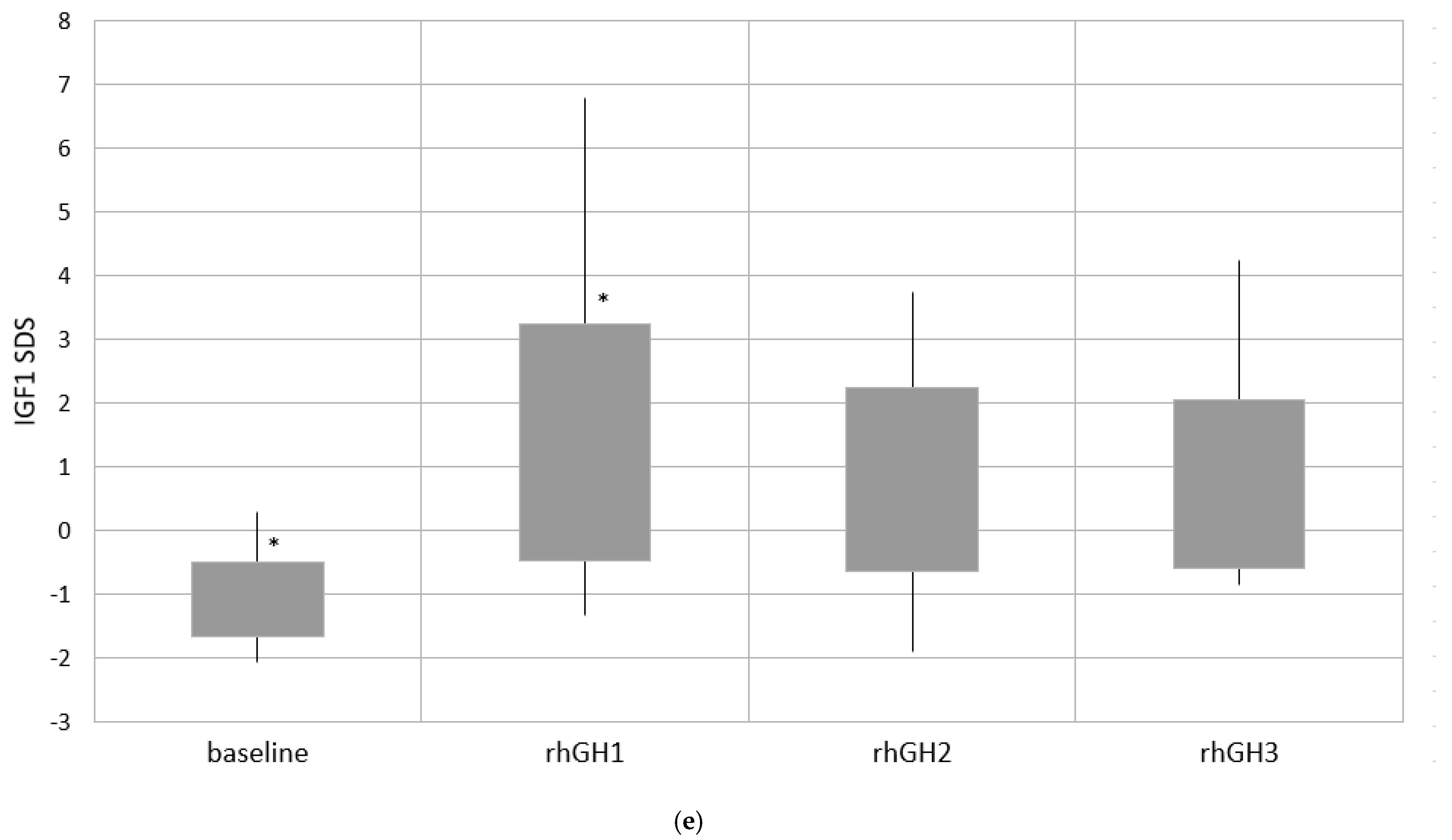
| IGF1 SDS | −0.89 ± 0.43 * −0.92 (−2.06–0.78) | 1.07 ± 1.58 * 0.79 (−1.33–6.8) | 1.69 ± 1.85 * 1.37 (−1.89–7.66) | 1.32 ± 1.59 * 1.13 (−1.86–6.36) |
| rhGH Therapy | Baseline | rhGH1 | rhGH2 | rhGH3 |
|---|---|---|---|---|
| Age of patients (years) (n, %) a | ||||
| Group 1 | 1.27 ± 0.40 (n = 45) | 2.52 ± 0.91 (n = 44, 98) | 4.13 ± 1.55 (n = 35, 78) | 5.70 ± 2.02 (n = 22, 49) |
| Group 2 | 2.93± 0.66 (n = 49) | 4.08 ± 1.11 (n = 46, 94) | 6.94 ± 2.56 (n = 45, 92) | 9.89 ± 3.08 (n = 41, 84) |
| Group 3 | 5.91 ± 0.98 (n = 21) | 7.44 ± 1.33 (n = 21, 100) | 10.92 ± 2.61 (n =19, 91) | 14.54 ± 2.96 (n = 15, 71) |
| Group 4 | 10.54 ± 2.44 (n = 32) | 11.97 ± 2.7 (n = 29, 91) | 13.94 ± 2.02 (n = 27, 84) | 16.47 ± 2.23 (n = 21, 66) |
| Time of rhGH therapy (years) | ||||
| Group 1 | NA | 1.24 ± 0.89 | 2.87 ± 1.6 | 4.32 ± 1.97 * |
| Group 2 | NA | 1.10 ± 0.76 | 3.98±2.35 | 6.85 ± 2.84 *,** |
| Group 3 | NA | 1.44 ± 0.89 | 4.93 ± 2.26 | 8.57 ± 2.8 *,**,*** |
| Group 4 | NA | 1.19 ± 0.73 | 3.77 ± 2.08 | 6.62 ± 2.72 *,*** |
| Height SDS | ||||
