
Dysfunctional High-Density Lipoproteins in Type 2 Diabetes Mellitus: Molecular Mechanisms and Therapeutic Implications

 , , , and
, , , and
Abstract
:1. Background
2. High Density Lipoproteins
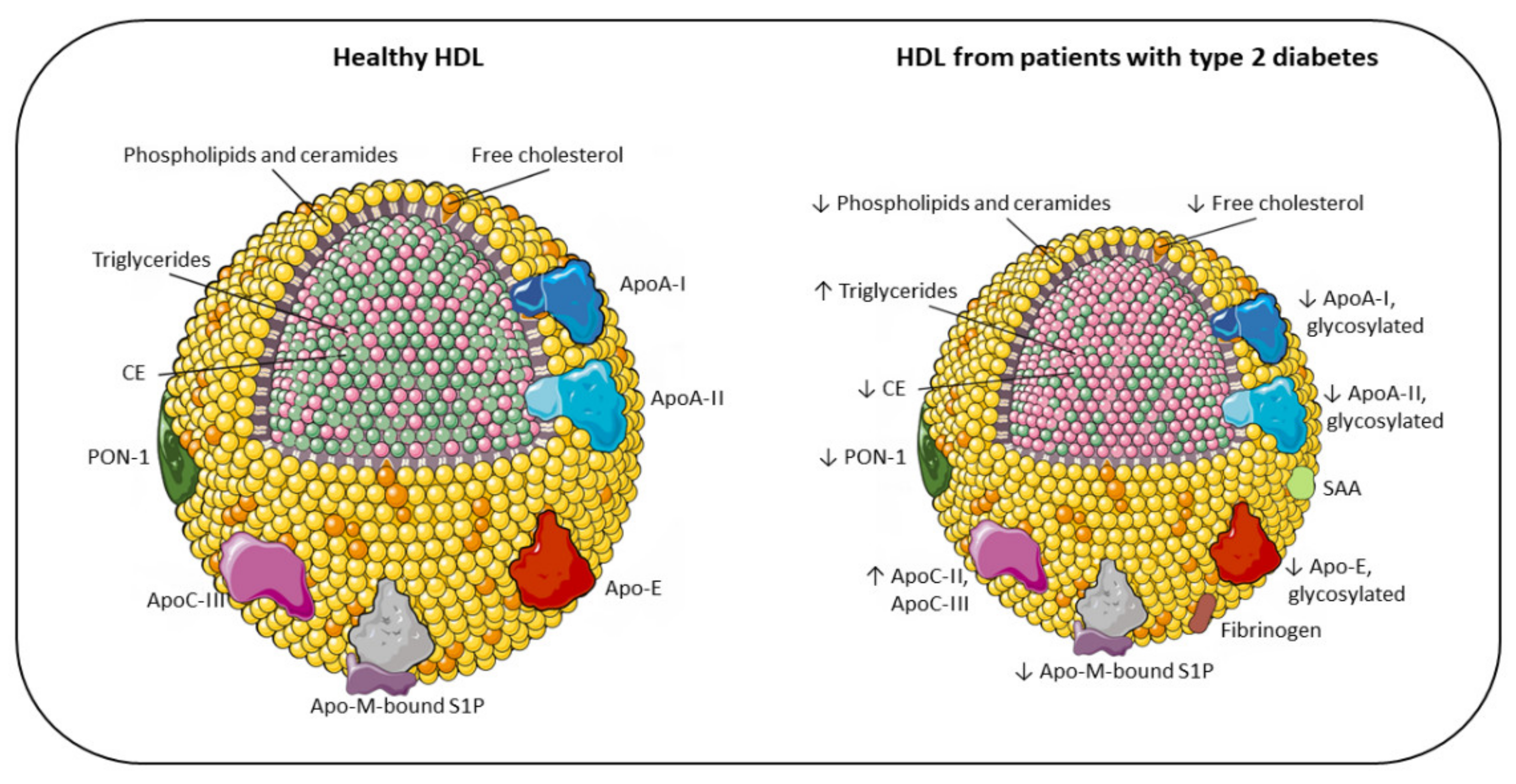
3. Alterations of HDL Plasma Level and Composition in T2DM
3.1. Modifications of HDL-C Plasma Levels
3.2. Modifications of HDL Size
3.3. Modifications of the Lipid Content
3.4. Modifications of the Protein Component
3.5. Modifications of HDL Due to Glycation and Oxidation
4. Alterations of HDL Functions in T2DM
4.1. Impaired Antioxidant Activity
4.2. Impaired Anti-Inflammatory Activity
4.3. Impaired Vasodilator Activity
4.4. Impaired Cholesterol Efflux Capacity
5. Therapeutic Interventions
5.1. Niacin
5.2. CETP Inhibitors
5.3. Peroxisome Proliferator-Activated Receptor (PPAR) Agonists and Fibrates
5.4. Metformin
5.5. Glucagon-Like Peptide-1 (GLP-1) Receptor Agonists
5.6. Dipeptidyl Peptidase-4 Inhibitors
5.7. Sodium-Glucose Cotransporter Type 2 Inhibitors (SGLT2is)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Williams, R.; Karuranga, S.; Malanda, B.; Saeedi, P.; Basit, A.; Besançon, S.; Bommer, C.; Esteghamati, A.; Ogurtsova, K.; Zhang, P.; et al. Global and regional estimates and projections of diabetes-related health expenditure: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2020, 162, 108072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.K.P.; Nicholls, S.J.; Tan, J.T.M.; Bursill, C.A. The Role of High-Density Lipoproteins in Diabetes and Its Vascular Complications. Int. J. Mol. Sci. 2018, 19, 1680. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, S.R.; Osme, A.; Ilchenko, S.; Golizeh, M.; Lee, K.; Wang, S.; Bena, J.; Previs, S.F.; Smith, J.D.; Kasumov, T. Glycation Reduces the Stability of ApoAI and Increases HDL Dysfunction in Diet-Controlled Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macheboeuf, M. Recherches sur les phosphoaminolipides du sérum sanguin. Nature des phospholipides liés aux albumines du sérum de Cheval à l’état de cenapses acido-précipitables. Chim. Biol. 1929, 11, 485–503. [Google Scholar]
- Hutchins, P.M.; Heinecke, J.W. Cholesterol efflux capacity, macrophage reverse cholesterol transport and cardioprotective HDL. Curr. Opin. Lipidol. 2015, 26, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. Handb. Exp. Pharmacol. 2015, 224, 3–51. [Google Scholar] [CrossRef] [Green Version]
- Calabresi, L.; Franceschini, G. Lecithin:cholesterol acyltransferase, high-density lipoproteins, and atheroprotection in humans. Trends Cardiovasc. Med. 2010, 20, 50–53. [Google Scholar] [CrossRef]
- Kujiraoka, T.; Iwasaki, T.; Ishihara, M.; Ito, M.; Nagano, M.; Kawaguchi, A.; Takahashi, S.; Ishi, J.; Tsuji, M.; Egashira, T.; et al. Altered distribution of plasma PAF-AH between HDLs and other lipoproteins in hyperlipidemia and diabetes mellitus. J. Lipid Res. 2003, 44, 2006–2014. [Google Scholar] [CrossRef] [Green Version]
- Lindgren, F.T.; Elliott, H.A.; Gofman, J.W. The ultracentrifugal characterization and isolation of human blood lipids and lipoproteins, with applications to the study of atherosclerosis. J. Phys. Colloid Chem. 1951, 55, 80–93. [Google Scholar] [CrossRef]
- Delalla, O.F.; Elliott, H.A.; Gofman, J.W. Ultracentrifugal studies of high density serum lipoproteins in clinically healthy adults. Am. J. Physiol. 1954, 179, 333–337. [Google Scholar] [CrossRef]
- Chapman, M.J.; Goldstein, S.; Lagrange, D.; Laplaud, P.M. A density gradient ultracentrifugal procedure for the isolation of the major lipoprotein classes from human serum. J. Lipid Res. 1981, 22, 339–358. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef] [PubMed]
- Gowri, M.S.; Van der Westhuyzen, D.R.; Bridges, S.R.; Anderson, J.W. Decreased protection by HDL from poorly controlled type 2 diabetic subjects against LDL oxidation may Be due to the abnormal composition of HDL. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2226–2233. [Google Scholar] [CrossRef] [Green Version]
- Milder, T.Y.; Stocker, S.L.; Abdel Shaheed, C.; McGrath-Cadell, L.; Samocha-Bonet, D.; Greenfield, J.R.; Day, R.O. Combination Therapy with an SGLT2 Inhibitor as Initial Treatment for Type 2 Diabetes: A Systematic Review and Meta-Analysis. J. Clin. Med. 2019, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barter, P.J. The causes and consequences of low levels of high density lipoproteins in patients with diabetes. Diabetes Metab. J. 2011, 35, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, C.C.; Thorpe, S.R.; Fu, M.X.; Harper, C.M.; Yoo, J.; Kim, S.M.; Wong, H.; Peters, A.L. Glycation impairs high-density lipoprotein function. Diabetologia 2000, 43, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Attia, N.; Nakbi, A.; Smaoui, M.; Chaaba, R.; Moulin, P.; Hammami, S.; Hamda, K.B.; Chanussot, F.; Hammami, M. Increased phospholipid transfer protein activity associated with the impaired cellular cholesterol efflux in type 2 diabetic subjects with coronary artery disease. Tohoku J. Exp. Med. 2007, 213, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Gomes Kjerulf, D.; Wang, S.; Omer, M.; Pathak, A.; Subramanian, S.; Han, C.Y.; Tang, C.; den Hartigh, L.J.; Shao, B.; Chait, A. Glycation of HDL blunts its anti-inflammatory and cholesterol efflux capacities in vitro, but has no effect in poorly controlled type 1 diabetes subjects. J. Diabetes Complicat. 2020, 34, 107693. [Google Scholar] [CrossRef]
