
Bringing GPCR Structural Biology to Medical Applications: Insights from Both V2 Vasopressin and Mu-Opioid Receptors

 ,
, 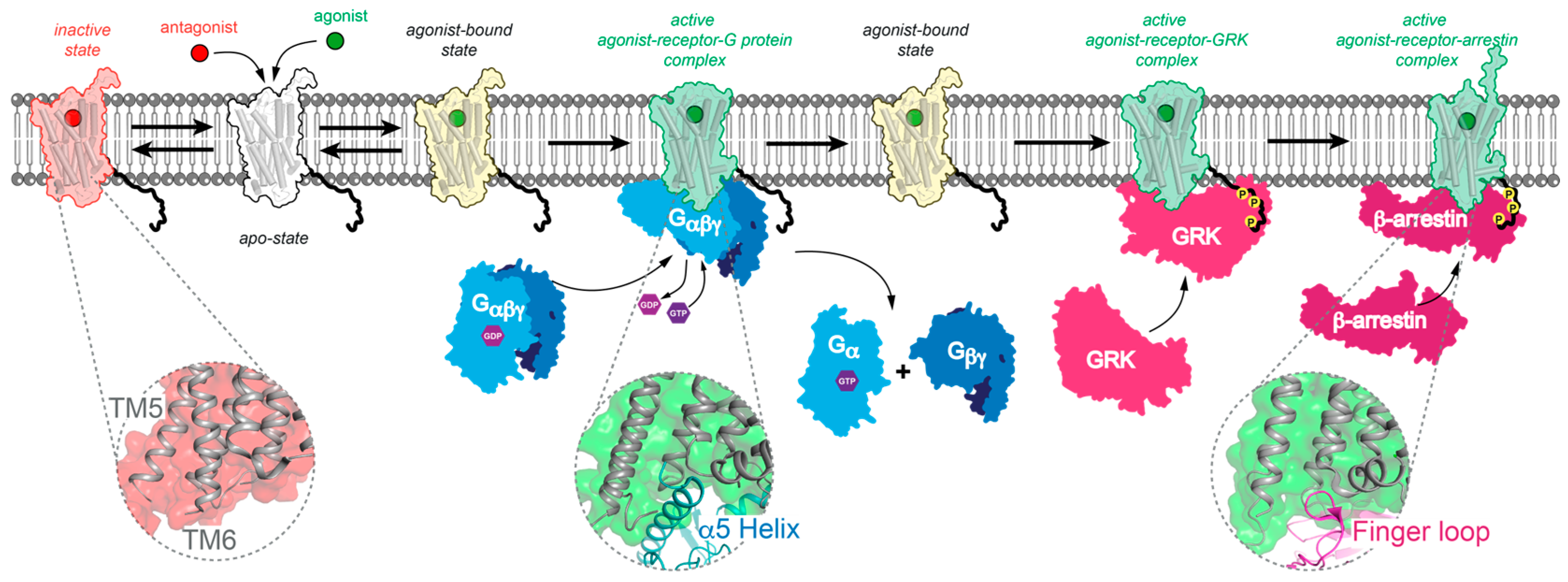{kind=link}
{kind=link}
{kind=link}
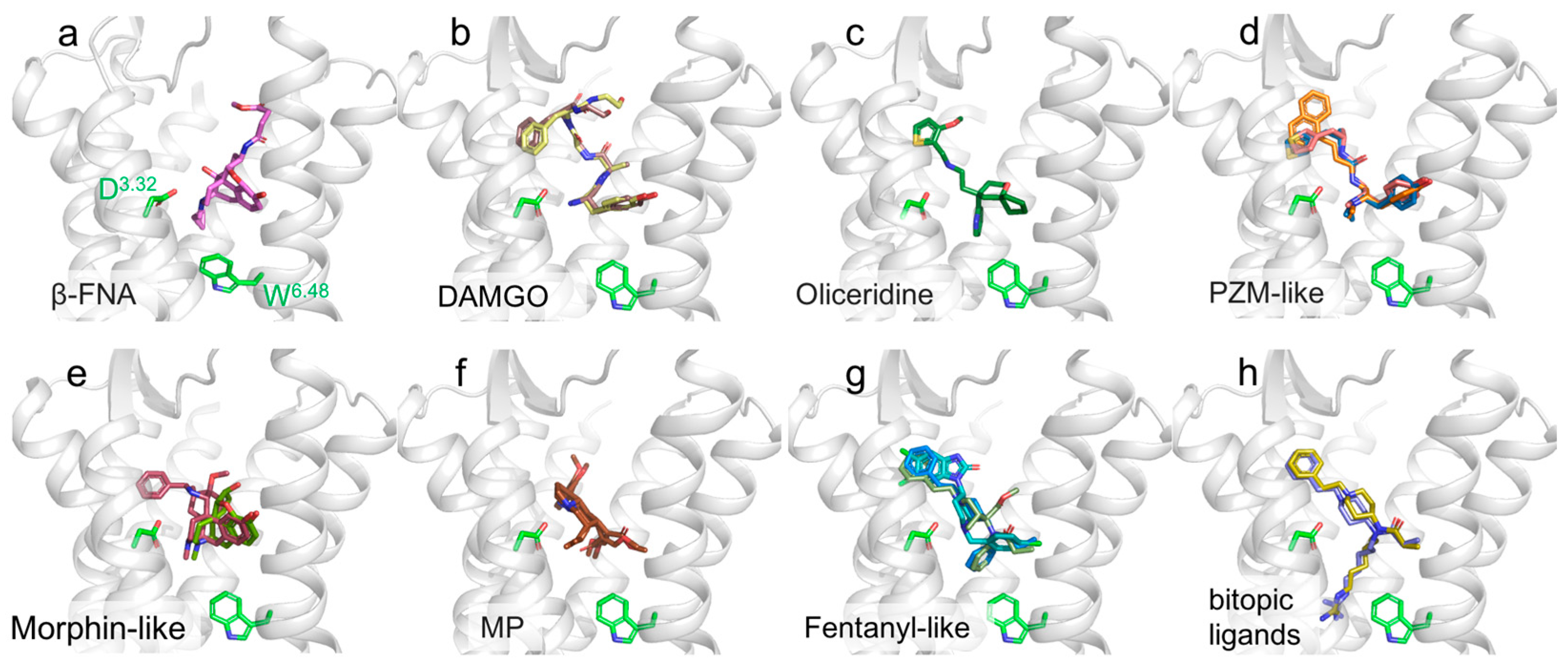{kind=link}
{kind=link}
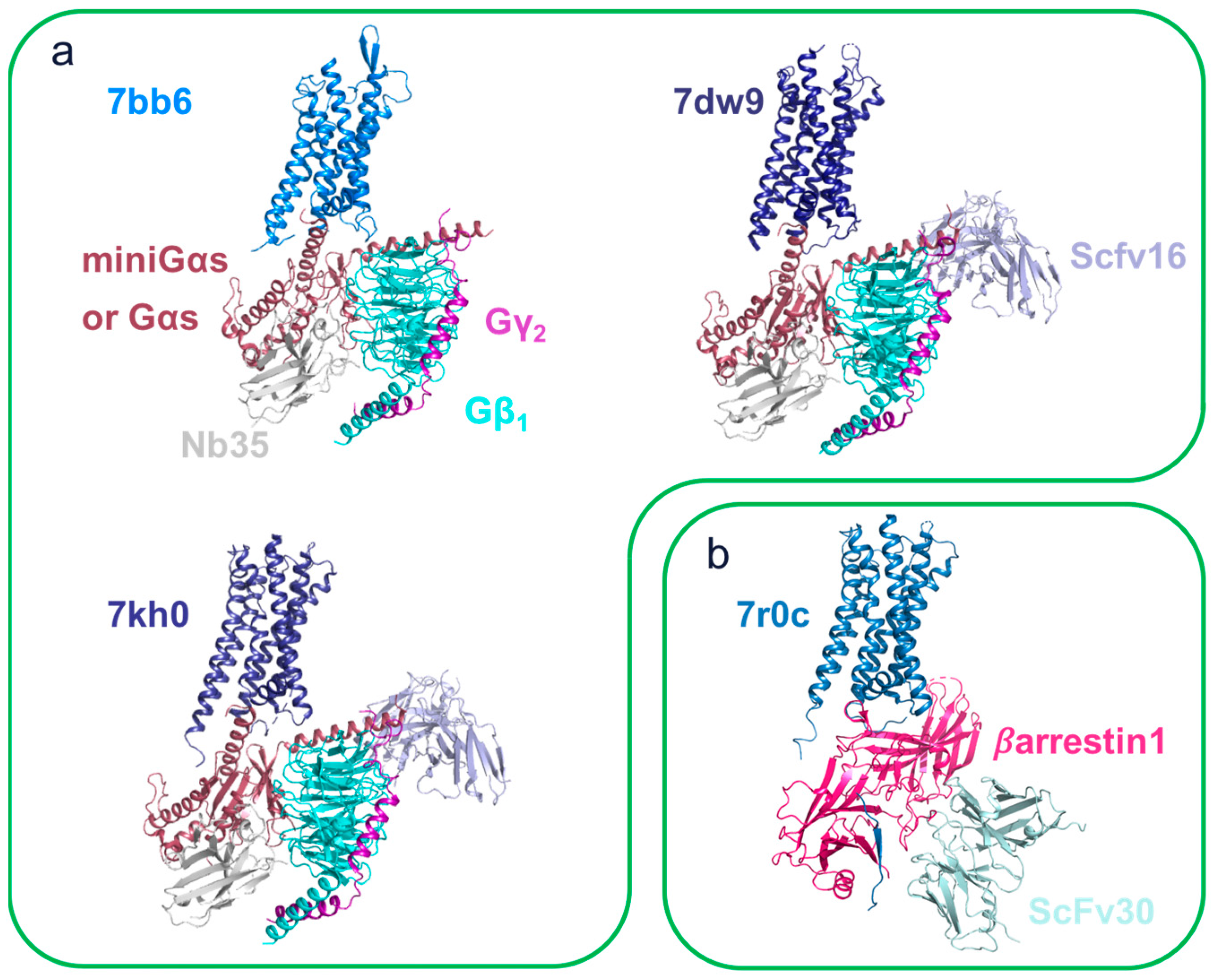{kind=link}
Abstract
:1. G Protein-Coupled Receptor (GPCR) Drug Targets
2. Structural Development Methods to Characterize GPCRs
3. µOR and V2R: Two Representative Peptide Class A GPCRs
4. Solving High-Resolution GPCRs Structures by X-ray Crystallography and Cryo-Electron Microscopy
4.1. X-ray Crystallography to Investigate µOR
4.2. Cryo-EM on µOR G-Protein Activated Structures
4.3. Cryo-EM, the Case of V2R
5. Solution NMR and Molecular Dynamics—Key Tools to Decipher the Dynamic of GPCRs
5.1. Solution NMR on µOR
5.2. Solution NMR on V2R Receptor, Cut Complementary Information
6. Concluding Thoughts
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gacasan, S.B.; Baker, D.L.; Parrill, A.L. G Protein-Coupled Receptors: The Evolution of Structural Insight. AIMS Biophys. 2017, 4, 491–527. [Google Scholar] [CrossRef]
- Munk, C.; Isberg, V.; Mordalski, S.; Harpsøe, K.; Rataj, K.; Hauser, A.S.; Kolb, P.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: The G Protein-Coupled Receptor Database—An Introduction. Br. J. Pharmacol. 2016, 173, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Tuteja, N. Signaling through G Protein Coupled Receptors. Plant Signal. Behav. 2009, 4, 942–947. [Google Scholar] [CrossRef] [Green Version]
- Keri, D.; Barth, P. Reprogramming G Protein Coupled Receptor Structure and Function. Curr. Opin. Struct. Biol. 2018, 51, 187–194. [Google Scholar] [CrossRef]
- Cook, J.L. G Protein-Coupled Receptors as Disease Targets: Emerging Paradigms. Ochsner J. 2010, 10, 2–7. [Google Scholar]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of Signalling and Biased Agonism in G Protein-Coupled Receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR Drug Discovery: Integrating Solution NMR Data with Crystal and Cryo-EM Structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Audet, M.; Garneau, P.; Pelletier, J.; Bouvier, M. High-Throughput Screening of G Protein-Coupled Receptor Antagonists Using a Bioluminescence Resonance Energy Transfer 1-Based Beta-Arrestin2 Recruitment Assay. J. Biomol. Screen. 2005, 10, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Albizu, L.; Teppaz, G.; Seyer, R.; Bazin, H.; Ansanay, H.; Manning, M.; Mouillac, B.; Durroux, T. Toward Efficient Drug Screening by Homogeneous Assays Based on the Development of New Fluorescent Vasopressin and Oxytocin Receptor Ligands. J. Med. Chem. 2007, 50, 4976–4985. [Google Scholar] [CrossRef] [PubMed]
- Mouillac, B.; Manning, M.; Durroux, T. Fluorescent Agonists and Antagonists for Vasopressin/Oxytocin G Protein-Coupled Receptors: Usefulness in Ligand Screening Assays and Receptor Studies. Mini Rev. Med. Chem. 2008, 8, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Zwier, J.M.; Roux, T.; Cottet, M.; Durroux, T.; Douzon, S.; Bdioui, S.; Gregor, N.; Bourrier, E.; Oueslati, N.; Nicolas, L.; et al. A Fluorescent Ligand-Binding Alternative Using Tag-Lite® Technology. J. Biomol. Screen. 2010, 15, 1248–1259. