
The Alternating Access Mechanism in Mammalian Multidrug Resistance Transporters and Their Bacterial Homologs

Abstract
:1. Introduction
1.1. Efflux Pumps
1.1.1. Major Facilitator Superfamily
1.1.2. Multi-Antimicrobial Extrusion
1.1.3. Small Multidrug Resistance
1.1.4. Resistance-Nodulation-Division
1.2. ABC Transporter
1.3. MDRs
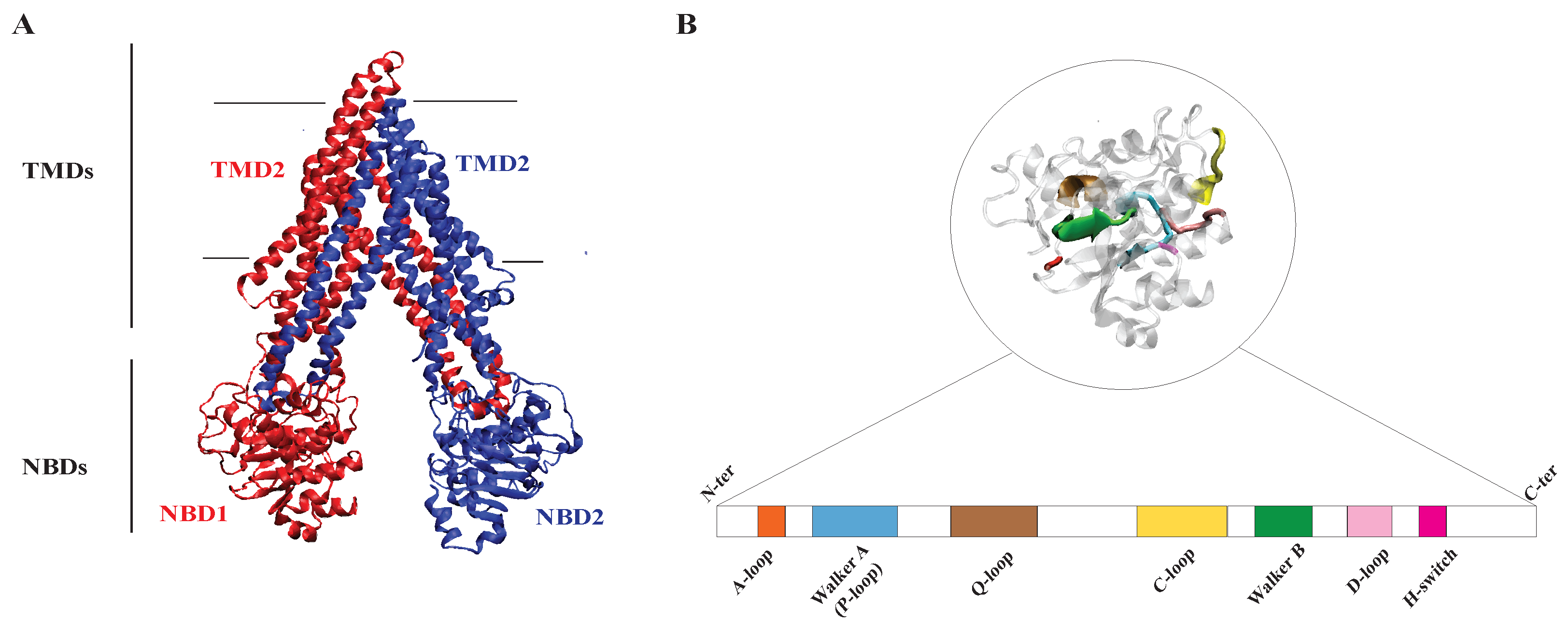
1.4. NBD and TMD
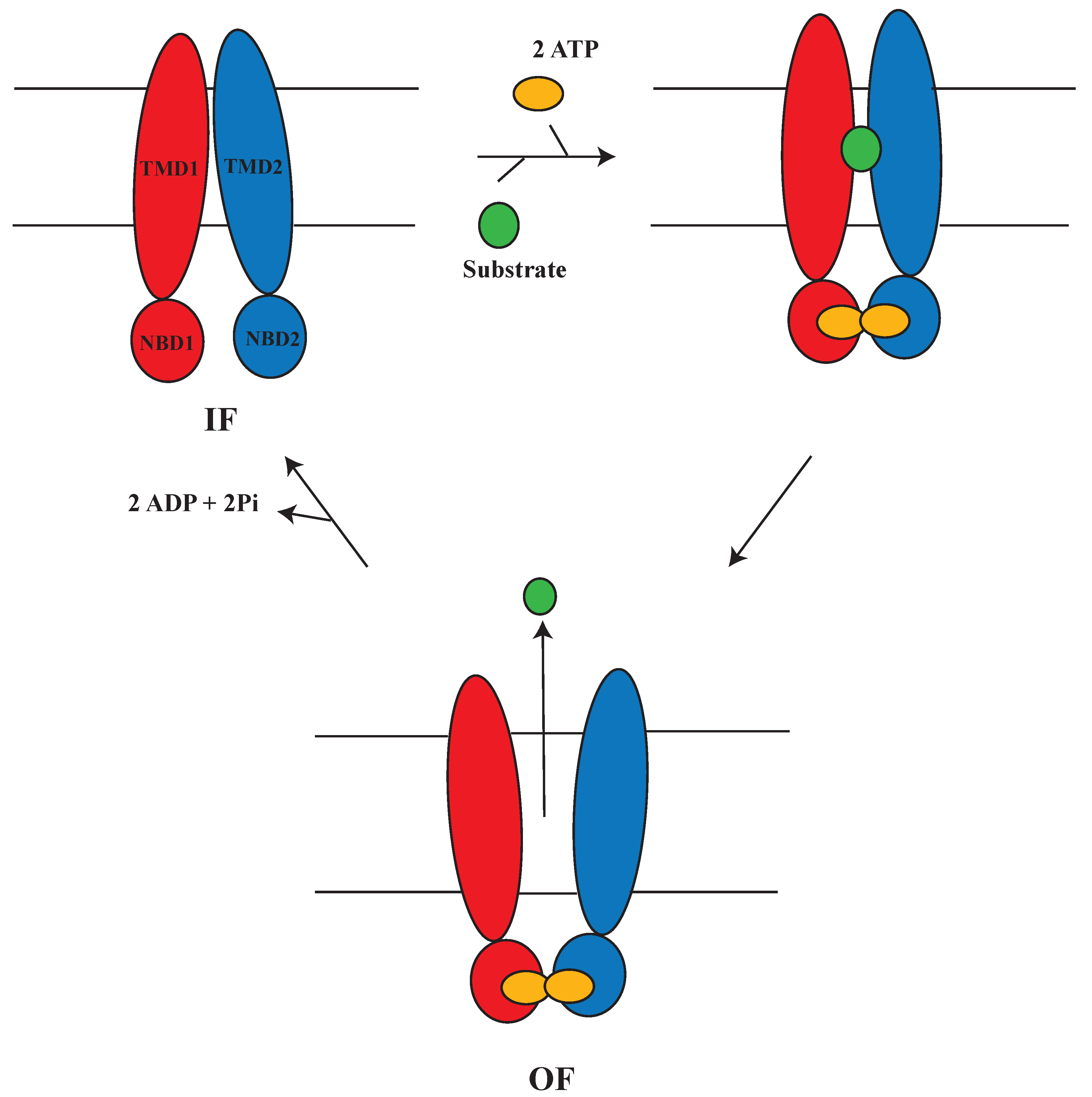
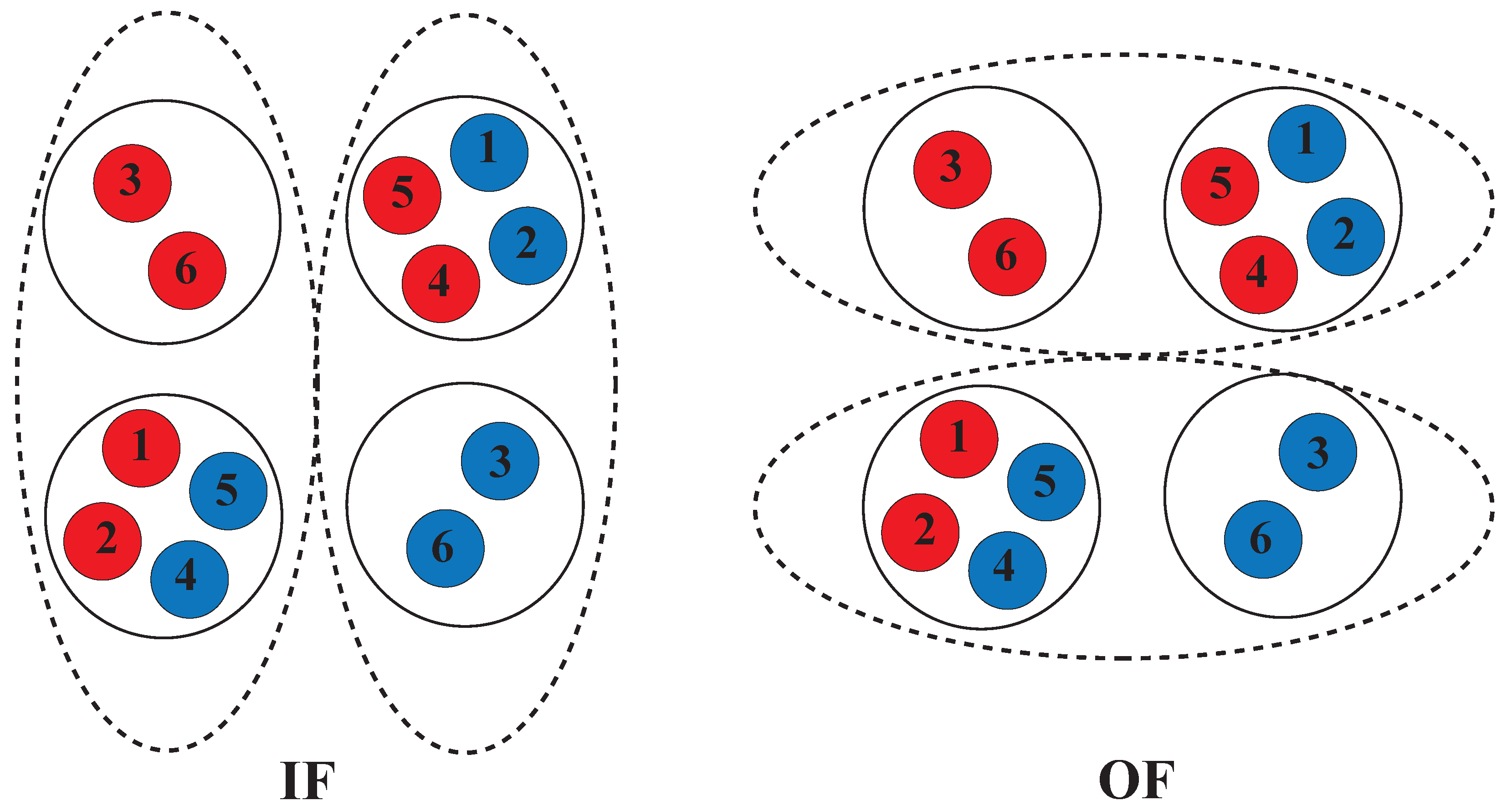
1.5. Alternating Access Mechanism (AAM)
2. Alternating Access Mechanism in Mammalian MDRs and Bacterial Homologs
2.1. MRP1
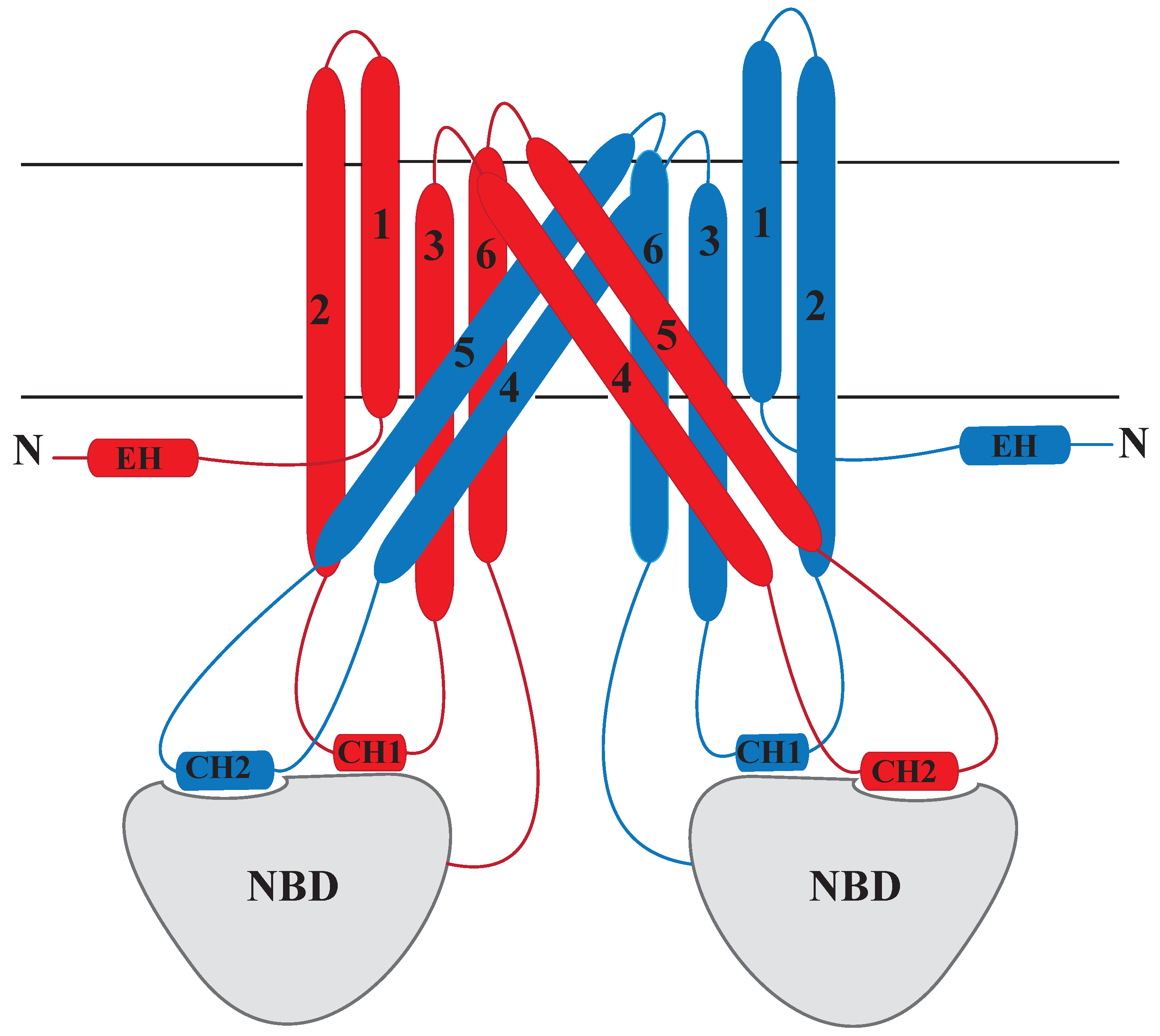
2.1.1. Structure of MRP1
2.1.2. Substrate Binding Site of MRP1
2.1.3. AAM in MRP1
2.2. P-glycoprotein
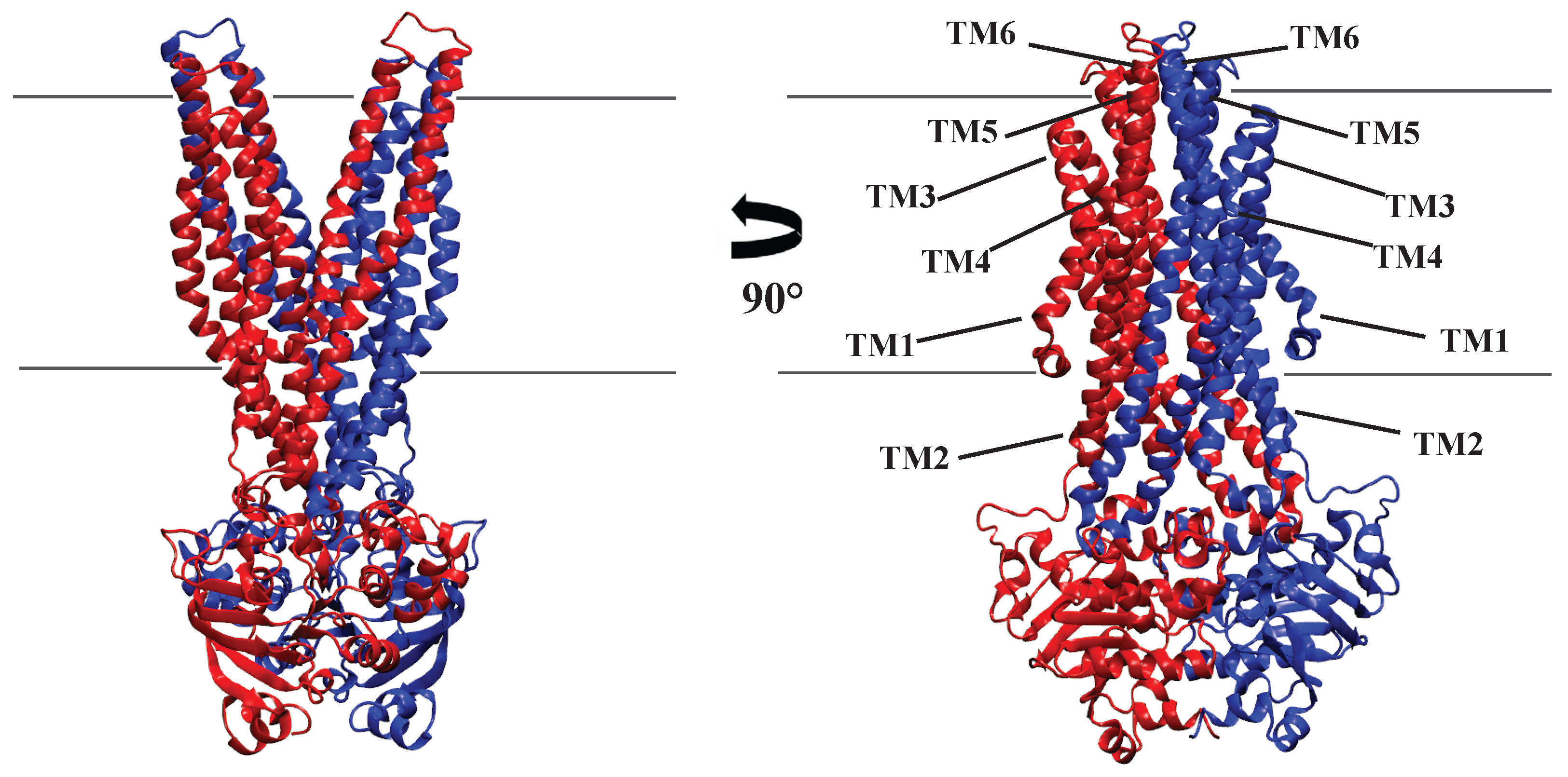
2.2.1. Structure of P-glycoprotein
2.2.2. Substrate Binding Site of P-glycoprotein
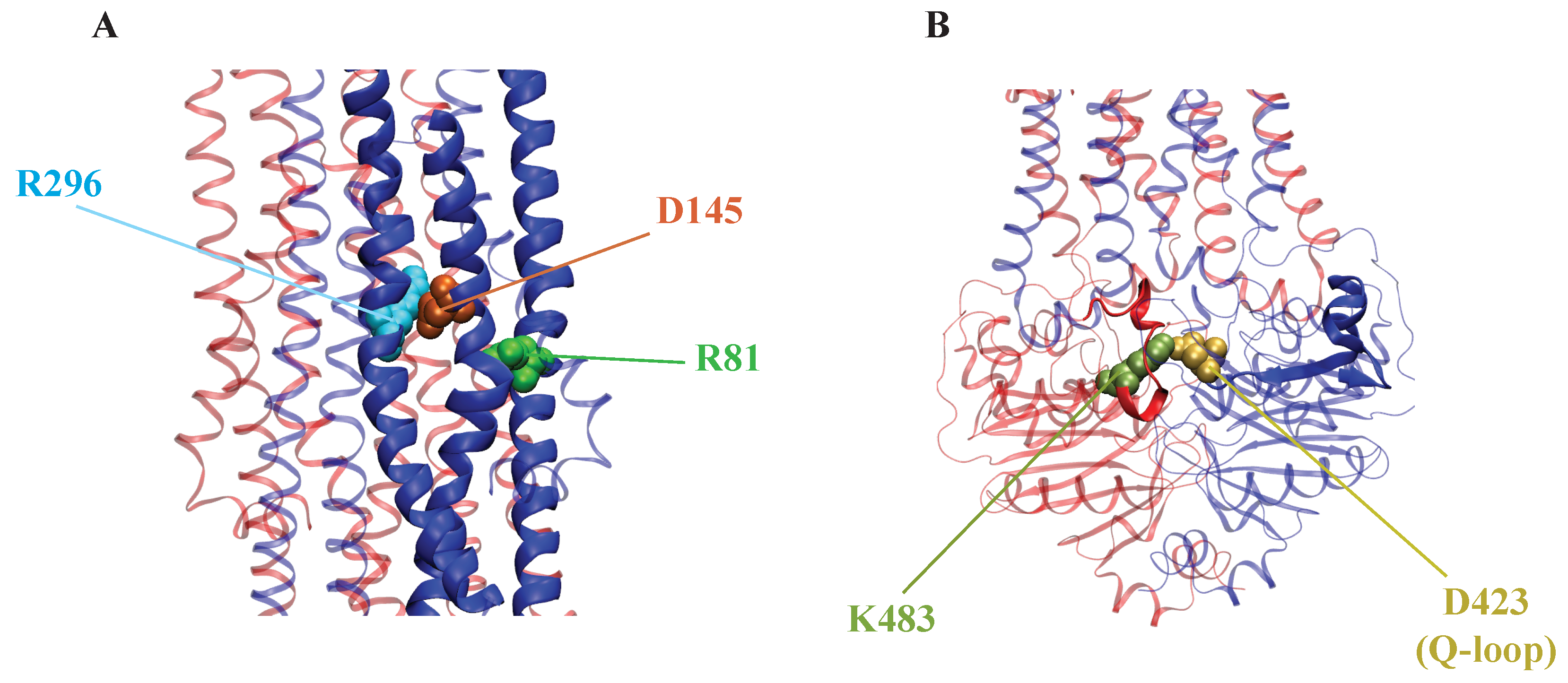
2.2.3. AAM in P-glycoprotein
2.3. Sav1866
2.3.1. Structure of Sav1866
2.3.2. AAM in Sav1866
2.4. MsbA
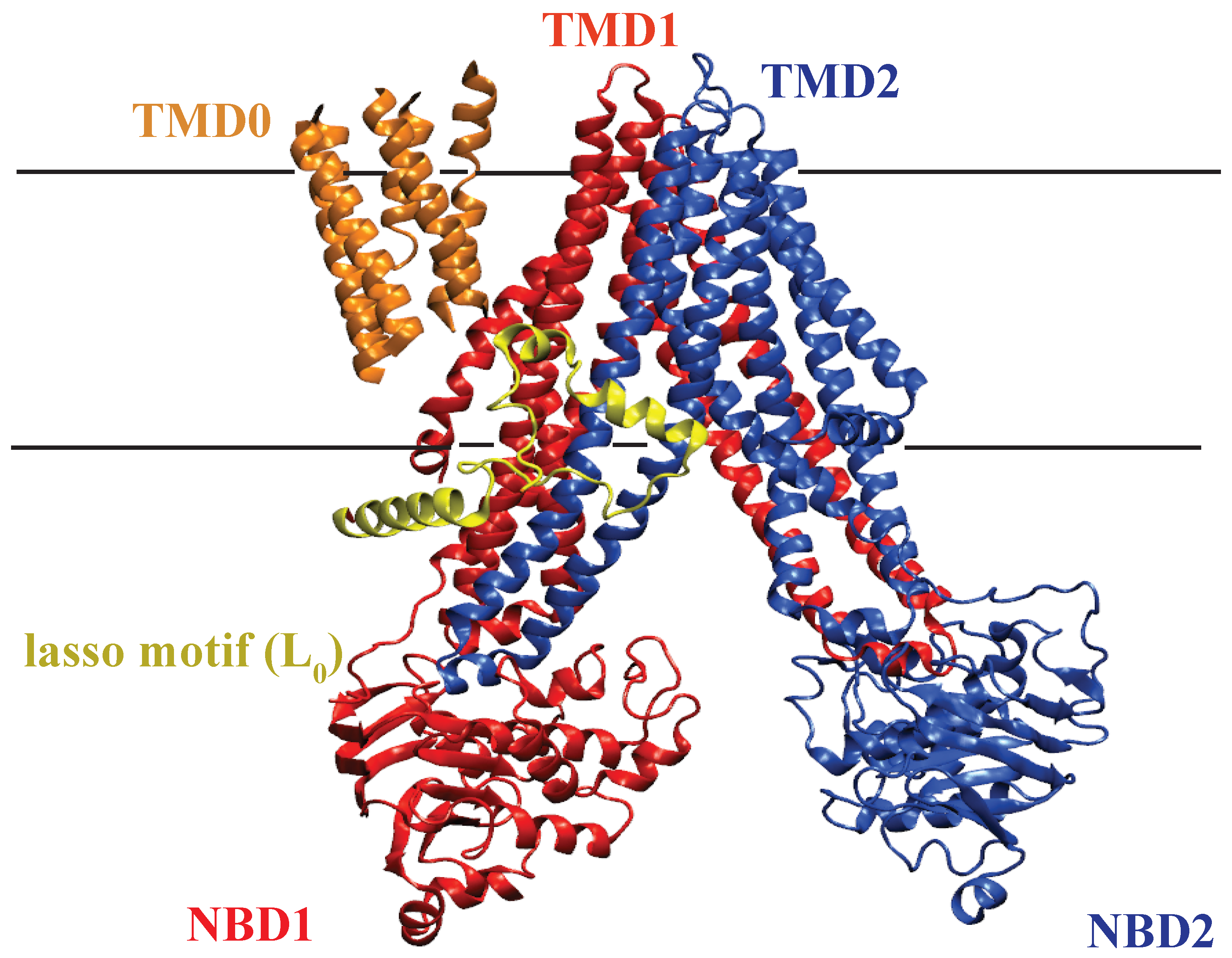
2.4.1. Structure of MsbA
2.4.2. Substrate Binding Site of MsbA
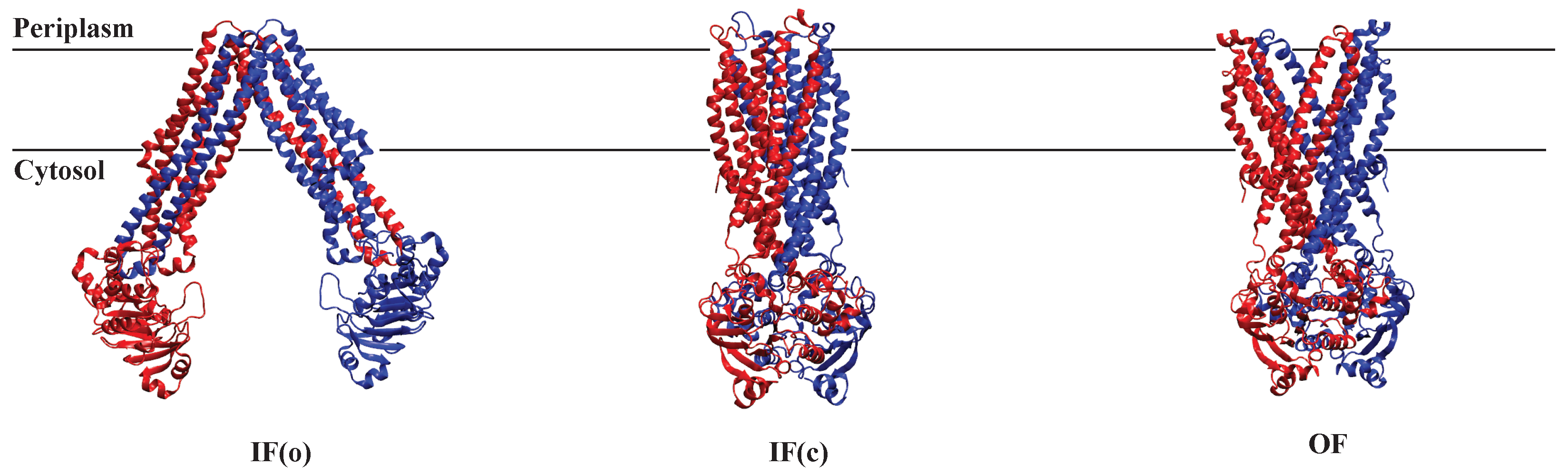
2.4.3. AAM in MsbA
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CRAC | Cholesterol Recognition/Interaction Amino Acid Consensus (CRAC) |
| ABC | Adenosine triphosphate binding cassette |
| GSH | Conjugated to antioxidant glutathione |
| Cryo-EM | Cryogenic Electron Microscopy |
| X-ray | X-ray Diffraction |
| MD(S) | Molecular dynamics (simulation) |
| MRP1 | Multidrug resistance protein 1 |
| AAM | Alternating Access Mechanism |
| MFS | Major facilitator superfamily |
| MATE | Multi-Antimicrobial Extrusion |
| AK | Adenylate kinase reaction |
| MDR | Multi-drug resistance |
| TMD | Transmembrane domain |
| MSD | membrane spanning domain |
| NBD | Nuclotide-binding domain |
| SMR | Small Multidrug Resistance |
| RND | Resistance-Nodulation-Division |
| ATP | Adenosine triphosphate |
| CV | Collective variable |
| TMH | Transmembrane helix |
| IH | Intracellular helix |
| ICL | Intracellular loop |
| CLR | Cholesterol |
| Pgp | P-glycoprotein |
| TM | Transmembrane |
| IF | Inward facing |
| OF | Outward facing |
| CH | Coupling helix |
| EH | Elbow helix |
| Pi | Inorganic phosphate |
| C | C terminus |
| N | N terminus |
References
- Muriithi, W.; Macharia, L.W.; Heming, C.P.; Echevarria, J.L.; Nyachieo, A.; Niemeyer Filho, P.; Neto, V.M. ABC transporters and the hallmarks of cancer: Roles in cancer aggressiveness beyond multidrug resistance. Cancer Biol. Med. 2020, 17, 253. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Fortunati, E.; Liu, D.X.; Li, Y. Pleiotropic roles of ABC transporters in breast cancer. Int. J. Mol. Sci. 2021, 22, 3199. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Wu, Z.X.; Yang, Y.; Teng, Q.X.; Li, Y.D.; Lei, Z.N.; Jani, K.A.; Kaushal, N.; Chen, Z.S. ATP-binding cassette (ABC) transporters in cancer: A review of recent updates. J. Evid.-Based Med. 2021, 14, 232–256. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-relevant ABC transporter for anti-cancer drug resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar] [CrossRef]