- Morgantini, C.; Natali, A.; Boldrini, B.; Imaizumi, S.; Navab, M.; Fogelman, A.M.; Ferrannini, E.; Reddy, S.T. Anti-inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes 2011, 60, 2617–2623. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horváth, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus: Atherosclerotic Cardiovascular Disease and Heart Failure in Type 2 Diabetes Mellitus—Mechanisms, Management, and Clinical Considerations. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Rye, K.A.; Duffy, S.J.; Barter, P.; Kingwell, B.A. The emerging role of HDL in glucose metabolism. Nat. Rev. Endocrinol. 2012, 8, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.S.; Toma, L.; Sima, A.V. Dual role of lipoproteins in endothelial cell dysfunction in atherosclerosis. Cell Tissue Res. 2012, 349, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Haase, C.L.; Tybjærg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. HDL Cholesterol and Risk of Type 2 Diabetes: A Mendelian Randomization Study. Diabetes 2015, 64, 3328–3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisar, T.; Couzens, E.; Hwang, A.; Russell, M.; Barlow, C.E.; DeFina, L.F.; Hoofnagle, A.N.; Kim, F. Type 2 diabetes is associated with loss of HDL endothelium protective functions. PLoS ONE 2018, 13, e0192616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikura, K.; Hanai, K.; Shinjyo, T.; Uchigata, Y. HDL cholesterol as a predictor for the incidence of lower extremity amputation and wound-related death in patients with diabetic foot ulcers. Atherosclerosis 2015, 239, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Russo, G.T.; De Cosmo, S.; Viazzi, F.; Pacilli, A.; Ceriello, A.; Genovese, S.; Guida, P.; Giorda, C.; Cucinotta, D.; Pontremoli, R.; et al. Plasma Triglycerides and HDL-C Levels Predict the Development of Diabetic Kidney Disease in Subjects With Type 2 Diabetes: The AMD Annals Initiative. Diabetes Care 2016, 39, 2278–2287. [Google Scholar] [CrossRef] [Green Version]
- Qi, Q.; Liang, L.; Doria, A.; Hu, F.B.; Qi, L. Genetic predisposition to dyslipidemia and type 2 diabetes risk in two prospective cohorts. Diabetes 2012, 61, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Bakogianni, M.C.; Kalofoutis, C.A.; Skenderi, K.I.; Kalofoutis, A.T. Clinical evaluation of plasma high-density lipoprotein subfractions (HDL2, HDL3) in non-insulin-dependent diabetics with coronary artery disease. J. Diabetes Complicat. 2001, 15, 265–269. [Google Scholar] [CrossRef]
- Russo, G.T.; Giandalia, A.; Romeo, E.L.; Alibrandi, A.; Horvath, K.V.; Asztalos, B.F.; Cucinotta, D. Markers of Systemic Inflammation and Apo-AI Containing HDL Subpopulations in Women with and without Diabetes. Int. J. Endocrinol. 2014, 2014, 607924. [Google Scholar] [CrossRef] [Green Version]
- Garvey, W.T.; Kwon, S.; Zheng, D.; Shaughnessy, S.; Wallace, P.; Hutto, A.; Pugh, K.; Jenkins, A.J.; Klein, R.L.; Liao, Y. Effects of insulin resistance and type 2 diabetes on lipoprotein subclass particle size and concentration determined by nuclear magnetic resonance. Diabetes 2003, 52, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, S.; Otvos, J.D.; Rosenson, R.S.; Pradhan, A.; Buring, J.E.; Ridker, P.M. Lipoprotein particle size and concentration by nuclear magnetic resonance and incident type 2 diabetes in women. Diabetes 2010, 59, 1153–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, I.J. Clinical review 124: Diabetic dyslipidemia: Causes and consequences. J. Clin. Endocrinol. Metab. 2001, 86, 965–971. [Google Scholar] [CrossRef]
- Awadallah, S.; Madkour, M.; Hamidi, R.A.; Alwafa, E.A.; Hattab, M.; Zakkour, B.; Al-Matroushi, A.; Ahmed, E.; Al-Kitbi, M. Plasma levels of Apolipoprotein A1 and Lecithin:Cholesterol Acyltransferase in type 2 diabetes mellitus: Correlations with haptoglobin phenotypes. Diabetes Metab. Syndr. 2017, 11 (Suppl. 2), S543–S546. [Google Scholar] [CrossRef]
- Fournier, N.; Myara, I.; Atger, V.; Moatti, N. Reactivity of lecithin-cholesterol acyl transferase (LCAT) towards glycated high-density lipoproteins (HDL). Clin. Chim. Acta 1995, 234, 47–61. [Google Scholar] [CrossRef]
- Rizzo, M.; Otvos, J.; Nikolic, D.; Montalto, G.; Toth, P.P.; Banach, M. Subfractions and subpopulations of HDL: An update. Curr. Med. Chem. 2014, 21, 2881–2891. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Why is HDL functionally deficient in type 2 diabetes? Curr. Diabetes Rep. 2008, 8, 51–59. [Google Scholar] [CrossRef]
- Cardner, M.; Yalcinkaya, M.; Goetze, S.; Luca, E.; Balaz, M.; Hunjadi, M.; Hartung, J.; Shemet, A.; Kränkel, N.; Radosavljevic, S.; et al. Structure-function relationships of HDL in diabetes and coronary heart disease. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Ståhlman, M.; Fagerberg, B.; Adiels, M.; Ekroos, K.; Chapman, J.M.; Kontush, A.; Borén, J. Dyslipidemia, but not hyperglycemia and insulin resistance, is associated with marked alterations in the HDL lipidome in type 2 diabetic subjects in the DIWA cohort: Impact on small HDL particles. Biochim. Biophys. Acta 2013, 1831, 1609–1617. [Google Scholar] [CrossRef]
- Brinck, J.W.; Thomas, A.; Lauer, E.; Jornayvaz, F.R.; Brulhart-Meynet, M.C.; Prost, J.C.; Pataky, Z.; Lofgren, P.; Hoffstedt, J.; Eriksson, M.; et al. Diabetes Mellitus Is Associated With Reduced High-Density Lipoprotein Sphingosine-1-Phosphate Content and Impaired High-Density Lipoprotein Cardiac Cell Protection. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R. Dyslipidemia in Diabetes. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, H.J., Kaltsas, G., et al., Eds.; Endotext: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Zhang, P.; Gao, J.; Pu, C.; Zhang, Y. Apolipoprotein status in type 2 diabetes mellitus and its complications (Review). Mol. Med. Rep. 2017, 16, 9279–9286. [Google Scholar] [CrossRef] [Green Version]