| Group 1 | −2.01 ± 1.70 | −0.85±1.35 | −0.65 ± 1.37 | −0.42 ± 1.40 |
| Group 2 | −2.42 ± 1.23 | −1.26±1.34 | −0.54 ± 1.41 | −0.06 ± 1.53 ** |
| Group 3 | −1.96 ± 1.37 | −0.74±1.48 | −0.12 ± 1.04 *** | -0.54 ± 1.27 |
| Group 4 | −1.88 ± 1.64 | −1.21 ± 1.34 | −0.98 ± 1.41 *** | -0.92 ± 1.41 ** |
| GV (cm/year) | ||||
| Group 1 | NA | 12.08 ± 3.66 * | 8.72 ± 2.19 * | 7.77 ± 2.06 * |
| Group 2 | NA | 10.45 ± 3.02 *,** | 8.17 ± 2.10 ** | 6.28±2.24 *,** |
| Group 3 | NA | 9.43 ± 1.34 * | 6.81 ± 1.3 *,**,*** | 4.89 ± 2.96 * |
| Group 4 | NA | 8.51 ± 2.59 *,** | 5.22 ± 2.23 *,**,*** | 3.29 ± 1.91 *,** |
| BMI SDS | ||||
| Group 1 | −0.62 ± 1.82 * | −0.20 ± 1.59 * | 0.04 ± 1.56 * | 0.16 ± 1.28 * |
| Group 2 | 0.46 ± 1.33 *,** | 0.55 ± 1.38 * | 0.75 ± 1.22 * | 0.95 ± 1.37 * |
| Group 3 | 1.03 ± 1.10 * | 0.80 ± 0.97 * | 0.93 ± 1.19 * | 0.93 ± 1.35 * |
| Group 4 | 1.36 ± 0.54 *,** | 0.8 ± 0.89 * | 1.12 ± 0.81 * | 1.45 ± 0.74 * |
| Age of patients at IGF1 measurement (years) (n) a | ||||
| Group 1 | 1.27 ± 0.41 (n = 44) | 2.48 ± 0.94 (n = 41) | 3.99 ± 1.38 (n = 29) | 6.21 ± 1.69 (n = 19) |