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Scumaci, D.; Giuzio, F.; Saturnino, C.; Aquaro, S.; Rosano, C.; Sinicropi, M.S. Multidrug resistance (MDR): A widespread phenomenon in pharmacological therapies. Molecules 2022, 27, 616. [Google Scholar] [CrossRef]
- Akhtar, A.A.; Turner, D.P. The role of bacterial ATP-binding cassette (ABC) transporters in pathogenesis and virulence: Therapeutic and vaccine potential. Microb. Pathog. 2022, 171, 105734. [Google Scholar] [CrossRef]
- Pote, M.S.; Gacche, R.N. ATP-binding cassette efflux transporters and MDR in cancer. Drug Discov. Today 2023, 28, 103537. [Google Scholar] [CrossRef]
- Dhakne, P.; Pillai, M.; Mishra, S.; Chatterjee, B.; Tekade, R.K.; Sengupta, P. Refinement of safety and efficacy of anti-cancer chemotherapeutics by tailoring their site-specific intracellular bioavailability through transporter modulation. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2023, 1878, 188906. [Google Scholar] [CrossRef]
- Duan, C.; Yu, M.; Xu, J.; Li, B.Y.; Zhao, Y.; Kankala, R.K. Overcoming Cancer Multi-drug Resistance (MDR): Reasons, mechanisms, nanotherapeutic solutions, and challenges. Biomed. Pharmacother. 2023, 162, 114643. [Google Scholar] [CrossRef]
- Pucelik, B.; Dąbrowski, J.M. Photodynamic inactivation (PDI) as a promising alternative to current pharmaceuticals for the treatment of resistant microorganisms. Adv. Inorg. Chem. 2022, 79, 65. [Google Scholar]
- Chitsaz, M.; Brown, M.H. The role played by drug efflux pumps in bacterial multidrug resistance. Essays Biochem. 2017, 61, 127–139. [Google Scholar]
- Huang, L.; Wu, C.; Gao, H.; Xu, C.; Dai, M.; Huang, L.; Hao, H.; Wang, X.; Cheng, G. Bacterial multidrug efflux pumps at the frontline of antimicrobial resistance: An overview. Antibiotics 2022, 11, 520. [Google Scholar] [CrossRef]
- da Silva, P.E.A.; Machado, D.; Ramos, D.; Couto, I.; Von Groll, A.; Viveiros, M. Efflux pumps in mycobacteria: Antimicrobial resistance, physiological functions, and role in pathogenicity. In Efflux-Mediated Antimicrobial Resistance in Bacteria: Mechanisms, Regulation and Clinical Implications; Adis: Cham, Switzerland, 2016; pp. 527–559. [Google Scholar]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: An update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef] [Green Version]
- Chetri, S. The culmination of multidrug-resistant efflux pumps vs. meager antibiotic arsenal era: Urgent need for an improved new generation of EPIs. Front. Microbiol. 2023, 14, 1149418. [Google Scholar] [CrossRef]
- Kumar, S.; He, G.; Kakarla, P.; Shrestha, U.; KC, R.; Ranaweera, I.; Mark Willmon, T.; R. Barr, S.; J. Hernandez, A.; F. Varela, M. Bacterial multidrug efflux pumps of the major facilitator superfamily as targets for modulation. Infect. Disord. Drug Targets (Former. Curr. Drug Targets Infect. Disord.) 2016, 16, 28–43. [Google Scholar]