- Caron, S.; Verrijken, A.; Mertens, I.; Samanez, C.H.; Mautino, G.; Haas, J.T.; Duran-Sandoval, D.; Prawitt, J.; Francque, S.; Vallez, E.; et al. Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Aroner, S.A.; Furtado, J.D.; Sacks, F.M.; Tsai, M.Y.; Mukamal, K.J.; McClelland, R.L.; Jensen, M.K. Apolipoprotein C-III and its defined lipoprotein subspecies in relation to incident diabetes: The Multi-Ethnic Study of Atherosclerosis. Diabetologia 2019, 62, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.M.; Davidson, W.S.; Urbina, E.M.; Dolan, L.M.; Heink, A.; Zang, H.; Lu, L.J.; Shah, A.S. Erratum. The Effects of Type 2 Diabetes on Lipoprotein Composition and Arterial Stiffness in Male Youth. Diabetes 2013;62:2958-2967. Diabetes 2016, 65, 2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.P. Diabetes and Protein Glycosylation, 1st ed.; York, S.-V.N., Ed.; Springer: New York, NY, USA, 1986; Volume 1, p. XII. [Google Scholar]
- Pan, B.; Ren, H.; Ma, Y.; Liu, D.; Yu, B.; Ji, L.; Pan, L.; Li, J.; Yang, L.; Lv, X.; et al. High-density lipoprotein of patients with type 2 diabetes mellitus elevates the capability of promoting migration and invasion of breast cancer cells. Int. J. Cancer 2012, 131, 70–82. [Google Scholar] [CrossRef]
- Hermo, R.; Mier, C.; Mazzotta, M.; Tsuji, M.; Kimura, S.; Gugliucci, A. Circulating levels of nitrated apolipoprotein A-I are increased in type 2 diabetic patients. Clin. Chem. Lab. Med. 2005, 43, 601–606. [Google Scholar] [CrossRef]
- Curtiss, L.K.; Witztum, J.L. Plasma apolipoproteins AI, AII, B, CI, and E are glucosylated in hyperglycemic diabetic subjects. Diabetes 1985, 34, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Calvo, C.; Talussot, C.; Ponsin, G.; Berthezene, F. Non enzymatic glycation of apolipoprotein A-I. Effects on its self-association and lipid binding properties. Biochem. Biophys. Res. Commun. 1988, 153, 1060–1067. [Google Scholar] [CrossRef]
- Lapolla, A.; Brioschi, M.; Banfi, C.; Tremoli, E.; Bonfante, L.; Cristoni, S.; Seraglia, R.; Traldi, P. On the search for glycated lipoprotein ApoA-I in the plasma of diabetic and nephropathic patients. J. Mass Spectrom. 2008, 43, 74–81. [Google Scholar] [CrossRef]
- Drew, B.G.; Duffy, S.J.; Formosa, M.F.; Natoli, A.K.; Henstridge, D.C.; Penfold, S.A.; Thomas, W.G.; Mukhamedova, N.; de Courten, B.; Forbes, J.M.; et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 2009, 119, 2103–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobécourt, E.; Tabet, F.; Lambert, G.; Puranik, R.; Bao, S.; Yan, L.; Davies, M.J.; Brown, B.E.; Jenkins, A.J.; Dusting, G.J.; et al. Nonenzymatic glycation impairs the antiinflammatory properties of apolipoprotein A-I. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 766–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Rojo, R.; Perez-Martinez, P.; Lopez-Moreno, J.; Martinez-Botas, J.; Delgado-Lista, J.; van-Ommen, B.; Yubero-Serrano, E.; Camargo, A.; Ordovas, J.M.; Perez-Jimenez, F.; et al. HDL cholesterol efflux normalised to apoA-I is associated with future development of type 2 diabetes: From the CORDIOPREV trial. Sci. Rep. 2017, 7, 12499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viktorinova, A.; Jurkovicova, I.; Fabryova, L.; Kinova, S.; Koren, M.; Stecova, A.; Svitekova, K. Abnormalities in the relationship of paraoxonase 1 with HDL and apolipoprotein A1 and their possible connection to HDL dysfunctionality in type 2 diabetes. Diabetes Res. Clin. Pract. 2018, 140, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [Green Version]
- Azizkhanian, I.; Trenchevska, O.; Bashawri, Y.; Hu, J.; Koska, J.; Reaven, P.D.; Nelson, R.W.; Nedelkov, D.; Yassine, H.N. Posttranslational modifications of apolipoprotein A-II proteoforms in type 2 diabetes. J. Clin. Lipidol. 2016, 10, 808–815. [Google Scholar] [CrossRef] [Green Version]
- Perségol, L.; Vergès, B.; Foissac, M.; Gambert, P.; Duvillard, L. Inability of HDL from type 2 diabetic patients to counteract the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia 2006, 49, 1380–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beliard, S.; Nogueira, J.P.; Maraninchi, M.; Lairon, D.; Nicolay, A.; Giral, P.; Portugal, H.; Vialettes, B.; Valero, R. Parallel increase of plasma apoproteins C-II and C-III in Type 2 diabetic patients. Diabet. Med. 2009, 26, 736–739. [Google Scholar] [CrossRef]
- Vu-Dac, N.; Schoonjans, K.; Kosykh, V.; Dallongeville, J.; Fruchart, J.C.; Staels, B.; Auwerx, J. Fibrates increase human apolipoprotein A-II expression through activation of the peroxisome proliferator-activated receptor. J. Clin. Investig. 1995, 96, 741–750. [Google Scholar] [CrossRef]
- Hiukka, A.; Stahlman, M.; Pettersson, C.; Levin, M.; Adiels, M.; Teneberg, S.; Leinonen, E.S.; Hulten, L.M.; Wiklund, O.; Oresic, M.; et al. ApoCIII-enriched LDL in type 2 diabetes displays altered lipid composition, increased susceptibility for sphingomyelinase, and increased binding to biglycan. Diabetes 2009, 58, 2018–2026. [Google Scholar] [CrossRef] [Green Version]
- Mackness, B.; Durrington, P.N.; Abuashia, B.; Boulton, A.J.; Mackness, M.I. Low paraoxonase activity in type II diabetes mellitus complicated by retinopathy. Clin. Sci. 2000, 98, 355–363. [Google Scholar] [CrossRef]