| Group 2 | 2.92 ± 0.67 (n = 47) | 4.31 ± 1.61 (n = 48) | 7.48 ± 2.77 (n = 44) | 9.56 ± 2.74 (n = 35) |
| Group 3 | 5.83 ± 0.93 (n = 20) | 7.20 ± 0.95 (n = 20) | 10.77 ± 2.92 (n = 19) | 14.28 ± 2.51 (n = 15) |
| Group 4 | 10.61 ± 2.45 (n = 31) | 11.82 ± 2.56 (n=31) | 13.47 ± 2.16 (n = 29) | 16.29 ± 2.12 (n = 23) |
| Time of rhGH therapy at IGF1 measurement (years) | ||||
| Group 1 | NA | 1.20 ± 0.91 | 2.72 ± 1.37 * | 4.87 ± 1.56 * |
| Group 2 | NA | 1.33 ± 1.28 | 4.44 ± 2.48 *,** | 6.61 ± 2.61 *,** |
| Group 3 | NA | 1.28 ± 0.70 | 4.70 ± 2.5 *,*** | 8.27 ± 2.54 *,**,*** |
| Group 4 | NA | 1.27 ± 0.84 | 3.02 ± 1.99 **,*** | 6.24 ± 2.54 *** |
| IGF1 SDS | ||||
| Group 1 | −0.85 ± 0.24 * | 0.43±1.32 * | 1.27 ± 1.41 * | 1.74±1.82 |
| Group 2 | −0.81 ± 0.35 ** | 1.28 ± 1.61 * | 2.12±1.93 *,** | 1.64 ± 1.63 ** |
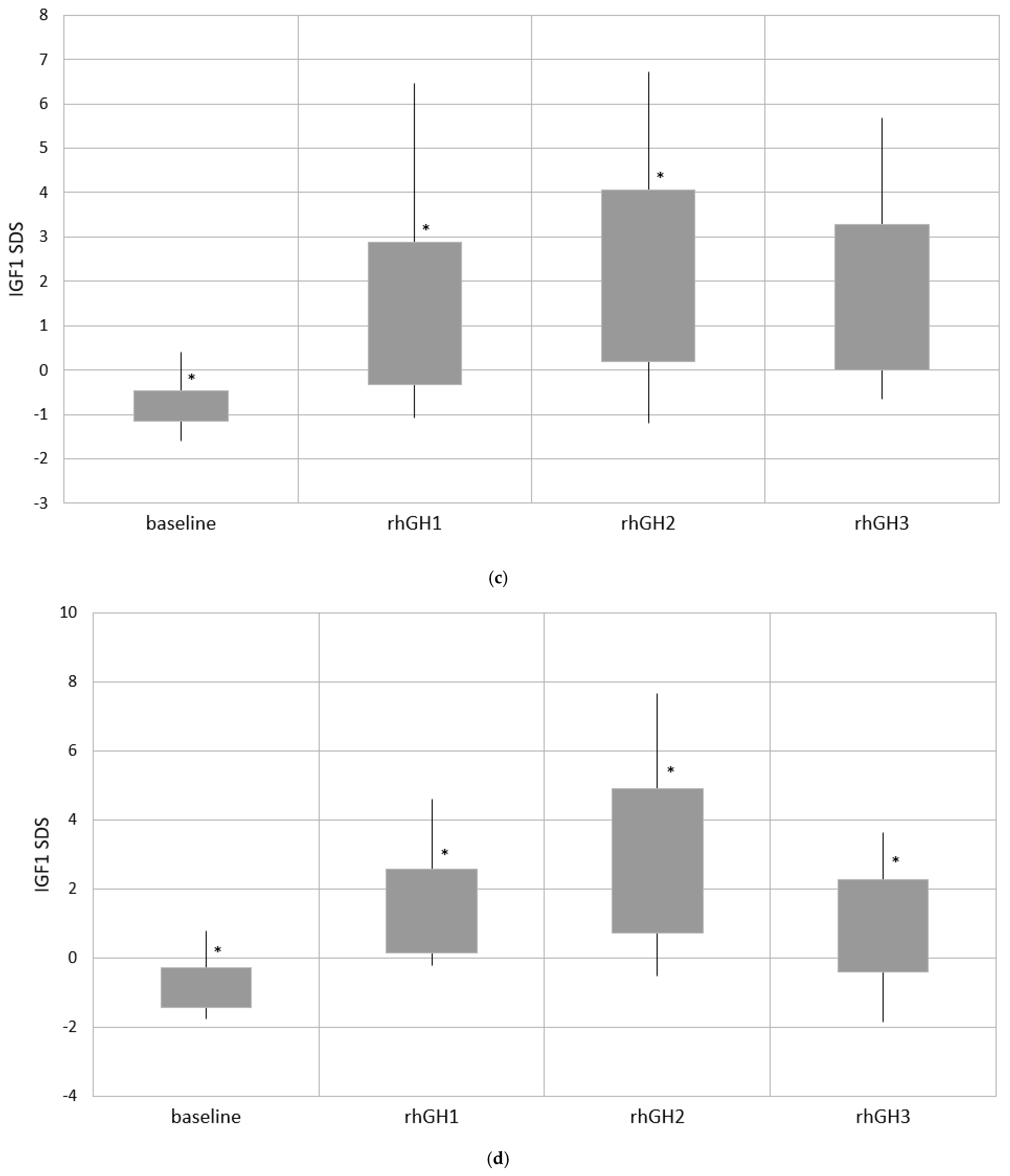
| Group 3 | −0.86 ± 0.58 | 1.36 ± 1.22 * | 2.82 ± 2.10 *,*** | 0.94 ± 1.33 |
| Group 4 | −1.08 ± 0.58 *,** | 1.40 ± 1.86 * | 0.81 ± 1.43 **,*** | 0.73 ± 1.32 ** |
| Results | Whole Cohort | Group 1 | Group 2 | Group 3 | Group 4 |