- Pasqua, M.; Grossi, M.; Zennaro, A.; Fanelli, G.; Micheli, G.; Barras, F.; Colonna, B.; Prosseda, G. The varied role of efflux pumps of the MFS family in the interplay of bacteria with animal and plant cells. Microorganisms 2019, 7, 285. [Google Scholar] [CrossRef] [Green Version]
- Varela, M.F.; Stephen, J.; Bharti, D.; Lekshmi, M.; Kumar, S. Inhibition of Multidrug Efflux Pumps Belonging to the Major Facilitator Superfamily in Bacterial Pathogens. Biomedicines 2023, 11, 1448. [Google Scholar] [CrossRef]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef]
- Kusakizako, T.; Miyauchi, H.; Ishitani, R.; Nureki, O. Structural biology of the multidrug and toxic compound extrusion superfamily transporters. Biochim. Biophys. Acta (BBA)—Biomembr. 2020, 1862, 183154. [Google Scholar] [CrossRef]
- Zhang, X.; He, X.; Baker, J.; Tama, F.; Chang, G.; Wright, S.H. Twelve transmembrane helices form the functional core of mammalian MATE1 (multidrug and toxin extruder 1) protein. J. Biol. Chem. 2012, 287, 27971–27982. [Google Scholar] [CrossRef] [Green Version]
- Jagessar, K.L.; Claxton, D.P.; Stein, R.A.; Mchaourab, H.S. Sequence and structural determinants of ligand-dependent alternating access of a MATE transporter. Proc. Natl. Acad. Sci. USA 2020, 117, 4732–4740. [Google Scholar] [CrossRef] [PubMed]
- Omote, H.; Hiasa, M.; Matsumoto, T.; Otsuka, M.; Moriyama, Y. The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol. Sci. 2006, 27, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Radchenko, M.; Symersky, J.; Nie, R.; Guo, Y. Structural insights into H + -coupled multidrug extrusion by a MATE transporter. Nat. Struct. Mol. Biol. 2013, 20, 1310–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, D.; Wang-Kan, X.; Neuberger, A.; Van Veen, H.W.; Pos, K.M.; Piddock, L.J.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Spengler, G.; Kincses, A.; Gajdács, M.; Amaral, L. New roads leading to old destinations: Efflux pumps as targets to reverse multidrug resistance in bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef] [Green Version]
- Handzlik, J.; Matys, A.; Kieć-Kononowicz, K. Recent advances in multi-drug resistance (MDR) efflux pump inhibitors of Gram-positive bacteria S. aureus. Antibiotics 2013, 2, 28–45. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H.; Takatsuka, Y. Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2009, 1794, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Daury, L.; Orange, F.; Taveau, J.C.; Verchere, A.; Monlezun, L.; Gounou, C.; Marreddy, R.K.; Picard, M.; Broutin, I.; Pos, K.M.; et al. Tripartite assembly of RND multidrug efflux pumps. Nat. Commun. 2016, 7, 10731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, J.; Ruggerone, P. Interaction of antibacterial compounds with RND efflux pumps in Pseudomonas aeruginosa. Front. Microbiol. 2015, 6, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klenotic, P.A.; Moseng, M.A.; Morgan, C.E.; Yu, E.W. Structural and functional diversity of resistance–nodulation–cell division transporters. Chem. Rev. 2020, 121, 5378–5416. [Google Scholar] [CrossRef]