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.T.; Pitis, P.; Liu, G.; Yuan, C.; Gotchev, D.; Cowan, C.L.; Rominger, D.H.; Koblish, M.; Dewire, S.M.; Crombie, A.L.; et al. Structure-Activity Relationships and Discovery of a G Protein Biased Mu Opioid Receptor Ligand, [(3-Methoxythiophen-2-Yl)Methyl]({2-[(9R)-9-(Pyridin-2-Yl)-6-Oxaspiro-[4.5]Decan- 9-Yl]Ethyl})Amine (TRV130), for the Treatment of Acute Severe Pain. J. Med. Chem. 2013, 56, 8019–8031. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L.; Irwin, J.J.; Shoichet, B.K. Discovery of New GPCR Ligands to Illuminate New Biology. Nat. Chem. Biol. 2017, 13, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korczynska, M.; Clark, M.J.; Valant, C.; Xu, J.; Moo, E.V.; Albold, S.; Weiss, D.R.; Torosyan, H.; Huang, W.; Kruse, A.C.; et al. Structure-Based Discovery of Selective Positive Allosteric Modulators of Antagonists for the M(2) Muscarinic Acetylcholine Receptor. Proc. Natl. Acad. Sci. USA 2018, 115, E2419–E2428. [Google Scholar] [CrossRef] [Green Version]
- Khoshouei, M.; Radjainia, M.; Baumeister, W.; Danev, R. Cryo-EM Structure of Haemoglobin at 3.2 Å Determined with the Volta Phase Plate. Nat. Commun. 2017, 8, 16099. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Wang, J.; Zhang, X.; Yang, Z.; Zhang, J.-C.; Zhao, L.; Peng, H.-L.; Lei, J.; Wang, H.-W. Single Particle Cryo-EM Reconstruction of 52 KDa Streptavidin at 3.2 Angstrom Resolution. Nat. Commun. 2019, 10, 2386. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Leiro, R.; Scheres, S.H.W. Unravelling Biological Macromolecules with Cryo-Electron Microscopy. Nature 2016, 537, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal Structure of Rhodopsin: A G Protein-Coupled Receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.G.F.; Choi, H.-J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.P.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal Structure of the Human Beta2 Adrenergic G-Protein-Coupled Receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR Structure Models and Ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.G.F.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal Structure of the Β2 Adrenergic Receptor-Gs Protein Complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal Structure of Rhodopsin Bound to Arrestin by Femtosecond X-Ray Laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the Neurotensin Receptor 1 in Complex with β-Arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef]
- Yin, W.; Li, Z.; Jin, M.; Yin, Y.-L.; de Waal, P.W.; Pal, K.; Yin, Y.; Gao, X.; He, Y.; Gao, J.; et al. A Complex Structure of Arrestin-2 Bound to a G Protein-Coupled Receptor. Cell Res. 2019, 29, 971–983. [Google Scholar] [CrossRef]
- Cao, C.; Barros-Álvarez, X.; Zhang, S.; Kim, K.; Dämgen, M.A.; Panova, O.; Suomivuori, C.-M.; Fay, J.F.; Zhong, X.; Krumm, B.E.; et al. Signaling Snapshots of a Serotonin Receptor Activated by the Prototypical Psychedelic LSD. Neuron 2022, 110, 3154–3167.e7. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Tate, C.G. Active State Structures of G Protein-Coupled Receptors Highlight the Similarities and Differences in the G Protein and Arrestin Coupling Interfaces. Curr. Opin. Struct. Biol. 2017, 45, 124–132. [Google Scholar] [CrossRef]