|---|---|---|---|---|---|
| Number of patients | n = 91 | n = 18 | n = 33 | n = 18 | n = 22 |
| Age of patients (years) a | 9.09 ± 4.46 | 3.62 ± 1.40 | 7.55 ± 2.72 | 11.00 ± 2.8 | 14.69 ± 2.04 |
| Time of rhGH therapy (years) | 4.13 ± 2.60 | 2.32 ± 1.30 * | 4.44 ± 2.45 * | 4.95 ± 3.00 * | 4.34 ± 2.79 * |
| Glucose 0 min (mg/dL) | 78.75 ± 10.72 | 74.61 ± 11.94 * | 75.54 ± 10.43 ** | 84.00 ± 7.90 *,** | 82.95 ± 9.33 *,** |
| Glucose 120 min (mg/dL) | 112.04 ± 25.02 | 100.44 ± 20.2 * | 120.39 ± 25.58 * | 108.67 ± 27.33 | 111.77 ± 22.65 |
| Insulin 0 min (uIU/mL) | 10.06 ± 7.28 | 5.39 ± 3.64 * | 9.66 ± 8.64 *,** | 11.14 ± 4.33 * | 13.23 ± 7.46 *,** |
| Insulin 120 min (uIU/mL) | 41.65 ± 33.13 | 18.60 ± 14.07 * | 46.79 ± 41.04 * | 49.77 ± 27.29 * | 46.55 ± 28.94 * |
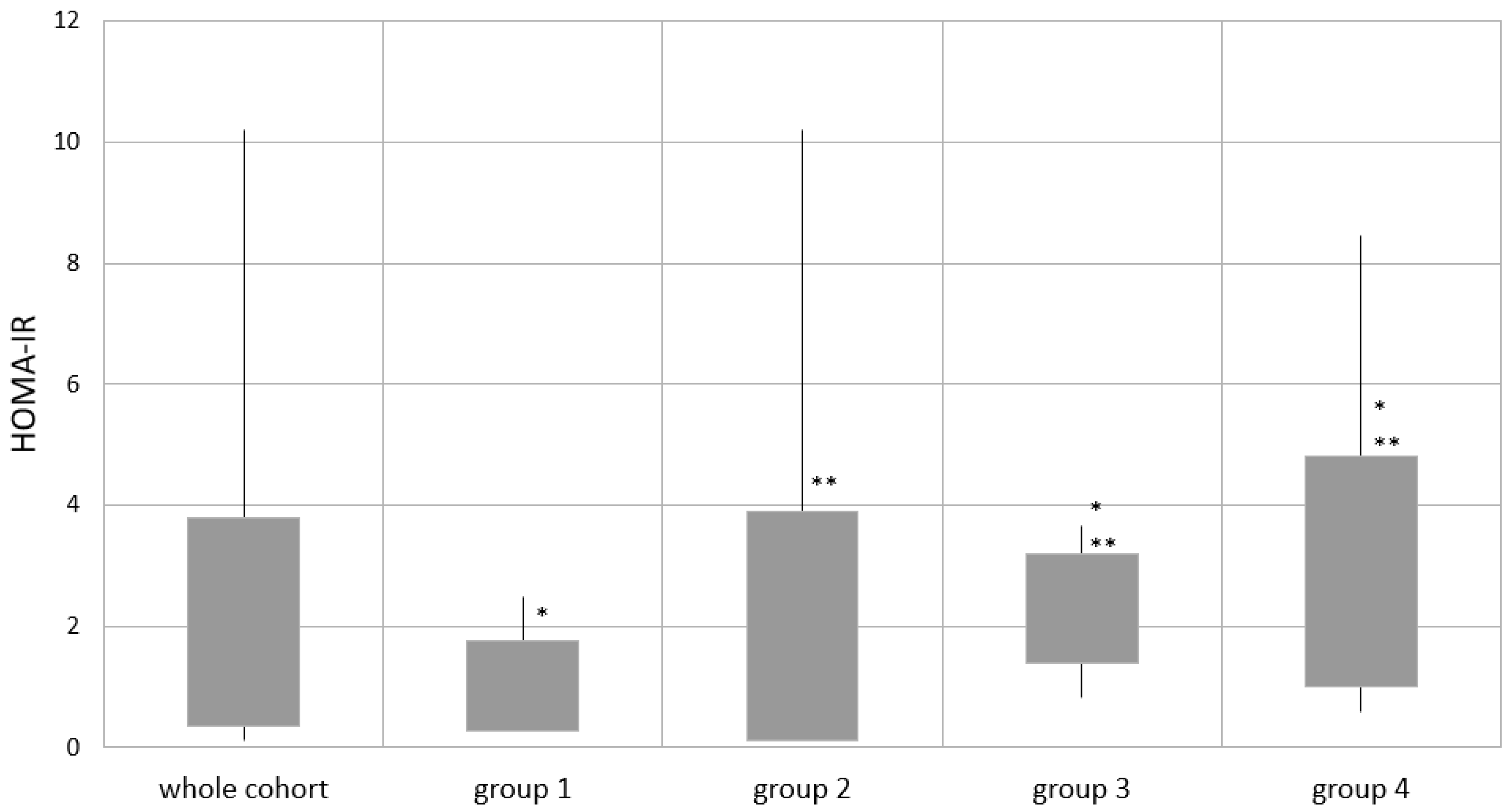
| HOMA IR | 2.07 ± 1.71 | 1.02 ± 0.74 * | 1.92 ± 1.97 ** | 2.30 ± 0.90 *,** | 2.91 ± 1.90 *,** |
| TC (mg/dL) | 184.3 ± 33.23 | 191.16 ± 39.40 | 177.87 ± 31.22 | 186.76 ± 28.83 | 186.28 ± 33.93 |
| HDL-C (mg/dL) | 56.36 ± 12.64 | 53.78±11.26 | 56.80 ± 12.75 | 55.26 ± 10.81 | 59.00 ± 15.23 |