- Ebtehaj, S.; Gruppen, E.G.; Parvizi, M.; Tietge, U.J.F.; Dullaart, R.P.F. The anti-inflammatory function of HDL is impaired in type 2 diabetes: Role of hyperglycemia, paraoxonase-1 and low grade inflammation. Cardiovasc. Diabetol. 2017, 16, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokri, Y.; Variji, A.; Nosrati, M.; Khonakdar-Tarsi, A.; Kianmehr, A.; Kashi, Z.; Bahar, A.; Bagheri, A.; Mahrooz, A. Importance of paraoxonase 1 (PON1) as an antioxidant and antiatherogenic enzyme in the cardiovascular complications of type 2 diabetes: Genotypic and phenotypic evaluation. Diabetes Res. Clin. Pract. 2020, 161, 108067. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, R.C.; Bisgaier, C.L.; Aviram, M.; Hsu, C.; Billecke, S.; La Du, B.N. Human serum Paraoxonase/Arylesterase’s retained hydrophobic N-terminal leader sequence associates with HDLs by binding phospholipids: Apolipoprotein A-I stabilizes activity. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2214–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastorikou, M.; Mackness, B.; Liu, Y.; Mackness, M. Glycation of paraoxonase-1 inhibits its activity and impairs the ability of high-density lipoprotein to metabolize membrane lipid hydroperoxides. Diabet. Med. 2008, 25, 1049–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Qian, M.M.; Liu, P.L.; Zhang, L.; Wang, Y.; Liu, D.H. Glycation of high-density lipoprotein triggers oxidative stress and promotes the proliferation and migration of vascular smooth muscle cells. J. Geriatr. Cardiol. 2017, 14, 473–480. [Google Scholar] [CrossRef]
- Nobécourt, E.; Jacqueminet, S.; Hansel, B.; Chantepie, S.; Grimaldi, A.; Chapman, M.J.; Kontush, A. Defective antioxidative activity of small dense HDL3 particles in type 2 diabetes: Relationship to elevated oxidative stress and hyperglycaemia. Diabetologia 2005, 48, 529–538. [Google Scholar] [CrossRef]
- Lotfollahi, Z.; Dawson, J.; Fitridge, R.; Bursill, C. The anti-inflammatory and pro-angiogenic properties of high-density lipoproteins (HDL): An emerging role in diabetic wound healing. Adv. Wound Care 2020. [Google Scholar] [CrossRef]
- Liu, D.; Ji, L.; Zhang, D.; Tong, X.; Pan, B.; Liu, P.; Zhang, Y.; Huang, Y.; Su, J.; Willard, B.; et al. Nonenzymatic glycation of high-density lipoprotein impairs its anti-inflammatory effects in innate immunity. Diabetes Metab. Res. Rev. 2012, 28, 186–195. [Google Scholar] [CrossRef]
- Mao, J.Y.; Sun, J.T.; Yang, K.; Shen, W.F.; Lu, L.; Zhang, R.Y.; Tong, X.; Liu, Y. Serum amyloid A enrichment impairs the anti-inflammatory ability of HDL from diabetic nephropathy patients. J. Diabetes Complicat. 2017, 31, 1538–1543. [Google Scholar] [CrossRef]
- Adedayo, A.; Eluwole, A.; Tedla, F.; Kremer, A.; Mastrogiovanni, N.; Khan, M.; Rosenberg, C.; Dreizen, P.; La Rosa, J.; Salciccioli, L.; et al. Association between nitrated lipoproteins and vascular function in type 2 diabetes. Front. Biosci. Landmark Ed. 2021, 26, 644–663. [Google Scholar] [CrossRef]
- Shao, B.; Pennathur, S.; Heinecke, J.W. Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. J. Biol. Chem. 2012, 287, 6375–6386. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Kurano, M.; Nanya, M.; Shimizu, T.; Ohkawa, R.; Tozuka, M.; Yatomi, Y. Glycation of HDL Polymerizes Apolipoprotein M and Attenuates Its Capacity to Bind to Sphingosine 1-Phosphate. J. Atheroscler. Thromb. 2020. [Google Scholar] [CrossRef] [PubMed]
- Whetzel, A.M.; Bolick, D.T.; Srinivasan, S.; Macdonald, T.L.; Morris, M.A.; Ley, K.; Hedrick, C.C. Sphingosine-1 phosphate prevents monocyte/endothelial interactions in type 1 diabetic NOD mice through activation of the S1P1 receptor. Circ. Res. 2006, 99, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Ebtehaj, S.; Gruppen, E.G.; Bakker, S.J.L.; Dullaart, R.P.F.; Tietge, U.J.F. HDL (High-Density Lipoprotein) Cholesterol Efflux Capacity Is Associated With Incident Cardiovascular Disease in the General Population. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1874–1883. [Google Scholar] [CrossRef] [PubMed]
- Hunjadi, M.; Lamina, C.; Kahler, P.; Bernscherer, T.; Viikari, J.; Lehtimaki, T.; Kahonen, M.; Hurme, M.; Juonala, M.; Taittonen, L.; et al. HDL cholesterol efflux capacity is inversely associated with subclinical cardiovascular risk markers in young adults: The cardiovascular risk in Young Finns study. Sci. Rep. 2020, 10, 19223. [Google Scholar] [CrossRef] [PubMed]
- Soria-Florido, M.T.; Castaner, O.; Lassale, C.; Estruch, R.; Salas-Salvado, J.; Martinez-Gonzalez, M.A.; Corella, D.; Ros, E.; Aros, F.; Elosua, R.; et al. Dysfunctional High-Density Lipoproteins Are Associated With a Greater Incidence of Acute Coronary Syndrome in a Population at High Cardiovascular Risk: A Nested Case-Control Study. Circulation 2020, 141, 444–453. [Google Scholar] [CrossRef]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Shiu, S.W.; Wong, Y.; Tan, K.C. Pre-beta1 HDL in type 2 diabetes mellitus. Atherosclerosis 2017, 263, 24–28. [Google Scholar] [CrossRef]
- He, Y.; Ronsein, G.E.; Tang, C.; Jarvik, G.P.; Davidson, W.S.; Kothari, V.; Song, H.D.; Segrest, J.P.; Bornfeldt, K.E.; Heinecke, J.W. Diabetes Impairs Cellular Cholesterol Efflux From ABCA1 to Small HDL Particles. Circ. Res. 2020, 127, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Apro, J.; Tietge, U.J.; Dikkers, A.; Parini, P.; Angelin, B.; Rudling, M. Impaired Cholesterol Efflux Capacity of High-Density Lipoprotein Isolated From Interstitial Fluid in Type 2 Diabetes Mellitus-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 787–791. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Tang, C.; Heinecke, J.W.; Oram, J.F. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J. Lipid Res. 2010, 51, 1849–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Lebedy, D.; Rasheed, E.; Kafoury, M.; Abd-El Haleem, D.; Awadallah, E.; Ashmawy, I. Anti-apolipoprotein A-1 autoantibodies as risk biomarker for cardiovascular diseases in type 2 diabetes mellitus. J. Diabetes Complicat. 2016, 30, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Dullaart, R.P.F.; Pagano, S.; Perton, F.G.; Vuilleumier, N. Antibodies Against the C-Terminus of ApoA-1 Are Inversely Associated with Cholesterol Efflux Capacity and HDL Metabolism in Subjects with and without Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2019, 20, 732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dullaart, R.P.; Annema, W.; de Boer, J.F.; Tietge, U.J. Pancreatic beta-cell function relates positively to HDL functionality in well-controlled type 2 diabetes mellitus. Atherosclerosis 2012, 222, 567–573. [Google Scholar] [CrossRef]