- Bous, J.; Fouillen, A.; Orcel, H.; Trapani, S.; Cong, X.; Fontanel, S.; Saint-Paul, J.; Lai-Kee-Him, J.; Urbach, S.; Sibille, N.; et al. Structure of the Vasopressin Hormone-V2 Receptor-β-Arrestin1 Ternary Complex. Sci. Adv. 2022, 8, eabo7761. [Google Scholar] [CrossRef]
- Casiraghi, M.; Point, E.; Pozza, A.; Moncoq, K.; Banères, J.-L.; Catoire, L.J. NMR Analysis of GPCR Conformational Landscapes and Dynamics. Mol. Cell. Endocrinol. 2019, 484, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Granier, S.; Bourguet, W.; Seyer, R.; Rahmeh, R.; Mouillac, B.; Pascal, R.; Mendre, C.; Déméné, H. Structure of the Third Intracellular Loop of the Vasopressin V2 Receptor and Conformational Changes upon Binding to GC1qR. J. Mol. Biol. 2009, 388, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Guillien, M.; Mouhand, A.; Fournet, A.; Gontier, A.; Martí Navia, A.; Cordeiro, T.N.; Allemand, F.; Thureau, A.; Banères, J.-L.; Bernadó, P.; et al. Structural Insights into the Intrinsically Disordered GPCR C-Terminal Region, Major Actor in Arrestin-GPCR Interaction. Biomolecules 2022, 12, 617. [Google Scholar] [CrossRef]
- Bokoch, M.P.; Zou, Y.; Rasmussen, S.G.F.; Liu, C.W.; Nygaard, R.; Rosenbaum, D.M.; Fung, J.J.; Choi, H.-J.; Thian, F.S.; Kobilka, T.S.; et al. Ligand-Specific Regulation of the Extracellular Surface of a G-Protein-Coupled Receptor. Nature 2010, 463, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Biased Signaling Pathways in Β2-Adrenergic Receptor Characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef] [Green Version]
- Kofuku, Y.; Ueda, T.; Okude, J.; Shiraishi, Y.; Kondo, K.; Mizumura, T.; Suzuki, S.; Shimada, I. Functional Dynamics of Deuterated Β2 -Adrenergic Receptor in Lipid Bilayers Revealed by NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 2014, 53, 13376–13379. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural Insights into the Dynamic Process of Β2-Adrenergic Receptor Signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Cottet, M.; Faklaris, O.; Maurel, D.; Scholler, P.; Doumazane, E.; Trinquet, E.; Pin, J.-P.; Durroux, T. BRET and Time-Resolved FRET Strategy to Study GPCR Oligomerization: From Cell Lines toward Native Tissues. Front. Endocrinol. 2012, 3, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghuraman, H.; Chatterjee, S.; Das, A. Site-Directed Fluorescence Approaches for Dynamic Structural Biology of Membrane Peptides and Proteins. Front. Mol. Biosci. 2019, 6, 96. [Google Scholar] [CrossRef] [Green Version]
- Fanucci, G.E.; Cafiso, D.S. Recent Advances and Applications of Site-Directed Spin Labeling. Curr. Opin. Struct. Biol. 2006, 16, 644–653. [Google Scholar] [CrossRef]
- Bordignon, E.; Bleicken, S. New Limits of Sensitivity of Site-Directed Spin Labeling Electron Paramagnetic Resonance for Membrane Proteins. Biochim. Biophys. Acta Biomembr. 2018, 1860, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Li, J.; Yuan, S.; Shui, W. Probing the Structures of G Protein-Coupled Receptors with Mass Spectrometry-Based Techniques. Int. J. Mass Spectrom. 2023, 483, 116968. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-Dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [Green Version]