- Callaghan, R.; Gelissen, I.C.; George, A.M.; Hartz, A.M. Mamma Mia, P-glycoprotein binds again. FEBS Lett. 2020, 594, 4076–4084. [Google Scholar] [CrossRef] [PubMed]
- Srikant, S.; Gaudet, R. Mechanics and pharmacology of substrate selection and transport by eukaryotic ABC exporters. Nat. Struct. Mol. Biol. 2019, 26, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Holland, I.B.; Cole, S.P.; Kuchler, K.; Higgins, C.F. ABC Proteins: From Bacteria to Man; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Huang, J.; Ecker, G.F. A Structure-Based View on ABC-Transporter Linked to Multidrug Resistance. Molecules 2023, 28, 495. [Google Scholar] [CrossRef] [PubMed]
- Hohl, M.; Briand, C.; Grütter, M.G.; Seeger, M.A. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat. Struct. Mol. Biol. 2012, 19, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Procko, E.; O’Mara, M.L.; Bennett, W.D.; Tieleman, D.P.; Gaudet, R. The mechanism of ABC transporters: General lessons from structural and functional studies of an antigenic peptide transporter. FASEB J. 2009, 23, 1287–1302. [Google Scholar] [CrossRef] [PubMed]
- Symmons, M.F.; Bokma, E.; Koronakis, E.; Hughes, C.; Koronakis, V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc. Natl. Acad. Sci. USA 2009, 106, 7173–7178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, R.C.; Beis, K. Learning the ABCs one at a time: Structure and mechanism of ABC transporters. Biochem. Soc. Trans. 2019, 47, 23–36. [Google Scholar] [CrossRef] [PubMed]
- George, A.M.; Jones, P.M. Perspectives on the structure–function of ABC transporters: The Switch and Constant Contact Models. Prog. Biophys. Mol. Biol. 2012, 109, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.L.; Dassa, E.; Orelle, C.; Chen, J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 2008, 72, 317–364. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Aller, S.G.; Beis, K.; Carpenter, E.P.; Chang, G.; Chen, L.; Dassa, E.; Dean, M.; Duong Van Hoa, F.; Ekiert, D.; et al. Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett. 2020, 594, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Verhalen, B.; Dastvan, R.; Thangapandian, S.; Peskova, Y.; Koteiche, H.A.; Nakamoto, R.K.; Tajkhorshid, E.; Mchaourab, H.S. Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature 2017, 543, 738–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef] [PubMed]
- Moitra, K.; Dean, M. Evolution of ABC transporters by gene duplication and their role in human disease. Biol. Chem. 2011, 392, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Saurin, W.; Hofnung, M.; Dassa, E. Getting in or out: Early segregation between importers and exporters in the evolution of ATP-binding cassette (ABC) transporters. J. Mol. Evol. 1999, 48, 22–41. [Google Scholar] [CrossRef]