- Low, H.; Hoang, A.; Forbes, J.; Thomas, M.; Lyons, J.G.; Nestel, P.; Bach, L.A.; Sviridov, D. Advanced glycation end-products (AGEs) and functionality of reverse cholesterol transport in patients with type 2 diabetes and in mouse models. Diabetologia 2012, 55, 2513–2521. [Google Scholar] [CrossRef] [Green Version]
- Yassine, H.N.; Belopolskaya, A.; Schall, C.; Stump, C.S.; Lau, S.S.; Reaven, P.D. Enhanced cholesterol efflux to HDL through the ABCA1 transporter in hypertriglyceridemia of type 2 diabetes. Metabolism 2014, 63, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.C.; Tai, E.S.; Sviridov, D.; Nestel, P.J.; Ng, C.; Chan, E.; Teo, Y.; Wai, D.C. Relationships between cholesterol efflux and high-density lipoprotein particles in patients with type 2 diabetes mellitus. J. Clin. Lipidol. 2011, 5, 467–473. [Google Scholar] [CrossRef]
- Linton, M.R.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, H.J., Kaltsas, G., et al., Eds.; Endotext: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Zhou, H.; Tan, K.C.; Shiu, S.W.; Wong, Y. Determinants of leukocyte adenosine triphosphate-binding cassette transporter G1 gene expression in type 2 diabetes mellitus. Metabolism 2008, 57, 1135–1140. [Google Scholar] [CrossRef]
- Machado-Lima, A.; Iborra, R.T.; Pinto, R.S.; Castilho, G.; Sartori, C.H.; Oliveira, E.R.; Okuda, L.S.; Nakandakare, E.R.; Giannella-Neto, D.; Machado, U.F.; et al. In type 2 diabetes mellitus glycated albumin alters macrophage gene expression impairing ABCA1-mediated cholesterol efflux. J. Cell. Physiol. 2015, 230, 1250–1257. [Google Scholar] [CrossRef]
- Gordon, D.J.; Probstfield, J.L.; Garrison, R.J.; Neaton, J.D.; Castelli, W.P.; Knoke, J.D.; Jacobs, D.R.; Bangdiwala, S.; Tyroler, H.A. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 1989, 79, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Ertek, S. High-density Lipoprotein (HDL) Dysfunction and the Future of HDL. Curr. Vasc. Pharmacol. 2018, 16, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Rye, K.A.; Tardif, J.C.; Waters, D.D.; Boekholdt, S.M.; Breazna, A.; Kastelein, J.J. Effect of torcetrapib on glucose, insulin, and hemoglobin A1c in subjects in the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial. Circulation 2011, 124, 555–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodor, E.T.; Offermanns, S. Nicotinic acid: An old drug with a promising future. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S68–S75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birjmohun, R.S.; Hutten, B.A.; Kastelein, J.J.; Stroes, E.S. Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds: A meta-analysis of randomized controlled trials. J. Am. Coll. Cardiol. 2005, 45, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, I.M.; Shishehbor, M.H.; Ansell, B.J. High-density lipoprotein as a therapeutic target: A systematic review. JAMA 2007, 298, 786–798. [Google Scholar] [CrossRef]
- van der Hoorn, J.W.; de Haan, W.; Berbee, J.F.; Havekes, L.M.; Jukema, J.W.; Rensen, P.C.; Princen, H.M. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden.CETP mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2016–2022. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.H.; Kamanna, V.S.; Ganji, S.H.; Xiong, X.M.; Kashyap, M.L. Niacin increases HDL biogenesis by enhancing DR4-dependent transcription of ABCA1 and lipidation of apolipoprotein A-I in HepG2 cells. J. Lipid Res. 2012, 53, 941–950. [Google Scholar] [CrossRef] [Green Version]
- Zakiev, E.; Feng, M.; Sukhorukov, V.; Kontush, A. HDL-Targeting Therapeutics: Past, Present and Future. Curr. Pharm. Des. 2017, 23, 1207–1215. [Google Scholar] [CrossRef]
- Franceschini, G.; Favari, E.; Calabresi, L.; Simonelli, S.; Bondioli, A.; Adorni, M.P.; Zimetti, F.; Gomaraschi, M.; Coutant, K.; Rossomanno, S.; et al. Differential effects of fenofibrate and extended-release niacin on high-density lipoprotein particle size distribution and cholesterol efflux capacity in dyslipidemic patients. J. Clin. Lipidol. 2013, 7, 414–422. [Google Scholar] [CrossRef]
- Gomaraschi, M.; Adorni, M.P.; Banach, M.; Bernini, F.; Franceschini, G.; Calabresi, L. Effects of established hypolipidemic drugs on HDL concentration, subclass distribution, and function. Handb. Exp. Pharmacol. 2015, 224, 593–615. [Google Scholar] [CrossRef] [Green Version]
- Khera, A.V.; Patel, P.J.; Reilly, M.P.; Rader, D.J. The addition of niacin to statin therapy improves high-density lipoprotein cholesterol levels but not metrics of functionality. J. Am. Coll. Cardiol. 2013, 62, 1909–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W.; Investigators, A.-H. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; Collins, R.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, J.; Chan, D.C.; Hamilton, S.J.; Tenneti, V.S.; Watts, G.F.; Barrett, P.H. Effect of niacin on high-density lipoprotein apolipoprotein A-I kinetics in statin-treated patients with type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 427–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, H.E.; Brinton, E.A.; Triscari, J.; Chen, E.; Maccubbin, D.; MacLean, A.A.; Gibson, K.L.; Ruck, R.A.; Johnson-Levonas, A.O.; O’Neill, E.A.; et al. Extended-release niacin/laropiprant significantly improves lipid levels in type 2 diabetes mellitus irrespective of baseline glycemic control. Vasc. Health Risk Manag. 2015, 11, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Li, Y.; Wen, A. Effect of niacin on lipids and glucose in patients with type 2 diabetes: A meta-analysis of randomized, controlled clinical trials. Clin. Nutr. 2015, 34, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Masana, L.; Cabre, A.; Heras, M.; Amigo, N.; Correig, X.; Martinez-Hervas, S.; Real, J.T.; Ascaso, J.F.; Quesada, H.; Julve, J.; et al. Remarkable quantitative and qualitative differences in HDL after niacin or fenofibrate therapy in type 2 diabetic patients. Atherosclerosis 2015, 238, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.; Holmes, M.V.; Preiss, D. Cholesteryl Ester Transfer Protein Inhibition for Preventing Cardiovascular Events: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 477–487. [Google Scholar] [CrossRef]