- Connor, M.; Christie, M.D. Opioid Receptor Signalling Mechanisms. Clin. Exp. Pharmacol. Physiol. 1999, 26, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Schmid, C.L.; Groer, C.E.; Bohn, L.M. Functional Selectivity at the μ-Opioid Receptor: Implications for Understanding Opioid Analgesia and Tolerance. Pharmacol. Rev. 2011, 63, 1001–1019. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, A.; Schmiedel, F.; Sianati, S.; Bailey, A.; Bateman, J.T.; Levitt, E.S.; Williams, J.T.; Christie, M.J.; Schulz, S. Phosphorylation-Deficient G-Protein-Biased μ-Opioid Receptors Improve Analgesia and Diminish Tolerance but Worsen Opioid Side Effects. Nat. Commun. 2019, 10, 367. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, A.; Gillis, A.; Hill, R.; Schmiedel, F.; Bailey, C.; Kelly, E.; Henderson, G.; Christie, M.J.; Schulz, S. Morphine-Induced Respiratory Depression Is Independent of β-Arrestin2 Signalling. Br. J. Pharmacol. 2020, 177, 2923–2931. [Google Scholar] [CrossRef] [Green Version]
- Treschan, T.A.; Peters, J. The Vasopressin System: Physiology and Clinical Strategies. Anesthesiology 2006, 105, 599–612; quiz 639–640. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, J.E.; Iezzi, M.; Vischer, U.M. Desmopressin (DDAVP) Induces NO Production in Human Endothelial Cells via V2 Receptor- and CAMP-Mediated Signaling. J. Thromb. Haemost. JTH 2003, 1, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Kumagami, H.; Loewenheim, H.; Beitz, E.; Wild, K.; Schwartz, H.; Yamashita, K.; Schultz, J.; Paysan, J.; Zenner, H.P.; Ruppersberg, J.P. The Effect of Anti-Diuretic Hormone on the Endolymphatic Sac of the Inner Ear. Pflugers Arch. 1998, 436, 970–975. [Google Scholar] [CrossRef]
- Goldvaser, H.; Rozen-Zvi, B.; Yerushalmi, R.; Gafter-Gvili, A.; Lahav, M.; Shepshelovich, D. Malignancy Associated SIADH: Characterization and Clinical Implications. Acta Oncol. Stockh. Swed. 2016, 55, 1190–1195. [Google Scholar] [CrossRef]
- Mentrasti, G.; Scortichini, L.; Torniai, M.; Giampieri, R.; Morgese, F.; Rinaldi, S.; Berardi, R. Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH): Optimal Management. Ther. Clin. Risk Manag. 2020, 16, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic Kidney Disease. Nat. Rev. Dis. Primer 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Seyedabadi, M.; Ghahremani, M.H.; Albert, P.R. Biased Signaling of G Protein Coupled Receptors (GPCRs): Molecular Determinants of GPCR/Transducer Selectivity and Therapeutic Potential. Pharmacol. Ther. 2019, 200, 148–178. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, E.; Kumari, P.; Jaiman, D.; Shukla, A.K. Methodological Advances: The Unsung Heroes of the GPCR Structural Revolution. Nat. Rev. Mol. Cell Biol. 2015, 16, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal Structure of the Μ-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. In Receptor Molecular Biology; Sealfon, S.C., Ed.; Methods in Neurosciences; Academic Press: Cambridge, MA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- He, L.; Fong, J.; von Zastrow, M.; Whistler, J.L. Regulation of Opioid Receptor Trafficking and Morphine Tolerance by Receptor Oligomerization. Cell 2002, 108, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Sun, X.; Bohn, L.M.; Sadée, W. Opioid Receptor Homo- and Heterodimerization in Living Cells by Quantitative Bioluminescence Resonance Energy Transfer. Mol. Pharmacol. 2005, 67, 2173–2184. [Google Scholar] [CrossRef] [Green Version]
