
Myeloid-Derived Suppressor Cells and Mesenchymal Stem/Stromal Cells in Myeloid Malignancies


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Myeloid-Derived Suppressor Cells
3. Mesenchymal Stromal/Stem Cells
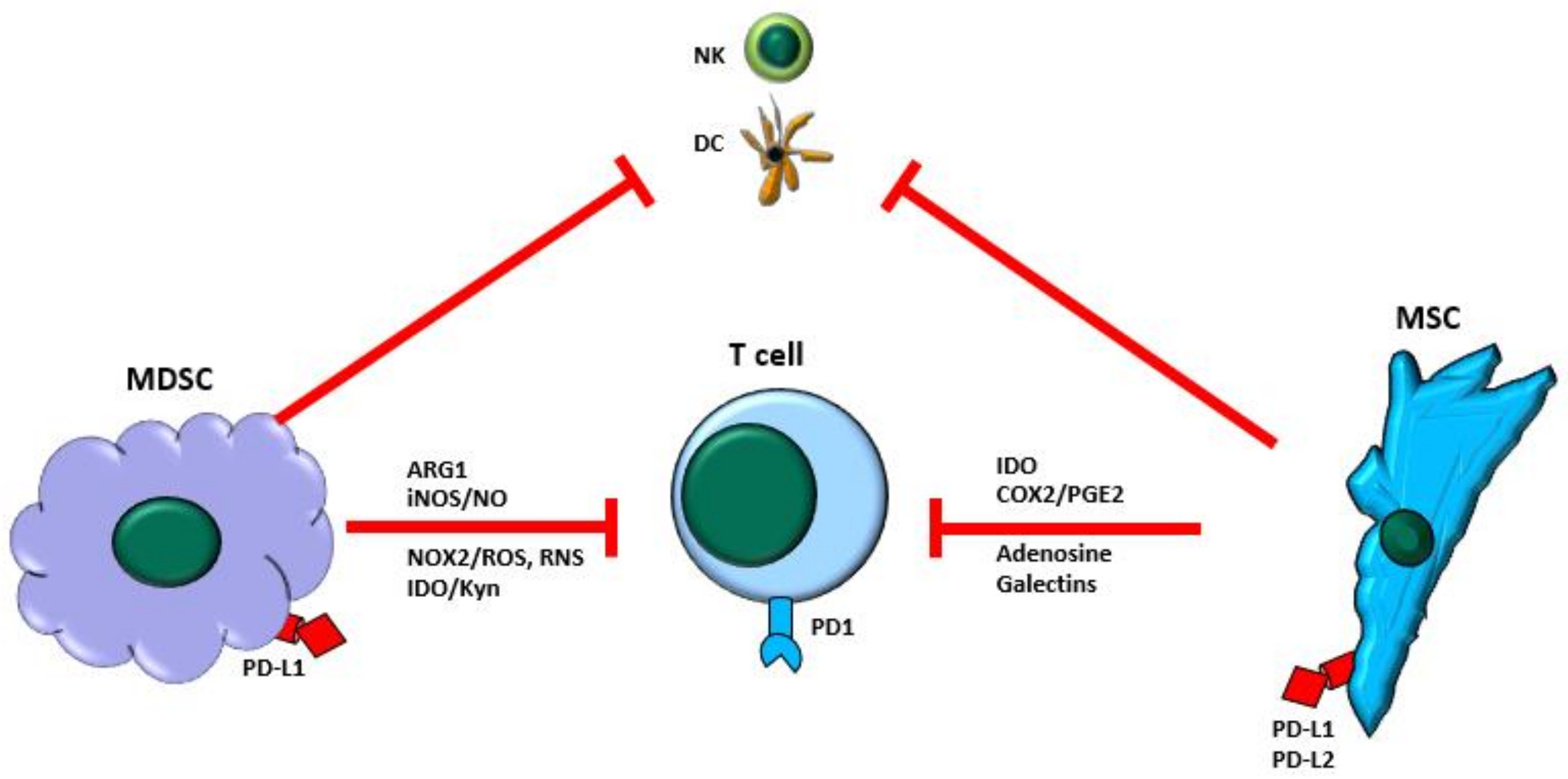
4. MDSCs and MSCs in T-Cell Immunosuppression Function and Mechanism
4.1. MDSC and T-Cell Immunosuppression
4.2. MSC and T-Cell Immunosuppression
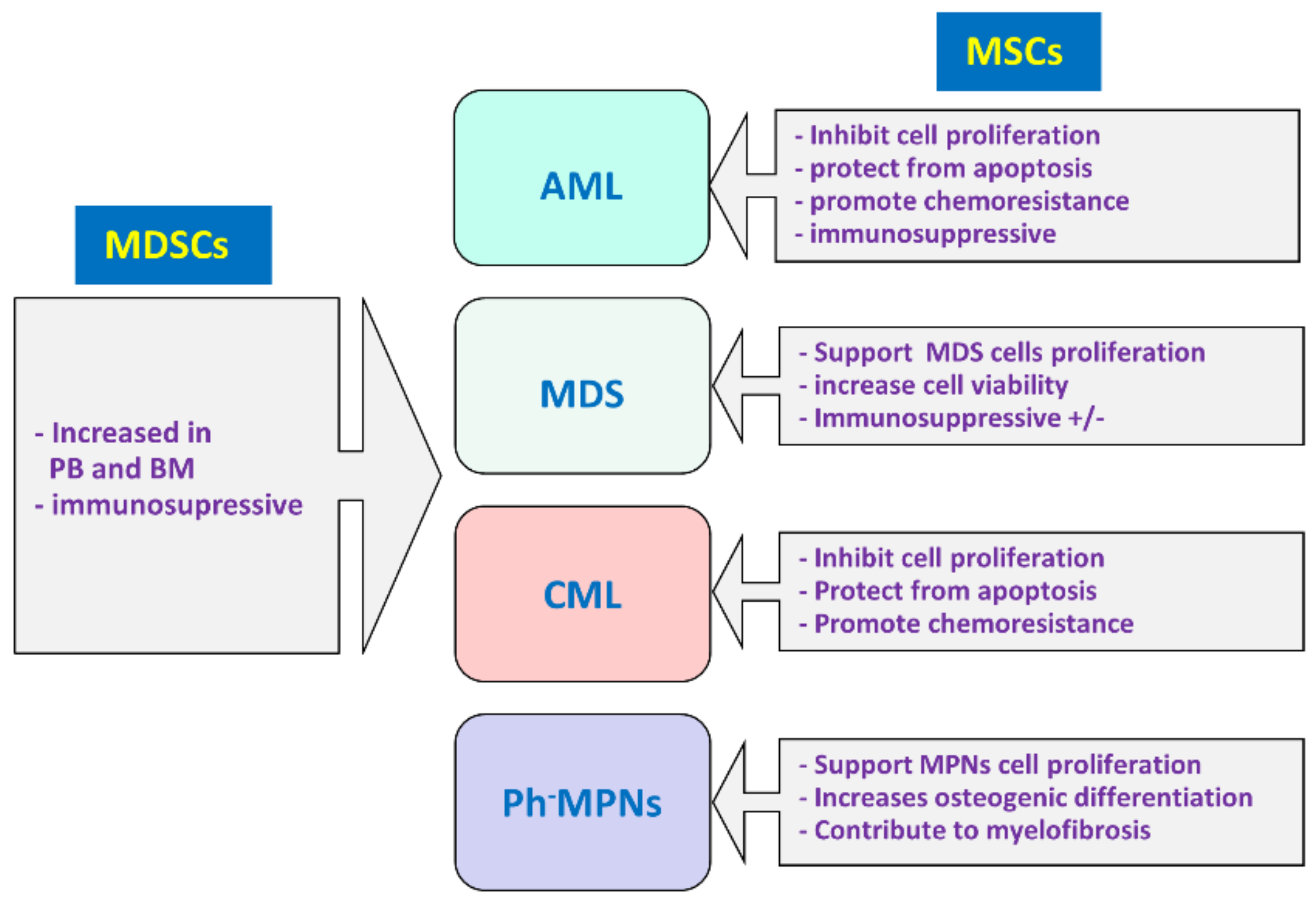
5. MDSCs and MSCs in Myeloid Malignancies
5.1. Acute Myeloid Leukemia
5.1.1. MDSCs in Acute Myeloid Leukemia
5.1.2. MSCs in Acute Myeloid Leukemia
5.2. Myelodysplastic Syndrome
5.2.1. MDSCs in Myelodysplastic Syndrome
5.2.2. MSCs in Myelodysplastic Syndrome
5.3. Myeloproliferative Neoplasms
5.3.1. Chronic Myeloid Leukemia
5.3.2. Philadelphia Chromosome-Negative Myeloproliferative Neoplasms
6. MDSC and MSC Interplay
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ng, A.P.; Alexander, W.S. Haematopoietic stem cells: Past, present and future. Cell Death Discov. 2017, 3, 17002. [Google Scholar] [CrossRef]
- Reagan, M.R.; Rosen, C.J. Navigating the bone marrow niche: Translational insights and cancer-driven dysfunction. Nat. Rev. Rheumatol. 2016, 12, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Behrmann, L.; Wellbrock, J.; Fiedler, W. The bone marrow stromal niche: A therapeutic target of hematological myeloid malignancies. Expert Opin. Ther. Targets 2020, 24, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Ma, T.; Li, X.; Wei, L.; Liu, Z.; Feng, B.; Dong, B.; Chen, X. New progress in the study of germline susceptibility genes of myeloid neoplasms. Oncol. Lett. 2021, 21, 317. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Y.; Guo, N.; Wang, S. MDSCs: Key criminals of tumor pre-metastatic niche formation. Front. Immunol. 2019, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Tumino, N.; Di Pace, A.L.; Besi, F.; Quatrini, L.; Vacca, P.; Moretta, L. Interaction Between MDSC and NK Cells in Solid and Hematological Malignancies: Impact on HSCT. Front. Immunol. 2021, 12, 638841. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.; Geyh, S.; Germing, U.; Haas, R. Mesenchymal stromal cells in myeloid malignancies. Blood Res. 2016, 51, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Younos, I.H.; Abe, F.; Talmadge, J.E. Myeloid-derived suppressor cells: Their role in the pathophysiology of hematologic malignancies and potential as therapeutic targets. Leuk. Lymphoma 2015, 56, 2251–2263. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Khaled, Y.S.; Ammori, B.J.; Elkord, E. Myeloid-derived suppressor cells in cancer: Recent progress and prospects. Immunol. Cell Biol. 2013, 91, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.X.; Kim, T.S.; Poh, C.L. Understanding the Differentiation, Expansion, Recruitment and Suppressive Activities of Myeloid-Derived Suppressor Cells in Cancers. Int. J. Mol. Sci. 2020, 21, 3599. [Google Scholar] [CrossRef] [PubMed]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wu, T.; Shao, S.; Shi, B.; Zhao, Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology 2015, 14, e1004983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eruslanov, E.; Neuberger, M.; Daurkin, I.; Perrin, G.Q.; Algood, C.; Dahm, P.; Rosser, C.; Vieweg, J.; Gilbert, S.M.; Kusmartsev, S. Circulating and tumor-infiltrating myeloid cell subsets in patients with bladder cancer. Int. J. Cancer 2012, 130, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Gebhardt, C.; Umansky, L.; Beckhove, P.; Schulze, T.J.; Utikal, J.; Umansky, V. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int. J. Cancer 2015, 136, 2352–2360. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala Cuellar, A.P.; Kang, J.H.; Jeung, E.B.; Choi, K.C. Roles of Mesenchymal Stem Cells in Tissue Regeneration and Immunomodulation. Biomol. Ther. 2019, 27, 25–33. [Google Scholar] [CrossRef]
- Medyouf, H. The microenvironment in human myeloid malignancies: Emerging concepts and therapeutic implications. Blood 2017, 129, 1617–1626. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef]
- Klyushnenkova, E.; Mosca, J.D.; Zernetkina, V.; Majumdar, M.K.; Beggs, K.J.; Simonetti, D.W.; Deans, R.J.; McIntosh, K.R. T cell responses to allogeneic human mesenchymal stem cells: Immunogenicity, tolerance, and suppression. J. Biomed. Sci. 2005, 12, 47–57. [Google Scholar] [CrossRef]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchman, S.M.; Ponchel, F.; Boxall, S.A.; Cuthbert, R.; Kouroupis, D.; Roshdy, T.; Giannoudis, P.V.; Emery, P.; McGonagle, D.; Jones, E.A. Transcriptional profile of native CD271+ multipotential stromal cells: Evidence for multiple fates, with prominent osteogenic and Wnt pathway signaling activity. Arthritis Rheum. 2012, 64, 2632–2643. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Nakatsuka, R.; Sumide, K.; Kawamura, H.; Takahashi, M.; Fujioka, T.; Uemura, Y.; Asano, H.; Sasaki, Y.; Inoue, M.; et al. Prospectively Isolated Human Bone Marrow Cell-Derived MSCs Support Primitive Human CD34-Negative Hematopoietic Stem Cells. Stem Cells 2015, 33, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Lacombe, J.; Hanoun, M.; Mizoguchi, T.; Bruns, I.; Kunisaki, Y.; Frenette, P.S. PDGFRα and CD51 mark human Nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013, 210, 1351–1367. [Google Scholar] [CrossRef] [PubMed]
- Rhee, K.J.; Lee, J.I.; Eom, Y.W. Mesenchymal Stem Cell-Mediated Effects of Tumor Support or Suppression. Int. J. Mol. Sci. 2015, 16, 30015–30033. [Google Scholar] [CrossRef] [Green Version]
- Barcellos-de-Souza, P.; Gori, V.; Bambi, F.; Chiarugi, P. Tumor microenvironment: Bone marrow-mesenchymal stem cells as key players. Biochim. Biophys. Acta 2013, 1836, 321–335. [Google Scholar] [CrossRef]
- Musiał-Wysocka, A.; Kot, M.; Majka, M. The Pros and Cons of Mesenchymal Stem Cell-Based Therapies. Cell Transplant. 2019, 28, 801–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papait, A.; Stefani, F.R.; Cargnoni, A.; Magatti, M.; Parolini, O.; Silini, A.R. The Multifaceted Roles of MSCs in the Tumor Microenvironment: Interactions with Immune Cells and Exploitation for Therapy. Front. Cell Dev. Biol. 2020, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastaki Khoshbin, A.; Eskian, M.; Keshavarz-Fathi, M.; Rezaei, N. Roles of Myeloid-Derived Suppressor Cells in Cancer Metastasis: Immunosuppression and Beyond. Arch. Immunol. Ther. Exp. 2019, 67, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Castro-Manrreza, M.E.; Montesinos, J.J. Immunoregulation by mesenchymal stem cells: Biological aspects and clinical applications. J. Immunol. Res. 2015, 2015, 394917. [Google Scholar] [CrossRef] [Green Version]