- Toth, P. Torcetrapib And Atherosclerosis: What Happened And Where Do We Go From Here? Future Lipidol. 2007, 2, 277–284. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Maranhão, R.; Freitas, F. HDL Metabolism and Atheroprotection: Predictive Value of Lipid Transfers. Adv. Clin. Chem. 2014, 65, 1–41. [Google Scholar] [PubMed]
- Stalenhoef, A.F.; Davidson, M.H.; Robinson, J.G.; Burgess, T.; Duttlinger-Maddux, R.; Kallend, D.; Goldberg, A.C.; Bays, H. Efficacy and safety of dalcetrapib in type 2 diabetes mellitus and/or metabolic syndrome patients, at high cardiovascular disease risk. Diabetes Obes. Metab. 2012, 14, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Kumar, A.; Patel, D.R.; St John, J.; Riesmeyer, J.; Weerakkody, G.; Ruotolo, G.; Wolski, K.E.; McErlean, E.; Cremer, P.C.; et al. Effect of CETP inhibition with evacetrapib in patients with diabetes mellitus enrolled in the ACCELERATE trial. BMJ Open Diabetes Res. Care 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, S.J.; Ray, K.K.; Ballantyne, C.M.; Beacham, L.A.; Miller, D.L.; Ruotolo, G.; Nissen, S.E.; Riesmeyer, J.S.; Investigators, A. Comparative effects of cholesteryl ester transfer protein inhibition, statin or ezetimibe on lipid factors: The ACCENTUATE trial. Atherosclerosis 2017, 261, 12–18. [Google Scholar] [CrossRef]
- Cannon, C.P.; Shah, S.; Dansky, H.M.; Davidson, M.; Brinton, E.A.; Gotto, A.M.; Stepanavage, M.; Liu, S.X.; Gibbons, P.; Ashraf, T.B.; et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N. Engl. J. Med. 2010, 363, 2406–2415. [Google Scholar] [CrossRef] [Green Version]
- Metzinger, M.P.; Saldanha, S.; Gulati, J.; Patel, K.V.; El-Ghazali, A.; Deodhar, S.; Joshi, P.H.; Ayers, C.; Rohatgi, A. Effect of Anacetrapib on Cholesterol Efflux Capacity: A Substudy of the DEFINE Trial. J. Am. Heart Assoc. 2020, 9, e018136. [Google Scholar] [CrossRef]
- Asleh, R.; Miller-Lotan, R.; Aviram, M.; Hayek, T.; Yulish, M.; Levy, J.E.; Miller, B.; Blum, S.; Milman, U.; Shapira, C.; et al. Haptoglobin genotype is a regulator of reverse cholesterol transport in diabetes in vitro and in vivo. Circ. Res. 2006, 99, 1419–1425. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.P.; Hochberg, I.; Jablonski, K.; Resnick, H.E.; Lee, E.T.; Best, L.; Howard, B.V.; Strong Heart, S. Haptoglobin phenotype is an independent risk factor for cardiovascular disease in individuals with diabetes: The Strong Heart Study. J. Am. Coll. Cardiol. 2002, 40, 1984–1990. [Google Scholar] [CrossRef] [Green Version]
- Bowman, L.; Chen, F.; Sammons, E.; Hopewell, J.C.; Wallendszus, K.; Stevens, W.; Valdes- Marquez, E.; Wiviott, S.; Cannon, C.P.; Braunwald, E.; et al. Randomized Evaluation of the Effects of Anacetrapib through Lipid-modification (REVEAL)-A large-scale, randomized, placebo-controlled trial of the clinical effects of anacetrapib among people with established vascular disease: Trial design, recruitment, and baseline characteristics. Am. Heart J. 2017, 187, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; Sammons, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Jakob, T.; Nordmann, A.J.; Schandelmaier, S.; Ferreira-Gonzalez, I.; Briel, M. Fibrates for primary prevention of cardiovascular disease events. Cochrane Database Syst. Rev. 2016, 11, CD009753. [Google Scholar] [CrossRef]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013, 549627. [Google Scholar] [CrossRef] [Green Version]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, H.N.; Elam, M.B.; Lovato, L.C.; Crouse, J.R.; Leiter, L.A.; Linz, P.; Friedewald, W.T.; Buse, J.B.; Gerstein, H.C.; Probstfield, J.; et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N. Engl. J. Med. 2010, 362, 1563–1574. [Google Scholar] [CrossRef]
- Linz, P.E.; Lovato, L.C.; Byington, R.P.; O’Connor, P.J.; Leiter, L.A.; Weiss, D.; Force, R.W.; Crouse, J.R.; Ismail-Beigi, F.; Simmons, D.L.; et al. Paradoxical reduction in HDL-C with fenofibrate and thiazolidinedione therapy in type 2 diabetes: The ACCORD Lipid Trial. Diabetes Care 2014, 37, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Tsunoda, F.; Asztalos, I.B.; Horvath, K.V.; Steiner, G.; Schaefer, E.J.; Asztalos, B.F. Fenofibrate, HDL, and cardiovascular disease in Type-2 diabetes: The DAIS trial. Atherosclerosis 2016, 247, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Maranghi, M.; Hiukka, A.; Badeau, R.; Sundvall, J.; Jauhiainen, M.; Taskinen, M.R. Macrophage cholesterol efflux to plasma and HDL in subjects with low and high homocysteine levels: A FIELD substudy. Atherosclerosis 2011, 219, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Triolo, M.; Annema, W.; de Boer, J.F.; Tietge, U.J.; Dullaart, R.P. Simvastatin and bezafibrate increase cholesterol efflux in men with type 2 diabetes. Eur. J. Clin. Investig. 2014, 44, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.A.; Arora, R.R. Fibrates in the prevention of cardiovascular disease in patients with type 2 diabetes mellitus--a pooled meta-analysis of randomized placebo-controlled clinical trials. Int. J. Cardiol. 