- Möller, J.; Isbilir, A.; Sungkaworn, T.; Osberg, B.; Karathanasis, C.; Sunkara, V.; Grushevskyi, E.O.; Bock, A.; Annibale, P.; Heilemann, M.; et al. Single-Molecule Analysis Reveals Agonist-Specific Dimer Formation of µ-Opioid Receptors. Nat. Chem. Biol. 2020, 16, 946–954. [Google Scholar] [CrossRef]
- Neilan, C.L.; Husbands, S.M.; Breeden, S.; Ko, M.C.H.; Aceto, M.D.; Lewis, J.W.; Woods, J.H.; Traynor, J.R. Characterization of the Complex Morphinan Derivative BU72 as a High Efficacy, Long-Lasting Mu-Opioid Receptor Agonist. Eur. J. Pharmacol. 2004, 499, 107–116. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural Insights into Μ-Opioid Receptor Activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the Μ-Opioid Receptor-G(i) Protein Complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Sounier, R.; Mas, C.; Steyaert, J.; Laeremans, T.; Manglik, A.; Huang, W.; Kobilka, B.K.; Déméné, H.; Granier, S. Propagation of Conformational Changes during μ-Opioid Receptor Activation. Nature 2015, 524, 375–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husbands, S.M.; Neilan, C.L.; Broadbear, J.; Grundt, P.; Breeden, S.; Aceto, M.D.; Woods, J.H.; Lewis, J.W.; Traynor, J.R. BU74, a Complex Oripavine Derivative with Potent Kappa Opioid Receptor Agonism and Delayed Opioid Antagonism. Eur. J. Pharmacol. 2005, 509, 117–125. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Liu, M.R.; Woods, J.H.; Medzihradsky, F. Binding Affinity and Selectivity of Opioids at Mu, Delta and Kappa Receptors in Monkey Brain Membranes. J. Pharmacol. Exp. Ther. 1994, 271, 1630–1637. [Google Scholar] [PubMed]
- Minami, M.; Onogi, T.; Nakagawa, T.; Katao, Y.; Aoki, Y.; Katsumata, S.; Satoh, M. DAMGO, a Mu-Opioid Receptor Selective Ligand, Distinguishes between Mu-and Kappa-Opioid Receptors at a Different Region from That for the Distinction between Mu- and Delta-Opioid Receptors. FEBS Lett. 1995, 364, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Nafría, J.; Tate, C.G. Cryo-EM Structures of GPCRs Coupled to G(s), G(i) and G(O). Mol. Cell. Endocrinol. 2019, 488, 1–13. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Pan, Y.-X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hetzer, F.; Huang, W.; Qu, Q.; Meyerowitz, J.; Kaindl, J.; Hübner, H.; Skiniotis, G.; Kobilka, B.K.; Gmeiner, P. Structure-Based Evolution of G Protein-Biased μ-Opioid Receptor Agonists. Angew. Chem. Int. Ed. Engl. 2022, 61, e202200269. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Wang, Y.; He, B.; He, X.; Zhou, X.E.; Guo, S.; Rao, Q.; Yang, J.; Liu, J.; Zhou, Q.; et al. Molecular Recognition of Morphine and Fentanyl by the Human μ-Opioid Receptor. Cell 2022, 185, 4361–4375.e19. [Google Scholar] [CrossRef]
- Qu, Q.; Huang, W.; Aydin, D.; Paggi, J.M.; Seven, A.B.; Wang, H.; Chakraborty, S.; Che, T.; DiBerto, J.F.; Robertson, M.J.; et al. Insights into Distinct Signaling Profiles of the ΜOR Activated by Diverse Agonists. Nat. Chem. Biol. 2023, 19, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, A.; Wang, H.; Zaidi, S.A.; DiBerto, J.F.; Che, T.; Qu, Q.; Robertson, M.J.; Madasu, M.K.; El Daibani, A.; Varga, B.R.; et al. Structure-Based Design of Bitopic Ligands for the µ-Opioid Receptor. Nature 2023, 613, 767–774. [Google Scholar] [CrossRef]