- Bouige, P.; Laurent, D.; Piloyan, L.; Dassa, E. Phylogenetic and functional classification of ATP-binding cassette (ABC) systems. Curr. Protein Pept. Sci. 2002, 3, 541–559. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the human lipid exporter ABCA1. Cell 2017, 169, 1228–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, N.M.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.B.; Hou, W.T.; Cheng, M.T.; Jiang, Y.L.; Chen, Y.; Zhou, C.Z. Structure of a MacAB-like efflux pump from Streptococcus pneumoniae. Nat. Commun. 2018, 9, 196. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Mann, E.; Whitfield, C.; Zimmer, J. Architecture of a channel-forming O-antigen polysaccharide ABC transporter. Nature 2018, 553, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hou, W.T.; Fan, T.; Liu, B.; Pan, T.; Li, Y.H.; Jiang, Y.L.; Wen, W.; Chen, Z.P.; Sun, L.; et al. Cryo-electron microscopy structure and transport mechanism of a wall teichoic acid ABC transporter. MBio 2020, 11, e02749-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, Z.L.; Chen, J. ATP binding enables substrate release from multidrug resistance protein 1. Cell 2018, 172, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Johnson, Z.L.; Wasserman, M.R.; Levring, J.; Chen, J.; Liu, S. Characterization of the kinetic cycle of an ABC transporter by single-molecule and cryo-EM analyses. eLife 2020, 9, e56451. [Google Scholar] [CrossRef] [PubMed]
- Pietz, H.L.; Abbas, A.; Johnson, Z.L.; Oldham, M.L.; Suga, H.; Chen, J. A macrocyclic peptide inhibitor traps MRP1 in a catalytically incompetent conformation. Proc. Natl. Acad. Sci. USA 2023, 120, e2220012120. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.L.; Chen, J. Structural basis of substrate recognition by the multidrug resistance protein MRP1. Cell 2017, 168, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Khandelwal, N.K.; Millan, C.R.; Zangari, S.I.; Avila, S.; Williams, D.; Thaker, T.M.; Tomasiak, T.M. The structural basis for regulation of the glutathione transporter Ycf1 by regulatory domain phosphorylation. Nat. Commun. 2022, 13, 1278. [Google Scholar] [CrossRef]
- Esser, L.; Zhou, F.; Pluchino, K.M.; Shiloach, J.; Ma, J.; Tang, W.k.; Gutierrez, C.; Zhang, A.; Shukla, S.; Madigan, J.P.; et al. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017, 292, 446–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckford, P.D.; Sharom, F.J. The reconstituted Escherichia coli MsbA protein displays lipid flippase activity. Biochem. J. 2010, 429, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicklisch, S.C.; Rees, S.D.; McGrath, A.P.; Gökirmak, T.; Bonito, L.T.; Vermeer, L.M.; Cregger, C.; Loewen, G.; Sandin, S.; Chang, G.; et al. Global marine pollutants inhibit P-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci. Adv. 2016, 2, e1600001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Le, C.A.; Harvey, D.S.; Aller, S.G. Structural definition of polyspecific compensatory ligand recognition by P-glycoprotein. IUCrJ 2020, 7, 663–672. [Google Scholar] [CrossRef]