- Müller, L.; Tunger, A.; Wobus, M.; von Bonin, M.; Towers, R.; Bornhäuser, M.; Dazzi, F.; Wehner, R.; Schmitz, M. Immunomodulatory Properties of Mesenchymal Stromal Cells: An Update. Front. Cell Dev. Biol. 2021, 9, 637725. [Google Scholar] [CrossRef]
- Khatami, M. Unresolved inflammation and cancer: Loss of natural immune surveillance as the correct “target” for therapy! Seeing the “Elephant” in the light of logic. Cell Biochem. Biophys. 2012, 62, 501–509. [Google Scholar] [CrossRef]
- Sivagnanalingam, U.; Beatty, P.L.; Finn, O.J. Myeloid derived suppressor cells in cancer, premalignancy and inflammation: A roadmap to cancer immunoprevention. Mol. Carcinog. 2020, 59, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Immunosenescence: The potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell. Mol. Life Sci. 2019, 76, 1901–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santibanez, J.F.; Bjelica, S. Transforming Growth Factor-Beta1 and Myeloid-Derived Suppressor Cells Interplay in Cancer. Open Cancer Immunol. J. 2017, 6, 1–14. [Google Scholar] [CrossRef]
- Wu, A.A.; Drake, V.; Huang, H.S.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology 2015, 4, e1016700. [Google Scholar] [CrossRef] [PubMed]
- Pyzer, A.R.; Cole, L.; Rosenblatt, J.; Avigan, D.E. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int. J. Cancer 2016, 139, 1915–1926. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [Green Version]
- Blesson, S.; Thiery, J.; Gaudin, C.; Stancou, R.; Kolb, J.P.; Moreau, J.L.; Theze, J.; Mami-Chouaib, F.; Chouaib, S. Analysis of the mechanisms of human cytotoxic T lymphocyte response inhibition by NO. Int. Immunol. 2002, 14, 1169–1178. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Monu, N.R.; Frey, A.B. Myeloid-derived suppressor cells and anti-tumor T cells: A complex relationship. Immunol. Investig. 2012, 41, 595–613. [Google Scholar] [CrossRef] [Green Version]
- Schmielau, J.; Finn, O.J. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001, 61, 4756–4760. [Google Scholar]
- Nagaraj, S.; Schrum, A.G.; Cho, H.I.; Celis, E.; Gabrilovich, D.I. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J. Immunol. 2010, 184, 3106–3116. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Guo, J.; Weng, L.; Tang, W.; Jin, S.; Ma, W. Myeloid-derived suppressor cells-new and exciting players in lung cancer. J. Hematol. Oncol. 2020, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rad Pour, S.; Morikawa, H.; Kiani, N.A.; Yang, M.; Azimi, A.; Shafi, G.; Shang, M.; Baumgartner, R.; Ketelhuth, D.F.J.; Kamleh, M.A.; et al. Exhaustion of CD4+ T-cells mediated by the Kynurenine Pathway in Melanoma. Sci. Rep. 2019, 9, 12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiang, Y.; Xin, V.W.; Wang, X.W.; Peng, X.C.; Liu, X.Q.; Wang, D.; Li, N.; Cheng, J.T.; Lyv, Y.N.; et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 2020, 13, 107. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 20, e1247135. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2+ myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Hanson, E.M.; Clements, V.K.; Sinha, P.; Ilkovitch, D.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J. Immunol. 2009, 183, 937–944. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.H.; Sinha, P.; Horn, L.A.; Clements, V.K.; Yang, H.; Li, J.; Tracey, K.J.; Ostrand-Rosenberg, S. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014, 74, 5723–5733. [Google Scholar] [CrossRef] [Green Version]
- Le Blanc, K.; Tammik, C.; Rosendahl, K.; Zetterberg, E.; Ringdén, O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp. Hematol. 2003, 31, 890–896. [Google Scholar] [CrossRef]
- Machado, C.V.; Telles, P.D.; Nascimento, I.L. Immunological characteristics of mesenchymal stem cells. Rev. Bras. Hematol. Hemoter. 2013, 35, 62–67. [Google Scholar] [CrossRef]
- Zhao, Q.; Ren, H.; Han, Z. Mesenchymal stem cells: Immunomodulatory capability and clinical potential in immune diseases. J. Cell. Immunother. 2016, 2, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciciarello, M.; Corradi, G.; Loscocco, F.; Visani, G.; Monaco, F.; Cavo, M.; Curti, A.; Isidori, A. The Yin and Yang of the Bone Marrow Microenvironment: Pros and Cons of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Chiu, S.M.; Motan, D.A.; Zhang, Z.; Chen, L.; Ji, H.L.; Tse, H.F.; Fu, Q.L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef] [Green Version]
- Rasmusson, I.; Ringdén, O.; Sundberg, B.; Le Blanc, K. Mesenchymal stem cells inhibit the formation of cytotoxic T lymphocytes, but not activated cytotoxic T lymphocytes or natural killer cells. Transplantation 2003, 76, 1208–1213. [Google Scholar] [CrossRef]
- Polchert, D.; Sobinsky, J.; Douglas, G.; Kidd, M.; Moadsiri, A.; Reina, E.; Genrich, K.; Mehrotra, S.; Setty, S.; Smith, B.; et al. IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur. J. Immunol. 2008, 38, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Jang, I.K.; Lee, M.W.; Ko, Y.J.; Lee, D.H.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Enhanced Immunosuppressive Properties of Human Mesenchymal Stem Cells Primed by Interferon-γ. EBioMedicine 2018, 28, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Meisel, R.; Zibert, A.; Laryea, M.; Gobel, U.; Daubener, W.; Dilloo, D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood 2004, 103, 4619–4621. [Google Scholar] [CrossRef] [Green Version]
- Harrell, C.R.; Djonov, V.; Volarevic, V. The Cross-Talk between Mesenchymal Stem Cells and Immune Cells in Tissue Repair and Regeneration. Int. J. Mol. Sci. 2021, 22, 2472. [Google Scholar] [CrossRef]
- Menta, R.; Mancheño-Corvo, P.; Del Río, B.; Ramírez, C.; DelaRosa, O.; Dalemans, W.; Lombardo, E. Tryptophan concentration is the main mediator of the capacity of adipose mesenchymal stromal cells to inhibit T-lymphocyte proliferation in vitro. Cytotherapy 2014, 16, 1679–1691. [Google Scholar] [CrossRef]
- Laing, A.G.; Fanelli, G.; Ramirez-Valdez, A.; Lechler, R.I.; Lombardi, G.; Sharpe, P.T. Mesenchymal stem cells inhibit T-cell function through conserved induction of cellular stress. PLoS ONE 2019, 14, e0213170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, W.; Zhang, J.; Yuan, Z.; Ren, G.; Zhang, L.; Chen, X.; Rabson, A.B.; Roberts, A.I.; Wang, Y.; Shi, Y. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014, 74, 1576–1587. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Chen, X.; Huang, Y.; Li, W.; Li, J.; Cao, K.; Cao, G.; Zhang, L.; Li, F.; Roberts, A.I.; et al. Phylogenetic distinction of iNOS and IDO function in mesenchymal stem cell-mediated immunosuppression in mammalian species. Cell Death Differ. 2014, 21, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Girotti, M.R.; Marais, R.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouffi, C.; Bony, C.; Courties, G.; Jorgensen, C.; Noël, D. IL-6-dependent PGE2 secretion by mesenchymal stem cells inhibits local inflammation in experimental arthritis. PLoS ONE 2010, 5, e14247. [Google Scholar] [CrossRef]