2010, 141, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.R.; Lincoff, A.M.; Mudaliar, S.; Rabbia, M.; Chognot, C.; Herz, M. Effect of the dual peroxisome proliferator-activated receptor-alpha/gamma agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): A phase II, randomised, dose-ranging study. Lancet 2009, 374, 126–135. [Google Scholar] [CrossRef]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; Group, K.S. Efficacy and Safety of Pemafibrate Versus Fenofibrate in Patients with High Triglyceride and Low HDL Cholesterol Levels: A Multicenter, Placebo-Controlled, Double-Blind, Randomized Trial. J. Atheroscler. Thromb. 2018, 25, 521–538. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, S.; Yamashita, S.; Arai, H.; Araki, E.; Yokote, K.; Suganami, H.; Fruchart, J.C.; Kodama, T.; Group, K.S. Effects of K-877, a novel selective PPARalpha modulator (SPPARMalpha), in dyslipidaemic patients: A randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis 2016, 249, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Pemafibrate, a New Selective PPARalpha Modulator: Drug Concept and Its Clinical Applications for Dyslipidemia and Metabolic Diseases. Curr. Atheroscler. Rep. 2020, 22, 5. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes, A. Standards of Medical Care in Diabetes-2020 Abridged for Primary Care Providers. Clin. Diabetes 2020, 38, 10–38. [Google Scholar] [CrossRef] [Green Version]
- Gebrie, D.; Getnet, D.; Manyazewal, T. Cardiovascular safety and efficacy of metformin-SGLT2i versus metformin-sulfonylureas in type 2 diabetes: Systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2021, 11, 137. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Anabtawi, A.; Miles, J.M. Metformin: Nonglycemic Effects and Potential Novel Indications. Endocr. Pract. 2016, 22, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Role of Glucose and Lipids in the Atherosclerotic Cardiovascular Disease of Patients with Diabetes. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; Endotext: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Buse, J.B.; Tan, M.H.; Prince, M.J.; Erickson, P.P. The effects of oral anti-hyperglycaemic medications on serum lipid profiles in patients with type 2 diabetes. Diabetes Obes. Metab. 2004, 6, 133–156. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Cheng, P.C.; Tu, S.T.; Hsu, S.R.; Cheng, Y.C.; Liu, Y.H. Effect of metformin monotherapy on serum lipid profile in statin-naive individuals with newly diagnosed type 2 diabetes mellitus: A cohort study. PeerJ 2018, 6, e4578. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742. [Google Scholar] [CrossRef]
- Scisciola, L.; Rizzo, M.R.; Cataldo, V.; Fontanella, R.A.; Balestrieri, M.L.; D’Onofrio, N.; Marfella, R.; Paolisso, G.; Barbieri, M. Incretin drugs effect on epigenetic machinery: New potential therapeutic implications in preventing vascular diabetic complications. FASEB J. 2020, 34, 16489–16503. [Google Scholar] [CrossRef]
- Sommese, L.; Zullo, A.; Mancini, F.P.; Fabbricini, R.; Soricelli, A.; Napoli, C. Clinical relevance of epigenetics in the onset and management of type 2 diabetes mellitus. Epigenetics 2017, 12, 401–415. [Google Scholar] [CrossRef]
- Bethel, M.A.; Patel, R.A.; Merrill, P.; Lokhnygina, Y.; Buse, J.B.; Mentz, R.J.; Pagidipati, N.J.; Chan, J.C.; Gustavson, S.M.; Iqbal, N.; et al. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: A meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 105–113. [Google Scholar] [CrossRef]
- Escobar, C.; Barrios, V.; Cosin, J.; Gamez Martinez, J.M.; Huelmos Rodrigo, A.I.; Ortiz Cortes, C.; Torres Llergo, J.; Requeijo, C.; Sola, I.; Martinez Zapata, M.J. SGLT2 inhibitors and GLP1 agonists administered without metformin compared to other glucose-lowering drugs in patients with type 2 diabetes mellitus to prevent cardiovascular events: A systematic review. Diabet. Med. 2021, 38, e14502. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Masson, W.; Lavalle-Cobo, A.; Lobo, M.; Masson, G.; Molinero, G. Novel antidiabetic drugs and risk of cardiovascular events in patients without baseline metformin use: A meta-analysis. Eur. J. Prev. Cardiol. 2021, 28, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, S.L.; Rorth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Kober, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Nowrouzi-Sohrabi, P.; Soroush, N.; Tabrizi, R.; Shabani-Borujeni, M.; Rezaei, S.; Jafari, F.; Hosseini-Bensenjan, M.; Stricker, B.H.; van Hoek, M.; Ahmadizar, F. Effect of Liraglutide on Cardiometabolic Risk Profile in People with Coronary Artery Disease with or without Type 2 Diabetes: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Pharmacol. 2021, 12, 618208. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Wu, S.; Wang, J.; Guo, S.; Chai, S.; Yang, Z.; Li, L.; Zhang, Y.; Ji, L.; Zhan, S. Effect of glucagon-like peptide-1 receptor agonists on lipid profiles among type 2 diabetes: A systematic review and network meta-analysis. Clin. Ther. 2015, 37, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Blonde, L.; Pencek, R.; MacConell, L. Association among weight change, glycemic control, and markers of cardiovascular risk with exenatide once weekly: A pooled analysis of patients with type 2 diabetes. Cardiovasc. Diabetol. 2015, 14, 12. [Google Scholar] [CrossRef] [Green Version]
- Tuttolomondo, A.; Cirrincione, A.; Casuccio, A.; Del Cuore, A.; Daidone, M.; Di Chiara, T.; Di Raimondo, D.; Corte, V.D.; Maida, C.; Simonetta, I.; et al. Efficacy of dulaglutide on vascular health indexes in subjects with type 2 diabetes: A randomized trial. Cardiovasc. Diabetol. 2021, 20, 1. [Google Scholar] [CrossRef]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Shanmugasundaram, M.; Pineda, J.R.E.; Murugapandian, S. Glucose-Lowering Medications and Cardiovascular Outcomes. Curr. Cardiol. Rep. 2021, 23, 24. [Google Scholar] [CrossRef]