- Dawson, R.J.P.; Locher, K.P. Structure of a bacterial multidrug ABC transporter. Nature 2006, 443, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.J.P.; Locher, K.P. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007, 581, 935–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.; Miu, A.; Alexander, M.K.; Garcia, N.K.; Oh, A.; Zilberleyb, I.; Reichelt, M.; Austin, C.D.; Tam, C.; Shriver, S.; et al. Structural basis for dual-mode inhibition of the ABC transporter MsbA. Nature 2018, 557, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Padayatti, P.S.; Lee, S.C.; Stanfield, R.L.; Wen, P.C.; Tajkhorshid, E.; Wilson, I.A.; Zhang, Q. Structural insights into the lipid A transport pathway in MsbA. Structure 2019, 27, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Reyes, C.L.; Yu, J.; Roth, C.B.; Chang, G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA 2007, 104, 19005–19010. [Google Scholar] [CrossRef] [Green Version]
- Mi, W.; Li, Y.; Yoon, S.H.; Ernst, R.K.; Walz, T.; Liao, M. Structural basis of MsbA-mediated lipopolysaccharide transport. Nature 2017, 549, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angiulli, G.; Dhupar, H.S.; Suzuki, H.; Wason, I.S.; Duong Van Hoa, F.; Walz, T. New approach for membrane protein reconstitution into peptidiscs and basis for their adaptability to different proteins. eLife 2020, 9, e53530. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 2016, 27, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.P.; Kaplan, E.; Crow, A.; Koronakis, V. Antibiotic resistance mediated by the MacB ABC transporter family: A structural and functional perspective. Front. Microbiol. 2018, 9, 950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Jernigan, J.A.; Hatfield, K.M.; Wolford, H.; Nelson, R.E.; Olubajo, B.; Reddy, S.C.; McCarthy, N.; Paul, P.; McDonald, L.C.; Kallen, A.; et al. Multidrug-resistant bacterial infections in US hospitalized patients, 2012–2017. N. Engl. J. Med. 2020, 382, 1309–1319. [Google Scholar] [CrossRef]
- Kumar, S.; Anwer, R.; Azzi, A. Virulence potential and treatment options of multidrug-resistant (MDR) Acinetobacter baumannii. Microorganisms 2021, 9, 2104. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. ABC family transporters. In Drug Transporters in Drug Disposition, Effects and Toxicity; Springer: Singapore, 2019; pp. 13–100. [Google Scholar]
- Zahreddine, H.; Borden, K. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, G.; Roth, C.B. Structure of MsbA from E. coli: A homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 2001, 293, 1793–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; De Feo, V.; Zia-Ul-Haq, M. Natural products as alternative choices for P-glycoprotein (P-gp) inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Feng, J.; Yuan, D.; Zhou, J.; Miao, W. Tracing the structural evolution of eukaryotic ATP binding cassette transporter superfamily. Sci. Rep. 2015, 5, 16724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, L.W.; Wang, I.X.; Nikaido, K.; Liu, P.Q.; Ames, G.F.L.; Kim, S.H. Crystal structure of the ATP-binding subunit of an ABC transporter. Nature 1998, 396, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Karcher, A.; Shin, D.S.; Craig, L.; Arthur, L.M.; Carney, J.P.; Tainer, J.A. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 2000, 101, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.C.; Karpowich, N.; Millen, L.; Moody, J.E.; Rosen, J.; Thomas, P.J.; Hunt, J.F. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 2002, 10, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolnerciks, J.K.; Andress, E.J.; Nicolaou, M.; Linton, K.J. Structure of ABC transporters. Essays Biochem. 2011, 50, 43. [Google Scholar] [PubMed] [Green Version]
- Seyffer, F.; Tampé, R. ABC transporters in adaptive immunity. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2015, 1850, 449–460. [Google Scholar] [CrossRef]
- Vetter, I.R.; Wittinghofer, A. Nucleoside triphosphate-binding proteins: Different scaffolds to achieve phosphoryl transfer. Q. Rev. Biophys. 1999, 32, 1–56. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; George, A.M. Subunit interactions in ABC transporters: Towards a functional architecture. FEMS Microbiol. Lett. 1999, 179, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F.; Hiles, I.D.; Salmond, G.P.; Gill, D.R.; Downie, J.A.; Evans, I.J.; Holland, I.B.; Gray, L.; Buckel, S.D.; Bell, A.W.; et al. A family of related ATP-binding subunits coupled to many distinct biological processes in bacteria. Nature 1986, 323, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Lu, G.; Lin, J.; Davidson, A.L.; Quiocho, F.A. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell 2003, 12, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Kunjachan, S.; Rychlik, B.; Storm, G.; Kiessling, F.; Lammers, T. Multidrug resistance: Physiological principles and nanomedical solutions. Adv. Drug Deliv. Rev. 2013, 65, 1852–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biemans-Oldehinkel, E.; Doeven, M.K.; Poolman, B. ABC transporter architecture and regulatory roles of accessory domains. FEBS Lett. 2006, 580, 1023–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Jardetzky, O. Simple allosteric model for membrane pumps. Nature 1966, 211, 969–970. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. A general theory of membrane transport from studies of bacteria. Nature 1957, 180, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Tanford, C. Simple model for the chemical potential change of a transported ion in active transport. Proc. Natl. Acad. Sci. USA 1982, 79, 2882–2884. [Google Scholar] [CrossRef] [Green Version]
- Patlak, C.S. Contributions to the theory of active transport: II. The gate type non-carrier mechanism and generalizations concerning tracer flow, efficiency, and measurement of energy expenditure. Bull. Math. Biophys. 1957, 19, 209–235. [Google Scholar] [CrossRef]