- Yang, F.Y.; Chen, R.; Zhang, X.; Huang, B.; Tsang, L.L.; Li, X.; Jiang, X. Preconditioning Enhances the Therapeutic Effects of Mesenchymal Stem Cells on Colitis Through PGE2-Mediated T-Cell Modulation. Cell Transplant. 2018, 27, 1352–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Jiang, E.; Yao, J.; Wang, M.; Chen, S.; Zhou, Z.; Zhai, W.; Ma, Q.; Feng, S.; Han, M. Interferon-γ mediates the immunosuppression of bone marrow mesenchymal stem cells on T-lymphocytes in vitro. Hematology 2018, 23, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, W.T.; Pendleton, J.D.; Beyer, W.M.; Egalka, M.C.; Guinan, E.C. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: Implications in transplantation. Transplantation 2003, 75, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Najar, M.; Raicevic, G.; Boufker, H.I.; Fayyad Kazan, H.; De Bruyn, C.; Meuleman, N.; Bron, D.; Toungouz, M.; Lagneaux, L. Mesenchymal stromal cells use PGE2 to modulate activation and proliferation of lymphocyte subsets: Combined comparison of adipose tissue, Wharton’s Jelly and bone marrow sources. Cell. Immunol. 2010, 264, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Kim, H.S.; Shin, T.H.; Kang, I.; Lee, J.Y.; Kim, J.J.; Kang, H.K.; Seo, Y.; Lee, S.; Yu, K.R.; et al. PGE2 maintains self-renewal of human adult stem cells via EP2-mediated autocrine signaling and its production is regulated by cell-to-cell contact. Sci. Rep. 2016, 6, 26298. [Google Scholar] [CrossRef] [Green Version]
- Saldanha-Araujo, F.; Ferreira, F.I.; Palma, P.V.; Araujo, A.G.; Queiroz, R.H.; Covas, D.T.; Zago, M.A.; Panepucci, R.A. Mesenchymal stromal cells up-regulate CD39 and increase adenosine production to suppress activated T-lymphocytes. Stem Cell Res. 2011, 7, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Sattler, C.; Steinsdoerfer, M.; Offers, M.; Fischer, E.; Schierl, R.; Heseler, K.; Däubener, W.; Seissler, J. Inhibition of T-cell proliferation by murine multipotent mesenchymal stromal cells is mediated by CD39 expression and adenosine generation. Cell Transplant. 2011, 20, 1221–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, A.I.; Sorrell, J.M. The MSC curtain that stops the immune system. Immunol. Lett. 2015, 168, 136–139. [Google Scholar] [CrossRef]
- Erdmann, A.A.; Gao, Z.G.; Jung, U.; Foley, J.; Borenstein, T.; Jacobson, K.A.; Fowler, D.H. Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood 2005, 105, 4707–4714. [Google Scholar] [CrossRef]
- Roh, M.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Zhang, B. Targeting CD73 to augment cancer immunotherapy. Curr. Opin. Pharm. 2020, 53, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Modenutti, C.P.; Capurro, J.I.B.; Di Lella, S.; Martí, M.A. The Structural Biology of Galectin-Ligand Recognition: Current Advances in Modeling Tools, Protein Engineering, and Inhibitor Design. Front. Chem. 2019, 7, 823. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M.; Mobergslien, A.; Boudabous, A.; Fløisand, Y. Mesenchymal stem cell-mediated T cell suppression occurs through secreted galectins. Int. J. Oncol. 2011, 38, 385–390. [Google Scholar] [CrossRef]
- Fajka-Boja, R.; Urbán, V.S.; Szebeni, G.J.; Szebeni, G.J.; Czibula, Á.; Blaskó, A.; Kriston-Pál, É.; Makra, I.; Hornung, Á.; Szabó, E.; et al. Galectin-1 is a local but not systemic immunomodulatory factor in mesenchymal stromal cells. Cytotherapy 2016, 18, 360–370. [Google Scholar] [CrossRef]
- Kim, S.N.; Lee, H.J.; Jeon, M.S.; Yi, T.; Song, S.U. Galectin-9 is Involved in Immunosuppression Mediated by Human Bone Marrow-derived Clonal Mesenchymal Stem Cells. Immune Netw. 2015, 15, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, L.C.; Heldring, N.; Kadri, N.; Le Blanc, K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cells 2017, 35, 766–776. [Google Scholar] [CrossRef]
- Augello, A.; Tasso, R.; Negrini, S.M.; Amateis, A.; Indiveri, F.; Cancedda, R.; Pennesi, G. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur. J. Immunol. 2005, 35, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Wang, Y.; Jin, Y.; Zhang, Q.; Zhang, Y.; Wang, L.; Shen, B.; Yin, S.; Liu, W.; Cui, L.; et al. A critical role of IFNgamma in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Res. 2008, 18, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, W.; Wang, G.; Hou, Y.; Xu, F.; Liu, R.; Wang, F.; Xue, J.; Hu, T.; Luan, X. Interferon-γ and tumor necrosis factor-a promote the ability of human placenta-derived mesenchymal stromal cells to express programmed death ligand-2 and induce the differentiation of CD4(+)interleukin-10(+) and CD8(+)interleukin-10(+)Treg subsets. Cytotherapy 2015, 17, 1560–1571. [Google Scholar] [CrossRef]
- Luan, X.; Li, G.; Wang, G.; Wang, F.; Lin, Y. Human placenta-derived mesenchymal stem cells suppress T cell proliferation and support the culture expansion of cord blood CD34? cells: A comparison with human bone marrow-derived mesenchymal stem cells. Tissue Cell 2013, 45, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Vgenopoulou, P.; Kousteni, S. The Bone Marrow Stromal Niche: A New Master Regulator of Hematological Myeloid Malignancies. In Encyclopedia of Bone Biology; Zaidi, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 225–235. [Google Scholar]
- Rodriguez-Abreu, D.; Bordoni, A.; Zucca, E. Epidemiology of hematological malignancies. Ann. Oncol. 2007, 18, i3–i8. [Google Scholar] [CrossRef] [PubMed]
- Sangiorgio, V.; Orazi, A. Update on the classification of myeloid neoplasms: The 2016 revised World Health Organization classification of hematopoietic and lymphoid neoplasms. Adv. Cell Gene Ther. 2020, 3, 78. [Google Scholar] [CrossRef]
- Loghavi, S.; Sui, D.; Wei, P.; Garcia-Manero, G.; Pierce, S.; Routbort, M.J.; Jabbour, E.J.; Pemmaraju, N.; Kanagal-Shamanna, R.; Gur, H.D.; et al. Validation of the 2017 revision of the WHO chronic myelomonocytic leukemia categories. Blood Adv. 2018, 14, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Goulard, M.; Dosquet, C.; Bonnet, D. Role of the microenvironment in myeloid malignancies. Cell. Mol. Life Sci. 2018, 75, 1377–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [PubMed]