- Zarzycka, B.; Zaidi, S.A.; Roth, B.L.; Katritch, V. Harnessing Ion-Binding Sites for GPCR Pharmacology. Pharmacol. Rev. 2019, 71, 571–595. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Y.; Hunkele, A.; Provasi, D.; Pasternak, G.W.; Filizola, M. Kinetic and Thermodynamic Insights into Sodium Ion Translocation through the μ-Opioid Receptor from Molecular Dynamics and Machine Learning Analysis. PLoS Comput. Biol. 2019, 15, e1006689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.; LeRouzic, V.; Schneider, S.; Bisignano, P.; Pasternak, G.W.; Filizola, M. Mechanistic Insights into the Allosteric Modulation of Opioid Receptors by Sodium Ions. Biochemistry 2014, 53, 5140–5149. [Google Scholar] [CrossRef] [Green Version]
- Bous, J.; Orcel, H.; Floquet, N.; Leyrat, C.; Lai-Kee-Him, J.; Gaibelet, G.; Ancelin, A.; Saint-Paul, J.; Trapani, S.; Louet, M.; et al. Cryo-Electron Microscopy Structure of the Antidiuretic Hormone Arginine-Vasopressin V2 Receptor Signaling Complex. Sci. Adv. 2021, 7, eabg5628. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, J.; Cao, S.; Sun, D.; Liu, H.; Lu, Q.; Liu, Z.; Du, Y.; Zhang, C. Cryo-EM Structure of the AVP-Vasopressin Receptor 2-G(s) Signaling Complex. Cell Res. 2021, 31, 932–934. [Google Scholar] [CrossRef]
- Zhou, F.; Ye, C.; Ma, X.; Yin, W.; Croll, T.I.; Zhou, Q.; He, X.; Zhang, X.; Yang, D.; Wang, P.; et al. Molecular Basis of Ligand Recognition and Activation of Human V2 Vasopressin Receptor. Cell Res. 2021, 31, 929–931. [Google Scholar] [CrossRef]
- Duan, J.; Shen, D.-D.; Zhou, X.E.; Bi, P.; Liu, Q.-F.; Tan, Y.-X.; Zhuang, Y.-W.; Zhang, H.-B.; Xu, P.-Y.; Huang, S.-J.; et al. Cryo-EM Structure of an Activated VIP1 Receptor-G Protein Complex Revealed by a NanoBiT Tethering Strategy. Nat. Commun. 2020, 11, 4121. [Google Scholar] [CrossRef]
- Waltenspühl, Y.; Schöppe, J.; Ehrenmann, J.; Kummer, L.; Plückthun, A. Crystal Structure of the Human Oxytocin Receptor. Sci. Adv. 2020, 6, eabb5419. [Google Scholar] [CrossRef]
- Bous, J.; Fouillen, A.; Orcel, H.; Granier, S.; Bron, P.; Mouillac, B. Structures of the Arginine-Vasopressin and Oxytocin Receptor Signaling Complexes. In Vitamins and Hormones; Academic Press: Cambridge, MA, USA, 2023. [Google Scholar]
- Filipek, S. Molecular Switches in GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 114–120. [Google Scholar] [CrossRef]
- Nakane, T.; Kimanius, D.; Lindahl, E.; Scheres, S.H. Characterisation of Molecular Motions in Cryo-EM Single-Particle Data by Multi-Body Refinement in RELION. eLife 2018, 7, e36861. [Google Scholar] [CrossRef]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: Reconstruction of Heterogeneous Cryo-EM Structures Using Neural Networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef]
- Punjani, A.; Fleet, D.J. 3D Variability Analysis: Resolving Continuous Flexibility and Discrete Heterogeneity from Single Particle Cryo-EM. J. Struct. Biol. 2021, 213, 107702. [Google Scholar] [CrossRef]
- Punjani, A.; Fleet, D.J. 3DFlex: Determining Structure and Motion of Flexible Proteins from Cryo-EM. Nat. Methods 2023, 20, 860–870. [Google Scholar] [CrossRef]
- Laporte, S.A.; Oakley, R.H.; Zhang, J.; Holt, J.A.; Ferguson, S.S.; Caron, M.G.; Barak, L.S. The Beta2-Adrenergic Receptor/Betaarrestin Complex Recruits the Clathrin Adaptor AP-2 during Endocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3712–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusach, A.; García-Nafría, J.; Tate, C.G. New Insights into GPCR Coupling and Dimerisation from Cryo-EM Structures. Curr. Opin. Struct. Biol. 2023, 80, 102574. [Google Scholar] [CrossRef] [PubMed]