- White, W.B.; Cannon, C.P.; Heller, S.R.; Nissen, S.E.; Bergenstal, R.M.; Bakris, G.L.; Perez, A.T.; Fleck, P.R.; Mehta, C.R.; Kupfer, S.; et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N. Engl. J. Med. 2013, 369, 1327–1335. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, M.; Katagiri, T.; Suzuki, H.; Matsunaga, S.; HYamada, M.; Ikarashi, T.; Yamamoto, M.; Furukawa, K.; Iwanaga, M.; Hatta, M.; et al. A 52-week randomized controlled trial of ipragliflozin or sitagliptin in type 2 diabetes combined with metformin: The N-ISM study. Diabetes Obes. Metab. 2021, 23, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Monami, M.; Vitale, V.; Ambrosio, M.L.; Bartoli, N.; Toffanello, G.; Ragghianti, B.; Monami, F.; Marchionni, N.; Mannucci, E. Effects on lipid profile of dipeptidyl peptidase 4 inhibitors, pioglitazone, acarbose, and sulfonylureas: Meta-analysis of placebo-controlled trials. Adv. Ther. 2012, 29, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.A.; Park, Y.M.; Yun, J.S.; Lim, T.S.; Song, K.H.; Yoo, K.D.; Ahn, Y.B.; Ko, S.H. A comparison of effects of DPP-4 inhibitor and SGLT2 inhibitor on lipid profile in patients with type 2 diabetes. Lipids Health Dis. 2017, 16, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchigami, A.; Shigiyama, F.; Kitazawa, T.; Okada, Y.; Ichijo, T.; Higa, M.; Hiyoshi, T.; Inoue, I.; Iso, K.; Yoshii, H.; et al. Efficacy of dapagliflozin versus sitagliptin on cardiometabolic risk factors in Japanese patients with type 2 diabetes: A prospective, randomized study (DIVERSITY-CVR). Cardiovasc. Diabetol. 2020, 19, 1. [Google Scholar] [CrossRef] [Green Version]
- Hsia, D.S.; Grove, O.; Cefalu, W.T. An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Solini, A.; Seghieri, M.; Giannini, L.; Biancalana, E.; Parolini, F.; Rossi, C.; Dardano, A.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. The Effects of Dapagliflozin on Systemic and Renal Vascular Function Display an Epigenetic Signature. J. Clin. Endocrinol. Metab. 2019, 104, 4253–4263. [Google Scholar] [CrossRef]
- Sanchez-Garcia, A.; Simental-Mendia, M.; Millan-Alanis, J.M.; Simental-Mendia, L.E. Effect of sodium-glucose co-transporter 2 inhibitors on lipid profile: A systematic review and meta-analysis of 48 randomized controlled trials. Pharmacol. Res. 2020, 160, 105068. [Google Scholar] [CrossRef]
- Filippas-Ntekouan, S.; Tsimihodimos, V.; Filippatos, T.; Dimitriou, T.; Elisaf, M. SGLT-2 inhibitors: Pharmacokinetics characteristics and effects on lipids. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1113–1121. [Google Scholar] [CrossRef]
- Fadini, G.P.; Bonora, B.M.; Zatti, G.; Vitturi, N.; Iori, E.; Marescotti, M.C.; Albiero, M.; Avogaro, A. Effects of the SGLT2 inhibitor dapagliflozin on HDL cholesterol, particle size, and cholesterol efflux capacity in patients with type 2 diabetes: A randomized placebo-controlled trial. Cardiovasc. Diabetol. 2017, 16, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Fukui, T.; Nakanishi, N.; Yamamoto, S.; Tomoyasu, M.; Osamura, A.; Ohara, M.; Yamamoto, T.; Ito, Y.; Hirano, T. Dapagliflozin decreases small dense low-density lipoprotein-cholesterol and increases high-density lipoprotein 2-cholesterol in patients with type 2 diabetes: Comparison with sitagliptin. Cardiovasc. Diabetol. 2017, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breder, I.; Cunha Breder, J.; Bonilha, I.; Munhoz, D.B.; Medorima, S.T.K.; Oliveira, D.C.; do Carmo, H.R.; Moreira, C.; Kontush, A.; Zimetti, F.; et al. Rationale and design of the expanded combination of evolocumab plus empagliflozin in diabetes: EXCEED-BHS3 trial. Ther. Adv. Chronic Dis. 2020, 11, 2040622320959248. [Google Scholar] [CrossRef] [PubMed]
- Barylski, M.; Toth, P.P.; Nikolic, D.; Banach, M.; Rizzo, M.; Montalto, G. Emerging therapies for raising high-density lipoprotein cholesterol (HDL-C) and augmenting HDL particle functionality. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Otocka-Kmiecik, A.; Mikhailidis, D.P.; Nicholls, S.J.; Davidson, M.; Rysz, J.; Banach, M. Dysfunctional HDL: A novel important diagnostic and therapeutic target in cardiovascular disease? Prog. Lipid Res. 2012, 51, 314–324. [Google Scholar] [CrossRef]


{kind=link}
| Changes in HDL Particle | Consequence in T2DM |
|---|---|
| Alterations in Plasma Level and HDL Phenotype | |
| 🡫 HDL plasma level | |
| Change in HDL size |
|
| Lipid content | |
| Changes in HDL Components | |
| 🡫 ApoA-I |
|
| |
| 🡫 ApoA-II |
|
| 🡫 ApoM-S1P |
|
| |
| 🡩 ApoC-II |
|
| 🡩 ApoC-III |
|
| |
| |
| 🡫 PON-1 |
|
| |
| |
| 🡫 PAF-AH |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonilha, I.; Zimetti, F.; Zanotti, I.; Papotti, B.; Sposito, A.C. Dysfunctional High-Density Lipoproteins in Type 2 Diabetes Mellitus: Molecular Mechanisms and Therapeutic Implications. J. Clin. Med. 2021, 10, 2233. https://doi.org/10.3390/jcm10112233
Bonilha I, Zimetti F, Zanotti I, Papotti B, Sposito AC. Dysfunctional High-Density Lipoproteins in Type 2 Diabetes Mellitus: Molecular Mechanisms and Therapeutic Implications. Journal of Clinical Medicine. 2021; 10(11):2233. https://doi.org/10.3390/jcm10112233
Chicago/Turabian StyleBonilha, Isabella, Francesca Zimetti, Ilaria Zanotti, Bianca Papotti, and Andrei C. Sposito. 2021. "Dysfunctional High-Density Lipoproteins in Type 2 Diabetes Mellitus: Molecular Mechanisms and Therapeutic Implications" Journal of Clinical Medicine 10, no. 11: 2233. https://doi.org/10.3390/jcm10112233