- Steffen, B.; Müller-Tidow, C.; Schwäble, J.; Berdel, W.E.; Serve, H. The molecular pathogenesis of acute myeloid leukemia. Crit. Rev. Oncol. 2005, 56, 195–221. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 73, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taghiloo, S.; Asgarian-Omran, H. Immune evasion mechanisms in Acute Myeloid Leukemia; a focus on immune checkpoint pathways. Crit. Rev. Oncol. 2021, 157, 103164. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Zhang, Z.F.; Ju, Y.; Li, L.; Zhang, B.C.; Liu, B. Increase in myeloid-derived suppressor cells (MDSCs) associated with minimal residual disease (MRD) detection in adult acute myeloid leukemia. Int. J. Hematol. 2015, 102, 579–586. [Google Scholar] [CrossRef]
- Wang, H.; Tao, Q.; Wang, Z.; Zhang, Q.; Xiao, H.; Zhou, M.; Dong, Y.; Zhai, Z. Circulating Monocytic Myeloid-Derived Suppressor Cells Are Elevated and Associated with Poor Prognosis in Acute Myeloid Leukemia. J. Immunol. Res. 2020, 2020, 7363084. [Google Scholar] [CrossRef]
- Lv, J.; Zhao, Y.; Zong, H.; Ma, G.; Wei, X.; Zhao, Y. Increased Levels of Circulating Monocytic- and Early-Stage Myeloid-Derived Suppressor Cells (MDSC) in Acute Myeloid Leukemia. Clin. Lab. 2021, 67, 10. [Google Scholar] [CrossRef]
- Hyun, S.Y.; Na, E.J.; Jang, J.E.; Chung, H.; Kim, S.J.; Kim, J.S.; Kong, J.H.; Shim, K.Y.; Lee, J.I.; Min, Y.H.; et al. Immunosuppressive role of CD11b+ CD33+ HLA-DR− myeloid-derived suppressor cells-like blast subpopulation in acute myeloid leukemia. Cancer Med. 2020, 9, 7007–7017. [Google Scholar] [CrossRef]
- Tohumeken, S.; Baur, R.; Böttcher, M.; Stoll, A.; Loschinski, R.; Panagiotidis, K.; Braun, M.; Saul, D.; Völkl, S.; Baur, A.S.; et al. Palmitoylated Proteins on AML-Derived Extracellular Vesicles Promote Myeloid-Derived Suppressor Cell Differentiation via TLR2/Akt/mTOR Signaling. Cancer Res. 2020, 80, 3663–3676. [Google Scholar] [CrossRef] [PubMed]
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K.; et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood 2017, 129, 1791–1802. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Huang, G.S.; Wang, Z.; Chen, X.Q.; Bai, Q.X.; Zhang, Y.Q.; Dong, B.X.; Wang, W.Q. Effects of human bone marrow stromal cell line (HFCL) on the proliferation, differentiation and apoptosis of acute myeloid leukemia cell lines U937, HL-60 and HL-60/VCR. Int. J. Hematol. 2008, 87, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Fonseka, M.; Ramasamy, R.; Tan, B.C.; Seow, H.F. Human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSC) inhibit the proliferation of K562 (human erythromyeloblastoid leukaemic cell line). Cell Biol. Int. 2012, 36, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Tian, C.; Guo, S.; Zhang, L.; Zhao, D.; Qu, F.; Zhao, W.; Wang, Y.; Wu, X.; Da, W.; et al. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk. Res. 2015, 39, 92–99. [Google Scholar] [CrossRef]
- Konopleva, M.; Konoplev, S.; Hu, W.; Zaritskey, A.; Afanasiev, B.; Andreeff, M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 2002, 16, 1713–1724. [Google Scholar] [CrossRef] [Green Version]
- Takam Kamga, P.; Bassi, G.; Cassaro, A.; Midolo, M.; Di Trapani, M.; Gatti, A.; Carusone, R.; Resci, F.; Perbellini, O.; Gottardi, M.; et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016, 7, 21713–21727. [Google Scholar] [CrossRef] [Green Version]
- Brenner, A.K.; Nepstad, I.; Bruserud, O. Mesenchymal stem cells support survival and proliferation of primary human acute myeloid leukemia cells through heterogeneous molecular mechanisms. Front. Immunol. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, S.M.; Appelbaum, F.R.; Willman, C.L.; Banker, D.E. Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Exp. Hematol. 2001, 29, 448–457. [Google Scholar] [CrossRef]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Huang, J.C.; Basu, S.K.; Zhao, X.; Chien, S.; Fang, M.; Oehler, V.G.; Appelbaum, F.R.; Becker, P.S. Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J. 2015, 5, e302. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Villar, O.; Garcia, J.L.; Sanchez-Guijo, F.M.; Robledo, C.; Villaron, E.M.; Hernández-Campo, P.; Lopez-Holgado, N.; Diez-Campelo, M.; Barbado, M.V.; Perez-Simon, J.A.; et al. Both expanded and uncultured mesenchymal stem cells from MDS patients are genomically abnormal, showing a specific genetic profile for the 5q-syndrome. Leukemia 2009, 23, 664–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe-Suzuki, S.; Kurata, M.; Abe, S.; Onishi, I.; Kirimura, S.; Nashimoto, M.; Murayama, S.; Hidaka, M.; Kitagawa, M. CXCL12+ stromal cells as bone marrow niche for CD34+ 129-hematopoietic cells and their association with disease progression in myelodysplastic syndromes. Lab. Investig. 2014, 94, 1212–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blau, O.; Baldus, C.D.; Hofmann, W.K.; Thiel, G.; Nolte, F.; Burmeister, T.; Türkmen, S.; Benlasfer, O.; Schümann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von der Heide, E.K.; Neumann, M.; Vosberg, S.; James, A.R.; Schroeder, M.P.; Ortiz-Tanchez, J.; Isaakidis, K.; Schlee, C.; Luther, M.; Jöhrens, K.; et al. Molecular alterations in bone marrow mesenchymal stromal cells derived from acute myeloid leukemia patients. Leukemia 2017, 31, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Rodríguez-Paredes, M.; Jäger, P.; Khandanpour, C.; Cadeddu, R.P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 2016, 30, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Shim, J.S.; Lee, G.Y.; Yim, H.W.; Kim, T.M.; Kim, M.; Leem, S.H.; Lee, J.W.; Min, C.K.; Oh, I.H. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015, 75, 2222–2231. [Google Scholar] [CrossRef] [Green Version]
- Diaz de la Guardia, R.; Lopez-Millan, B.; Lavoie, J.R.; Bueno, C.; Castaño, J.; Gómez-Casares, M.; Vives, S.; Palomo, L.; Juan, M.; Delgado, J.; et al. Detailed Characterization of Mesenchymal Stem/Stromal Cells from a Large Cohort of AML Patients Demonstrates a Definitive Link to Treatment Outcomes. Stem Cell Rep. 2017, 8, 1573–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A.; et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Visconte, V.; Tiu, R.V.; Rogers, H.J. Pathogenesis of myelodysplastic syndromes: An overview of molecular and non-molecular aspects of the disease. Blood Res. 2014, 49, 216–227. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2406. [Google Scholar] [CrossRef]
- Pleyer, L.; Neureiter, D.; Faber, V.; Greil, R. Myelodysplastic syndromes (MDS). In Chronic Myeloid Neoplasias and Clonal Overlap Syndromes: Epidemiology, Pathophysiology and Treatment Options; Greil, R., Pleyer, L., Neureiter, D., Faber, V., Eds.; Springer: Vienna, Austria, 2010; pp. 153–222. [Google Scholar]