- Drube, J.; Haider, R.S.; Matthees, E.S.F.; Reichel, M.; Zeiner, J.; Fritzwanker, S.; Ziegler, C.; Barz, S.; Klement, L.; Filor, J.; et al. GPCR Kinase Knockout Cells Reveal the Impact of Individual GRKs on Arrestin Binding and GPCR Regulation. Nat. Commun. 2022, 13, 540. [Google Scholar] [CrossRef] [PubMed]
- Isaikina, P.; Petrovic, I.; Jakob, R.P.; Sarma, P.; Ranjan, A.; Baruah, M.; Panwalkar, V.; Maier, T.; Shukla, A.K.; Grzesiek, S. A Key GPCR Phosphorylation Motif Discovered in Arrestin2-CCR5 Phosphopeptide Complexes. Mol. Cell 2023, in press. [Google Scholar] [CrossRef]
- Maharana, J.; Sarma, P.; Yadav, M.K.; Saha, S.; Singh, V.; Saha, S.; Chami, M.; Banerjee, R.; Shukla, A.K. Structural Snapshots Uncover a Key Phosphorylation Motif in GPCRs Driving β-Arrestin Activation. Mol. Cell 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Cahill, T.J., 3rd; Thomsen, A.R.B.; Tarrasch, J.T.; Plouffe, B.; Nguyen, A.H.; Yang, F.; Huang, L.-Y.; Kahsai, A.W.; Bassoni, D.L.; Gavino, B.J.; et al. Distinct Conformations of GPCR-β-Arrestin Complexes Mediate Desensitization, Signaling, and Endocytosis. Proc. Natl. Acad. Sci. USA 2017, 114, 2562–2567. [Google Scholar] [CrossRef] [Green Version]
- Eichel, K.; Jullié, D.; Barsi-Rhyne, B.; Latorraca, N.R.; Masureel, M.; Sibarita, J.-B.; Dror, R.O.; von Zastrow, M. Catalytic Activation of β-Arrestin by GPCRs. Nature 2018, 557, 381–386. [Google Scholar] [CrossRef]
- Eichel, K.; Jullié, D.; von Zastrow, M. β-Arrestin Drives MAP Kinase Signalling from Clathrin-Coated Structures after GPCR Dissociation. Nat. Cell Biol. 2016, 18, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, Y.; Kofuku, Y.; Ueda, T.; Pandey, S.; Dwivedi-Agnihotri, H.; Shukla, A.K.; Shimada, I. Biphasic Activation of β-Arrestin 1 upon Interaction with a GPCR Revealed by Methyl-TROSY NMR. Nat. Commun. 2021, 12, 7158. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Tesmer, J.J.G. G Protein-Coupled Receptor Interactions with Arrestins and GPCR Kinases: The Unresolved Issue of Signal Bias. J. Biol. Chem. 2022, 298, 102279. [Google Scholar] [CrossRef]
- Bostock, M.J.; Solt, A.S.; Nietlispach, D. The Role of NMR Spectroscopy in Mapping the Conformational Landscape of GPCRs. Curr. Opin. Struct. Biol. 2019, 57, 145–156. [Google Scholar] [CrossRef]
- Yang, L.; Liu, D.; Wüthrich, K. GPCR Structural Characterization by NMR Spectroscopy in Solution. Acta Biochim. Biophys. Sin. 2022, 54, 1207–1212. [Google Scholar] [CrossRef]
- Takeuchi, K.; Kofuku, Y.; Imai, S.; Ueda, T.; Tokunaga, Y.; Toyama, Y.; Shiraishi, Y.; Shimada, I. Function-Related Dynamics in Multi-Spanning Helical Membrane Proteins Revealed by Solution NMR. Membranes 2021, 11, 604. [Google Scholar] [CrossRef]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural Insights into G-Protein-Coupled Receptor Allostery. Nature 2018, 559, 45–53. [Google Scholar] [CrossRef]
- Okude, J.; Ueda, T.; Kofuku, Y.; Sato, M.; Nobuyama, N.; Kondo, K.; Shiraishi, Y.; Mizumura, T.; Onishi, K.; Natsume, M.; et al. Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the μ-Opioid Receptor. Angew. Chem. Int. Ed. Engl. 2015, 54, 15771–15776. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, S.; Imai, S.; Asao, N.; Kofuku, Y.; Ueda, T.; Shimada, I. Activation Mechanism of the μ-Opioid Receptor by an Allosteric Modulator. Proc. Natl. Acad. Sci. USA 2022, 119, e2121918119. [Google Scholar] [CrossRef] [PubMed]