| LDL-C (mg/dL) | 111.60 ± 32.28 | 120.81 ± 32.05 | 106.36 ± 33.69 | 115.14 ± 28.33 | 109.22 ± 33.70 |
| TG (mg/dL) | 89.85 ± 39.24 | 83.97 ± 32.92 | 84.66 ± 39.08 | 97.83 ± 38.90 | 98.98 ± 46.16 |
| Results | Whole Cohort | Group 1 | Group 2 | Group 3 | Group 4 |
|---|---|---|---|---|---|
| Number of patients | n = 89 | n = 23 | n = 29 | n = 16 | n = 21 |
| Age of patients (years) a | 8.58 ± 4.79 | 3.32 ± 1.37 | 7.22 ± 2.86 | 10.91 ± 2.68 | 14.44 ± 2.66 |
| Time of rhGH therapy (years) | 3.69 ± 2.61 | 2.02 ± 1.29 * | 4.29 ± 2.67 * | 4.81 ± 2.70 * | 3.85 ± 2.83 * |
| BA/CA | 0.90 ± 0.22 | 0.66 ± 0.16 * | 0.96 ± 0.21 * | 1.05 ± 0.14 * | 1.00 ± 0.10 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lecka-Ambroziak, A.; Wysocka-Mincewicz, M.; Doleżal-Ołtarzewska, K.; Zygmunt-Górska, A.; Wędrychowicz, A.; Żak, T.; Noczyńska, A.; Birkholz-Walerzak, D.; Stawerska, R.; Hilczer, M.; et al. Effects of Recombinant Human Growth Hormone Treatment, Depending on the Therapy Start in Different Nutritional Phases in Paediatric Patients with Prader–Willi Syndrome: A Polish Multicentre Study. J. Clin. Med. 2021, 10, 3176. https://doi.org/10.3390/jcm10143176
Lecka-Ambroziak A, Wysocka-Mincewicz M, Doleżal-Ołtarzewska K, Zygmunt-Górska A, Wędrychowicz A, Żak T, Noczyńska A, Birkholz-Walerzak D, Stawerska R, Hilczer M, et al. Effects of Recombinant Human Growth Hormone Treatment, Depending on the Therapy Start in Different Nutritional Phases in Paediatric Patients with Prader–Willi Syndrome: A Polish Multicentre Study. Journal of Clinical Medicine. 2021; 10(14):3176. https://doi.org/10.3390/jcm10143176
Chicago/Turabian StyleLecka-Ambroziak, Agnieszka, Marta Wysocka-Mincewicz, Katarzyna Doleżal-Ołtarzewska, Agata Zygmunt-Górska, Anna Wędrychowicz, Teresa Żak, Anna Noczyńska, Dorota Birkholz-Walerzak, Renata Stawerska, Maciej Hilczer, and et al. 2021. "Effects of Recombinant Human Growth Hormone Treatment, Depending on the Therapy Start in Different Nutritional Phases in Paediatric Patients with Prader–Willi Syndrome: A Polish Multicentre Study" Journal of Clinical Medicine 10, no. 14: 3176. https://doi.org/10.3390/jcm10143176