- Pang, W.W.; Pluvinage, J.V.; Price, E.A.; Sridhar, K.; Arber, D.A.; Greenberg, P.L.; Schrier, S.L.; Park, C.Y.; Weissman, I.L. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc. Natl. Acad. Sci. USA 2013, 110, 3011–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayyani, F.; Conley, A.P.; Strom, S.S.; Stevenson, W.; Cortes, J.E.; Borthakur, G.; Faderl, S.; O’Brien, S.; Pierce, S.; Kantarjian, H.; et al. Cause of death in patients with lower-risk myelodysplastic syndrome. Cancer 2010, 116, 2174–2179. [Google Scholar] [CrossRef]
- Chang, H.Y.; Rodriguez, V.; Narboni, G.; Bodey, G.P.; Luna, M.A.; Freireich, E.J. Causes of death in adults with acute leukemia. Medicine 1976, 55, 259–268. [Google Scholar] [CrossRef]
- Ma, X.; Does, M.; Raza, A.; Mayne, S.T. Myelodysplastic syndromes: Incidence and survival in the United States. Cancer 2007, 109, 1536–1542. [Google Scholar] [CrossRef] [Green Version]
- Pleyer, L.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; Sill, H.; Schlick, K.; Thaler, J.; Halter, B.; Machherndl-Spandl, S.; Zebisch, A.; et al. Azacitidine front-line in 339 patients with myelodysplastic syndromes and acute myeloid leukaemia: Comparison of French-American-British and World Health Organization classifications. J. Hematol. Oncol. 2016, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Solé, F.; Espinet, B.; Sanz, G.; Cervera, J.; Calasanz, M.J.; Luño, E.; Prieto, F.; Granada, I.; Hernández, J.M.; Cigudosa, J.C.; et al. Incidence, characterization and prognostic significance of chromosomal abnormalities in 640 patients with primary myelodysplastic syndromes. Br. J. Haematol. 2000, 108, 346–356. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzbecker, U.; Meredyth-Stewart, M.; Ehninger, G. The pathogenesis of myelodysplastic syndromes (MDS). Cancer Treat. Rev. 2007, 33, S53–S58. [Google Scholar] [CrossRef]
- Varney, M.E.; Melgar, K.; Niederkorn, M.; Smith, M.A.; Barreyro, L.; Starczynowski, D.T. Deconstructing innate immune signaling in myelodysplastic syndromes. Exp. Hematol. 2015, 43, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Kordasti, S.Y.; Afzali, B.; Lim, Z.; Ingram, W.; Hayden, J.; Barber, L.; Matthews, K.; Chelliah, R.; Guinn, B.; Lombardi, G.; et al. IL-17-producing CD4+ T cells, pro-inflammatory cytokines and apoptosis are increased in low risk myelodysplastic syndrome. Br. J. Haematol. 2009, 145, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. Oncoimmunology 2016, 5, e1062208. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef]
- Zhao, F.; Hoechst, B.; Duffy, A.; Gamrekelashvili, J.; Fioravanti, S.; Manns, M.P.; Greten, T.F.; Korangy, F. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology 2012, 136, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Jiang, H.; Liu, P.; Xie, N.; Fu, R.; Wang, H.; Liu, C.; Zhang, T.; Wang, H.; Shao, Z. Increased myeloid-derived suppressor cells in patients with myelodysplastic syndromes suppress CD8+ T lymphocyte function through the STAT3-ARG1 pathway. Leuk. Lymphoma 2021, 62, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Tao, J.; Fu, R.; Shao, Z. Myeloid-derived suppressor cell cytokine secretion as prognostic factor in myelodysplastic syndromes. Innate Immun. 2020, 26, 703–715. [Google Scholar] [CrossRef]
- Johnson, R.C.; Kurzer, J.H.; Greenberg, P.L.; Gratzinger, D. Mesenchymal stromal cell density is increased in higher grade myelodysplastic syndromes and independently predicts survival. Am. J. Clin. Pathol. 2014, 142, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Corradi, G.; Baldazzi, C.; Očadlíková, D.; Marconi, G.; Parisi, S.; Testoni, N.; Finelli, C.; Cavo, M.; Curti, A.; Ciciarello, M. Mesenchymal stromal cells from myelodysplastic and acute myeloid leukemia patients display in vitro reduced proliferative potential and similar capacity to support leukemia cell survival. Stem Cell Res. Ther. 2018, 9, 271. [Google Scholar] [CrossRef] [Green Version]
- Zhi-Gang, Z.; Wei-Ming, L.; Zhi-Chao, C.; Yong, Y.; Ping, Z. Immunosuppressive properties of mesenchymal stem cells derived from bone marrow of patient with hematological malignant diseases. Leuk. Lymphoma 2008, 49, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Gul-Uludag, H.; Valencia-Serna, J.; Kucharski, C.; Marquez-Curtis, L.A.; Jiang, X.; Larratt, L.; Janowska-Wieczorek, A.; Uludağ, H. Polymeric nanoparticle-mediated silencing of CD44 receptor in CD34 acute myeloid leukemia cells. Leuk. Res. 2014, 38, 1299–1308. [Google Scholar] [CrossRef]
- Soenen-Cornu, V.; Tourino, C.; Bonnet, M.L.; Guillier, M.; Flamant, S.; Kotb, R.; Bernheim, A.; Bourhis, J.H.; Preudhomme, C.; Fenaux, P.; et al. Mesenchymal cells generated from patients with myelodysplastic syndromes are devoid of chromosomal clonal markers and support short- and long-term hematopoiesis in vitro. Oncogene 2005, 24, 2441–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.G.; Xu, W.; Yu, H.P.; Fang, B.L.; Wu, S.H.; Li, F.; Li, W.M.; Li, Q.B.; Chen, Z.C.; Zou, P. Functional characteristics of mesenchymal stem cells derived from bone marrow of patients with myelodysplastic syndromes. Cancer Lett. 2012, 317, 136–143. [Google Scholar] [CrossRef]
- Aanei, C.M.; Flandrin, P.; Eloae, F.Z.; Carasevici, E.; Guyotat, D.; Wattel, E.; Campos, L. Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem Cells Dev. 2012, 21, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Oz, S.; Cadeddu, R.P.; Frobel, J.; Bruckner, B.; Kundgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, S.; Kini, A.; Srivastava, V.M.; Therese, M.M.; Nair, S.C.; Abraham, A.; Mathews, V.; George, B.; Kumar, S.; Venkatraman, A.; et al. Coexistence of aberrant hematopoietic and stromal elements in myelodysplastic syndromes. Blood Cells Mol. Dis. 2017, 66, 37–46. [Google Scholar] [CrossRef]
- Poon, Z.; Dighe, N.; Venkatesan, S.S.; Cheung, A.M.S.; Fan, X.; Bari, S.; Hota, M.; Ghosh, S.; Hwang, W.Y.K. Bone marrow MSCs in MDS: Contribution towards dysfunctional hematopoiesis and potential targets for disease response to hypomethylating therapy. Leukemia 2019, 33, 1487–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaus, M.; Stavroulaki, E.; Kastrinaki, M.C.; Fragioudaki, P.; Giannikou, K.; Psyllaki, M.; Pontikoglou, C.; Tsoukatou, D.; Mamalaki, C.; Papadaki, H.A. Reserves, functional, immunoregulatory, and cytogenetic properties of bone marrow mesenchymal stem cells in patients with myelodysplastic syndromes. Stem Cells Dev. 2010, 19, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Pavlaki, K.; Pontikoglou, C.G.; Demetriadou, A.; Batsali, A.K.; Damianaki, A.; Simantirakis, E.; Kontakis, M.; Galanopoulos, A.; Kotsianidis, I.; Kastrinaki, M.C.; et al. Impaired proliferative potential of bone marrow mesenchymal stromal cells in patients with myelodysplastic syndromes is associated with abnormal WNT signaling pathway. Stem Cells Dev. 2014, 23, 1568–1581. [Google Scholar] [CrossRef]