- Burford, N.T.; Clark, M.J.; Wehrman, T.S.; Gerritz, S.W.; Banks, M.; O’Connell, J.; Traynor, J.R.; Alt, A. Discovery of Positive Allosteric Modulators and Silent Allosteric Modulators of the μ-Opioid Receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 10830–10835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, X.; Maurel, D.; Déméné, H.; Vasiliauskaité-Brooks, I.; Hagelberger, J.; Peysson, F.; Saint-Paul, J.; Golebiowski, J.; Granier, S.; Sounier, R. Molecular Insights into the Biased Signaling Mechanism of the μ-Opioid Receptor. Mol. Cell 2021, 81, 4165–4175.e6. [Google Scholar] [CrossRef] [PubMed]
- Erlenbach, I.; Wess, J. Molecular Basis of V2 Vasopressin Receptor/Gs Coupling Selectivity. J. Biol. Chem. 1998, 273, 26549–26558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.-R.; Reiter, E.; Ahn, S.; Kim, J.; Chen, W.; Lefkowitz, R.J. Different G Protein-Coupled Receptor Kinases Govern G Protein and Beta-Arrestin-Mediated Signaling of V2 Vasopressin Receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.-Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct Phosphorylation Sites on the β(2)-Adrenergic Receptor Establish a Barcode That Encodes Differential Functions of β-Arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellot, G.; Pascal, R.; Mendre, C.; Urbach, S.; Mouillac, B.; Déméné, H. Expression, Purification and NMR Characterization of the Cyclic Recombinant Form of the Third Intracellular Loop of the Vasopressin Type 2 Receptor. Protein Expr. Purif. 2011, 78, 131–138. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Unfolded Proteins and Protein Folding Studied by NMR. Chem. Rev. 2004, 104, 3607–3622. [Google Scholar] [CrossRef]
- Sibille, N.; Bernadó, P. Structural Characterization of Intrinsically Disordered Proteins by the Combined Use of NMR and SAXS. Biochem. Soc. Trans. 2012, 40, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milles, S.; Salvi, N.; Blackledge, M.; Jensen, M.R. Characterization of Intrinsically Disordered Proteins and Their Dynamic Complexes: From in Vitro to Cell-like Environments. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 109, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. The Structural Basis of the Arrestin Binding to GPCRs. Mol. Cell. Endocrinol. 2019, 484, 34–41. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fouillen, A.; Bous, J.; Granier, S.; Mouillac, B.; Sounier, R. Bringing GPCR Structural Biology to Medical Applications: Insights from Both V2 Vasopressin and Mu-Opioid Receptors. Membranes 2023, 13, 606. https://doi.org/10.3390/membranes13060606
Fouillen A, Bous J, Granier S, Mouillac B, Sounier R. Bringing GPCR Structural Biology to Medical Applications: Insights from Both V2 Vasopressin and Mu-Opioid Receptors. Membranes. 2023; 13(6):606. https://doi.org/10.3390/membranes13060606
Chicago/Turabian StyleFouillen, Aurélien, Julien Bous, Sébastien Granier, Bernard Mouillac, and Remy Sounier. 2023. "Bringing GPCR Structural Biology to Medical Applications: Insights from Both V2 Vasopressin and Mu-Opioid Receptors" Membranes 13, no. 6: 606. https://doi.org/10.3390/membranes13060606