- Fei, C.; Zhao, Y.; Guo, J.; Gu, S.; Li, X.; Chang, C. Senescence of bone marrow mesenchymal stromal cells is accompanied by activation of p53/p21 pathway in myelodysplastic syndromes. Eur. J. Haematol. 2014, 93, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Fattizzo, B.; Giannotta, J.A.; Barcellini, W. Mesenchymal Stem Cells in Aplastic Anemia and Myelodysplastic Syndromes: The “Seed and Soil” Crosstalk. Int. J. Mol. Sci. 2020, 21, 5438. [Google Scholar] [CrossRef]
- Han, Q.; Sun, Z.; Liu, L.; Chen, B.; Cao, Y.; Li, K.; Zhao, R.C. Impairment in immuno-modulatory function of Flk1(+)CD31(−)CD34(−) MSCs from MDS-RA patients. Leuk. Res. 2007, 31, 1469–1478. [Google Scholar] [CrossRef]
- Wu, L.; Amarachintha, S.; Xu, J.; Oley, F., Jr.; Du, W. Mesenchymal COX2-PG secretome engages NR4A-WNT signalling axis in haematopoietic progenitors to suppress anti-leukaemia immunity. Br. J. Haematol. 2018, 183, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Mora, B.; Maffioli, M. New molecular genetics in the diagnosis and treatment of myeloproliferative neoplasms. Curr. Opin. Hematol. 2016, 23, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 Revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Minciacchi, V.R.; Kumar, R.; Krause, D.S. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells 2021, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Medeiros, L.J.; Hu, S. Chronic Myeloid Leukemia: Beyond BCR-ABL1. Curr. Hematol. Malig. Rep. 2018, 13, 435–445. [Google Scholar] [CrossRef]
- Höglund, M.; Sandin, F.; Simonsson, B. Epidemiology of chronic myeloid leukaemia: An update. Ann. Hematol. 2015, 94 (Suppl. S2), 241–247. [Google Scholar] [CrossRef]
- Cramer, K.; Nieborowska-Skorska, M.; Koptyra, M.; Slupianek, A.; Penserga, E.T.; Eaves, C.J.; Aulitzky, W.; Skorski, T. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008, 68, 6884–6888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Cortes, J.; Kantarjian, H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer 2012, 118, 3123–3127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apperley, J.F. Chronic myeloid leukaemia. Lancet 2015, 385, 1447–1459. [Google Scholar] [CrossRef]
- Kaleem, B.; Shahab, S.; Ahmed, N.; Shamsi, T.S. Chronic Myeloid Leukemia—Prognostic Value of Mutations. Asian Pac. J. Cancer Prev. 2015, 16, 7415–7423. [Google Scholar] [CrossRef] [Green Version]
- Makishima, H.; Jankowska, A.M.; McDevitt, M.A.; O’Keefe, C.; Dujardin, S.; Cazzolli, H.; Przychodzen, B.; Prince, C.; Nicoll, J.; Siddaiah, H.; et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood 2011, 117, e198–e206. [Google Scholar] [CrossRef]
- Loscocco, F.; Visani, G.; Galimberti, S.; Curti, A.; Isidori, A. BCR-ABL independent mechanisms of resistance in chronic myeloid leukemia. Front. Oncol. 2019, 9, 939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.; Clarson, J.; Tang, C.; Vidovic, L.; White, D.L.; Hughes, T.P.; Yong, A.S. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood 2017, 129, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, C.; Romano, A.; Parrinello, N.L.; La Cava, P.; Brundo, M.V.; Bramanti, V.; Stagno, F.; Vigneri, P.; Chiarenza, A.; Palumbo, G.A.; et al. Mesenchymal Stem Cells (MSC) regulate activation of Granulocyte-like Myeloid Derived Suppressor Cells (G-MDSC) in chronic myeloid leukemia patients. PLoS ONE 2016, 11, e0158392. [Google Scholar] [CrossRef]
- Xu, H.; Liu, J.; Shen, N.; Zhao, Z.; Cui, J.; Zhou, S.; Jiang, L.; Zhu, X.; Tang, L.; Liang, H.; et al. The interaction of tumor cells and myeloid-derived suppressor cells in chronic myelogenous leukemia. Leuk. Lymphoma 2020, 61, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Christiansson, L.; Soderlund, S.; Svensson, E.; Mustjoki, S.; Bengtsson, M.; Simonsson, B.; Olsson-Strömberg, U.; Loskog, A.S. Increased level of myeloid-derived suppressor cells, programmed death receptor ligand 1/programmed death receptor 1, and soluble CD25 in Sokal high risk chronic myeloid leukemia. PLoS ONE 2013, 8, e55818. [Google Scholar] [CrossRef] [Green Version]
- Giallongo, C.; Parrinello, N.L.; La Cava, P.; Camiolo, G.; Romano, A.; Scalia, M.; Stagno, F.; Palumbo, G.A.; Avola, R.; Li Volti, G.; et al. Monocytic myeloid-derived suppressor cells as prognostic factor in chronic myeloid leukaemia patients treated with dasatinib. J. Cell. Mol. Med. 2018, 22, 1070–1080. [Google Scholar] [CrossRef] [Green Version]
- Giallongo, C.; Parrinello, N.; Tibullo, D.; La Cava, P.; Romano, A.; Chiarenza, A.; Barbagallo, I.; Palumbo, G.A.; Stagno, F.; Vigneri, P.; et al. Myeloid derived suppressor cells (MDSCs) are increased and exert immunosuppressive activity together with polymorphonuclear leukocytes (PMNs) in chronic myeloid leukemia patients. PLoS ONE 2014, 9, e101848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ureshino, H. Treatment-free remission and immunity in chronic myeloid leukemia. Int. J. Hematol. 2021, 113, 642–647. [Google Scholar] [CrossRef]
- Zhang, X.; Tu, H.; Yang, Y.; Wan, Q.; Fang, L.; Wu, Q.; Li, J. High IL-7 levels in the bone marrow microenvironment mediate imatinib resistance and predict disease progression in chronic myeloid leukemia. Int. J. Hematol. 2016, 104, 358–367. [Google Scholar] [CrossRef]
- Zhang, X.; Tu, H.; Yang, Y.; Jiang, X.; Hu, X.; Luo, Q.; Li, J. Bone marrow-derived mesenchymal stromal cells promote resistance to tyrosine kinase inhibitors in chronic myeloid leukemia via the IL-7/JAK1/STAT5 pathway. J. Biol. Chem. 2019, 294, 12167–12179. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Tabe, Y.; Konoplev, S.; Xu, Y.; Leysath, C.E.; Lu, H.; Kimura, S.; Ohsaka, A.; Rios, M.B.; Calvert, L.; et al. CXCR4 up-regulation by Imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol. Cancer 2008, 7, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vianello, F.; Villanova, F.; Tisato, V.; Lymperi, S.; Ho, K.K.; Gomes, A.R.; Marin, D.; Bonnet, D.; Apperley, J.; Lam, E.W.; et al. Bone marrow mesenchymal stromal cells non-selectively protect chronic myeloid leukemia cells from imatinib-induced apoptosis via the CXCR4/CXCL12 axis. Haematologica 2010, 95, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Lam, E.W.; Soeiro, I.; Tisato, V.; Bonnet, D.; Dazzi, F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: Impact on in vivo tumor growth. Leukemia 2007, 21, 304–310. [Google Scholar] [CrossRef]
- Zhang, H.M.; Zhang, L.S. Influence of human bone marrow mesenchymal stem cells on proliferation of chronic myeloid leukemia cells. Chin. J. Cancer 2009, 28, 29–32. [Google Scholar]
- Talpaz, M.; Mercer, J.; Hehlmann, R. The interferon-alpha revival in CML. In Chronic Myeloid Leukemia; Hehlmann, R., Ed.; Hematologic Malignancies; Springer: Cham, Switzerland, 2016; Volume 94, pp. 207–230. [Google Scholar]
- Han, Y.; Wang, Y.; Xu, Z.; Li, J.; Yang, J.; Li, Y.; Shang, Y.; Luo, J. Effect of bone marrow mesenchymal stem cells from blastic phase chronic myelogenous leukemia on the growth and apoptosis of leukemia cells. Oncol. Rep. 2013, 30, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO Classification and Diagnostic Criteria for Myeloproliferative Neoplasms: Document Summary and in-Depth Discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [Green Version]
- Nasillo, V.; Riva, G.; Paolini, A.; Forghieri, F.; Roncati, L.; Lusenti, B.; Maccaferri, M.; Messerotti, A.; Pioli, V.; Gilioli, A.; et al. Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies. Int. J. Mol. Sci. 2021, 22, 1906. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-Term Survival and Blast Transformation in Molecularly Annotated Essential Thrombocythemia, Polycythemia Vera, and Myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Podoltsev, N.A.; Zeidan, A.M. Epidemiology of the Classical Myeloproliferative Neoplasms: The Four Corners of an Expansive and Complex Map. Blood Rev. 2020, 42, 100706. [Google Scholar] [CrossRef] [PubMed]
- Zoi, K.; Cross, N.C.P. Genomics of Myeloproliferative Neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef]
- Broséus, J.; Alpermann, T.; Wulfert, M.; Brichs, L.F.; Jeromin, S.; Lippert, E.; Rozman, M.; Lifermann, F.; Grossmann, V.; Haferlach, T.; et al. Age, JAK2 V617F and SF3B1 mutations are the main predicting factors for survival in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia 2013, 27, 1826–1831. [Google Scholar] [CrossRef] [Green Version]
- Hermouet, S.; Bigot-Corbel, E.; Gardie, B. Pathogenesis of myeloproliferative neoplasms: Role and mechanisms of chronic inflammation. Mediat. Inflamm. 2015, 2015, 145293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barosi, G. An immune dysregulation in MPN. Curr. Hematol. Malig. Rep. 2014, 9, 331–339. [Google Scholar] [CrossRef]
- Wang, J.C.; Kundra, A.; Andrei, M.; Baptiste, S.; Chen, C.; Wong, C.; Sindhu, H. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk. Res. 2016, 43, 39–43. [Google Scholar] [CrossRef]
- Decker, M.; Martinez-Morentin, L.; Wang, G.; Lee, Y.; Liu, Q.; Leslie, J.; Ding, L. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 2017, 19, 677–688. [Google Scholar] [CrossRef]
- Ramos, T.L.; Sánchez-Abarca, L.I.; Rosón-Burgo, B. Mesenchymal stromal cells (MSC) from JAK2+ myeloproliferative neoplasms differ from normal MSC and contribute to the maintenance of neoplastic hematopoiesis. PLoS ONE 2017, 12, e0182470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, R.K.; Ziegler, S.; Leisten, I.; Ferreira, M.S.; Schumacher, A.; Rath, B.; Fahrenkamp, D.; Müller-Newen, G.; Crysandt, M.; Wilop, S.; et al. Activated fibronectin-secretory phenotype of mesenchymal stromal cells in pre-fibrotic myeloproliferative neoplasms. J. Hematol. Oncol. 2014, 7, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinaud, C.; Desterke, C.; Konopacki, J.; Pieri, L.; Torossian, F.; Golub, R.; Schmutz, S.; Anginot, A.; Guerton, B.; Rochet, N.; et al. Osteogenic Potential of Mesenchymal Stromal Cells Contributes to Primary Myelofibrosis. Cancer Res. 2015, 75, 4753–4765. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Campbell, T.B.; Passegue, E. Normal and Leukemic Stem Cell Niches: Insights and Therapeutic Opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Schmitt-Graeff, A.H.; Nitschke, R.; Zeiser, R. The hematopoietic niche in myeloproliferative neoplasms. Mediat. Inflamm. 2015, 2015, 347270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Manshouri, T.; Estrov, Z.; Quintás-Cardama, A.; Burger, J.; Zhang, Y.; Livun, A.; Knez, L.; Harris, D.; Creighton, C.; Kantarjian, H.M.; et al. Bone marrow stroma-secreted cytokines protect JAK2(V617F)-mutated cells from the effects of a JAK2 inhibitor. Cancer Res. 2011, 71, 3831–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, B.L.; Yen, M.L.; Hsu, P.J.; Liu, K.J.; Wang, C.J.; Bai, C.H.; Sytwu, H. K Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-met and STAT3. Stem Cell Rep. 2013, 1, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.W.; Chen, H.Y.; Wang, L.T.; Wang, F.H.; Fang, L.W.; Lai, H.Y.; Chen, H.H.; Lu, J.; Hung, M.S.; Cheng, Y.; et al. Mesenchymal stem cells tune the development of monocyte-derived dendritic cells toward a myeloid-derived suppressive phenotype through growth-regulated oncogene chemokines. J. Immunol. 2013, 190, 5065–5077. [Google Scholar] [CrossRef] [Green Version]
- Giallongo, C.; Tibullo, D.; Parrinello, N.L.; La Cava, P.; Di Rosa, M.; Bramanti, V.; Di Raimondo, C.; Conticello, C.; Chiarenza, A.; Palumbo, G.A.; et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget 2016, 7, 85764–85775. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.M.; Zhuang, J.; Mundy, G.R. The pathogenesis of the bone disease of multiple myeloma. Bone 2008, 42, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, D.; Wang, J.; Arvindam, S.U.; Hallstrom, C.; Verneris, M.R.; Grzywacz, B.; Warlick, E.; Blazar, B.R.; Miller, J.S. Mesenchymal stromal cells shape the MDS microenvironment by inducing suppressive monocytes that dampen NK cell function. JCI Insight 2020, 5, 130155. [Google Scholar] [CrossRef] [Green Version]
- Kanterman, J.; Sade-Feldman, M.; Baniyash, M. New insights into chronic inflammation-induced immunosuppression. Semin. Cancer Biol. 2012, 22, 307–318. [Google Scholar] [CrossRef]
- Bizymi, N.; Bjelica, S.; Kittang, A.O.; Mojsilovic, S.; Velegraki, M.; Pontikoglou, C.; Roussel, M.; Ersvær, E.; Santibañez, J.F.; Lipoldová, M.; et al. Myeloid-Derived Suppressor Cells in Hematologic Diseases: Promising Biomarkers and Treatment Targets. Hemasphere 2019, 3, e168. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Fulzele, K.; Catic, A.; Sun, C.C.; Dombkowski, D.; Hurley, M.P.; Lezeau, S.; Attar, E.; Wu, J.Y.; Lin, H.Y.; et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. 2013, 19, 1513–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takizawa, H.; Manz, M.G. Impact of inflammation on early hematopoiesis and the microenvironment. Int. J. Hematol. 2017, 106, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Craver, B.M.; El Alaoui, K.; Scherber, R.M.; Fleischman, A.G. The Critical Role of Inflammation in the Pathogenesis and Progression of Myeloid Malignancies. Cancers 2018, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Jitschin, R.; Saul, D.; Braun, M.; Tohumeken, S.; Völkl, S.; Kischel, R.; Lutteropp, M.; Dos Santos, C.; Mackensen, A.; Mougiakakos, D. CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J. Immunother. Cancer 2018, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Aliperta, R.; Cartellieri, M.; Feldmann, A.; Arndt, C.; Koristka, S.; Michalk, I.; von Bonin, M.; Ehninger, A.; Bachmann, J.; Ehninger, G.; et al. Bispecific antibody releasing-mesenchymal stromal cell machinery for retargeting T cells towards acute myeloid leukemia blasts. Blood Cancer J. 2015, 5, e348. [Google Scholar] [CrossRef] [Green Version]
- Leung, S.Y.; Niimi, A.; Noble, A.; Oates, T.; Williams, A.S.; Medicherla, S.; Protter, A.A.; Chung, K.F. Effect of transforming growth factor-beta receptor I kinase inhibitor 2,4-disubstituted pteridine (SD-208) in chronic allergic airway inflammation and remodeling. J. Pharm. Exp. 2006, 319, 586–594. [Google Scholar] [CrossRef]
- Geyh, S.; Rodríguez-Paredes, M.; Jäger, P.; Koch, A.; Bormann, F.; Gutekunst, J.; Zilkens, C.; Germing, U.; Kobbe, G.; Lyko, F.; et al. Transforming growth factor β1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2018, 103, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Blobe, G.C. Role of transforming growth factor-beta in hematologic malignancies. Blood 2006, 107, 4589–4596. [Google Scholar] [CrossRef] [Green Version]
- Mojsilovic, S.; Mojsilovic, S.S.; Bjelica, S.; Santibanez, J.F. Transforming Growth Factor-Beta1 and Myeloid-Derived Suppressor Cells: A Cancerous Partnership. Dev. Dyn. 2021. [Google Scholar] [CrossRef] [PubMed]
- Larson, C.; Oronsky, B.; Carter, C.A.; Oronsky, A.; Knox, S.J.; Sher, D.; Reid, T.R. TGF-beta: A master immune regulator. Expert Opin. Ther. Targets 2020, 24, 427–438. [Google Scholar] [CrossRef]
- Tian, T.; Yu, S.; Liu, L.; Xue, F.; Yuan, C.; Wang, M.; Ji, C.; Ma, D. The profile of T helper subsets in bone marrow microenvironment is distinct for different stages of acute myeloid leukemia patients and chemotherapy partly ameliorates these variations. PLoS ONE 2015, 10, e0131761. [Google Scholar] [CrossRef] [Green Version]
- Tsushima, F.; Yao, S.; Shin, T.; Flies, A.; Flies, S.; Xu, H.; Tamada, K.; Pardoll, D.M.; Chen, L. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood 2007, 110, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Holmström, M.O.; Hasselbalch, H.C.; Andersen, M.H. Cancer Immune Therapy for Philadelphia Chromosome-Negative Chronic Myeloproliferative Neoplasms. Cancers 2020, 12, 1763. [Google Scholar] [CrossRef]
- Wang, H.; Kaur, G.; Sankin, A.I.; Chen, F.; Guan, F.; Zang, X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Ravandi-Kashani, F.; Garcia-Manero, G.; Ning, J.; Alhamal, Z.; DiNardo, C.D.; Pierce, S.A.; Pemmaraju, N.; Patel, K.P.; Blando, J.; Alfayez, M.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Non-randomized, Open-label, Phase 2 Study. Cancer Discov. 2018, 9, 370–383. [Google Scholar]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2V617F Causes PD-L1 Expression, Mediating Immune Escape in Myeloproliferative Neoplasms. Sci. Transl. Med. 2018, 10, eaam7729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, H.; Wu, D. Current status of leukemia cytotherapy—Exploitation with immune cells. Curr. Stem Cell Res. Ther. 2017, 12, 188–196. [Google Scholar] [CrossRef]
- Elsallab, M.; Levine, B.L.; Wayne, A.S.; Abou-El-Enein, M. CAR T-Cell Product Performance in Haematological Malignancies before and after Marketing Authorisation. Lancet Oncol. 2020, 21, e104–e116. [Google Scholar] [CrossRef]
- Lindo, L.; Wilkinson, L.H.; Hay, K.A. Befriending the Hostile Tumor Microenvironment in CAR T-Cell Therapy. Front. Immunol. 2021, 11, 618387. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, L.; Nomdedéu, J.F.; López, O.; Carnicer, M.J.; Bellido, M.; Aventín, A.; Brunet, S.; Sierra, J. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica 2001, 86, 1261–1269. [Google Scholar]
- Mardiros, A.; Dos Santos, C.; McDonald, T.; Brown, C.E.; Wang, X.; Budde, L.E.; Hoffman, L.; Aguilar, B.; Chang, W.C.; Bretzlaff, W.; et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood 2013, 122, 3138–3148. [Google Scholar] [CrossRef] [Green Version]
- Pizzitola, I.; Anjos-Afonso, F.; Rouault-Pierre, K.; Lassailly, F.; Tettamanti, S.; Spinelli, O.; Biondi, A.; Biagi, E.; Bonnet, D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 2014, 28, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Stevens, B.M.; Budde, E.E.; Forman, S.J.; Jordan, C.T.; Purev, E. Anti-CD123 CAR T-cell therapy for the treatment of myelodysplastic syndrome. Blood 2017, 130, 1917. [Google Scholar]
- Askmyr, M.; Agerstam, H.; Hansen, N.; Gordon, S.; Arvanitakis, A.; Rissler, M.; Juliusson, G.; Richter, J.; Järås, M.; Fioretos, T. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood 2013, 121, 3709–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Järås, M.; Johnels, P.; Hansen, N.; Agerstam, H.; Tsapogas, P.; Rissler, M.; Lassen, C.; Olofsson, T.; Bjerrum, O.W.; Richter, J.; et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc. Natl. Acad. Sci. USA 2010, 107, 16280–16285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warda, W.; Larosa, F.; Neto Da Rocha, M.; Trad, R.; Deconinck, E.; Fajloun, Z.; Faure, C.; Caillot, D.; Moldovan, M.; Valmary-Degano, S.; et al. CML Hematopoietic Stem Cells Expressing IL1RAP Can Be Targeted by Chimeric Antigen Receptor-Engineered T Cells. Cancer Res. 2019, 79, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Fultang, L.; Panetti, S.; Ng, M.; Collins, P.; Graef, S.; Rizkalla, N.; Booth, S.; Lenton, R.; Noyvert, B.; Shannon-Lowe, C.; et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine 2019, 47, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Zhang, Y.; Hu, Y.; Mei, H. T Cell Exhaustion and CAR-T Immunotherapy in Hematological Malignancies. BioMed Res. Int. 2021, 2021, 6616391. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.A.; Geumann, U.; Günther, C.; Hermann, F.G.; Abken, H. IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells. Cells 2020, 9, 873. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapor, S.; Santibanez, J.F. Myeloid-Derived Suppressor Cells and Mesenchymal Stem/Stromal Cells in Myeloid Malignancies. J. Clin. Med. 2021, 10, 2788. https://doi.org/10.3390/jcm10132788
Kapor S, Santibanez JF. Myeloid-Derived Suppressor Cells and Mesenchymal Stem/Stromal Cells in Myeloid Malignancies. Journal of Clinical Medicine. 2021; 10(13):2788. https://doi.org/10.3390/jcm10132788
Chicago/Turabian StyleKapor, Suncica, and Juan F. Santibanez. 2021. "Myeloid-Derived Suppressor Cells and Mesenchymal Stem/Stromal Cells in Myeloid Malignancies" Journal of Clinical Medicine 10, no. 13: 2788. https://doi.org/10.3390/jcm10132788