- Yan, N. Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 2015, 44, 257–283. [Google Scholar] [CrossRef]
- Drew, D.; Boudker, O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 2016, 85, 543–572. [Google Scholar] [CrossRef]
- Kroll, T.; Prescher, M.; Smits, S.H.J.; Schmitt, L. Structure and Function of Hepatobiliary ATP Binding Cassette Transporters. Chem. Rev. 2021, 121, 5240–5288. [Google Scholar] [CrossRef]
- Locher, K.P. Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Oldham, M.L.; Davidson, A.L.; Chen, J. Structural insights into ABC transporter mechanism. Curr. Opin. Struct. Biol. 2008, 18, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Enkavi, G.; Li, J.; Wen, P.; Thangapandian, S.; Moradi, M.; Jiang, T.; Han, W.; Tajkhorshid, E. Chapter Four—A Microscopic View of the Mechanisms of Active Transport Across the Cellular Membrane. Annu. Rep. Comput. Chem. 2014, 10, 77–125. [Google Scholar] [CrossRef]
- Deeley, R.G.; Westlake, C.; Cole, S.P. Transmembrane transport of endo-and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev. 2006, 86, 849–899. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.K.; Wang, Y.J.; Gupta, P.; Chen, Z.S. Multidrug resistance proteins (MRPs) and cancer therapy. AAPS J. 2015, 17, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Ivanyuk, A.; Livio, F.; Biollaz, J.; Buclin, T. Renal drug transporters and drug interactions. Clin. Pharmacokinet. 2017, 56, 825–892. [Google Scholar] [CrossRef]
- Wang, J.Q.; Cui, Q.; Lei, Z.N.; Teng, Q.X.; Ji, N.; Lin, L.; Liu, Z.; Chen, Z.S. Insights on the structure–function relationship of human multidrug resistance protein 7 (MRP7/ABCC10) from molecular dynamics simulations and docking studies. MedComm 2021, 2, 221–235. [Google Scholar] [CrossRef]
- Roy, U.; Barber, P.; Tse-Dinh, Y.C.; Batrakova, E.V.; Mondal, D.; Nair, M. Role of MRP transporters in regulating antimicrobial drug inefficacy and oxidative stress-induced pathogenesis during HIV-1 and TB infections. Front. Microbiol. 2015, 6, 948. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, K.; Xie, Y.; Baer, M.R.; Ross, D.D. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem. Pharmacol. 2012, 83, 1084–1103. [Google Scholar] [CrossRef] [Green Version]
- Shukalek, C.B.; Swanlund, D.P.; Rousseau, R.K.; Weigl, K.E.; Marensi, V.; Cole, S.P.; Leslie, E.M. Arsenic triglutathione [As (GS) 3] transport by multidrug resistance protein 1 (MRP1/ABCC1) is selectively modified by phosphorylation of Tyr920/Ser921 and glycosylation of Asn19/Asn23. Mol. Pharmacol. 2016, 90, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Zhang, J. Multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphism: From discovery to clinical application. Zhong Nan Xue Xue Bao Xue Ban J. Cent. South Univ. Med. Sci. 2011, 36, 927. [Google Scholar]
- Grube, M.; Hagen, P.; Jedlitschky, G. Neurosteroid transport in the brain: Role of ABC and SLC transporters. Front. Pharmacol. 2018, 9, 354. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, X.; Huang, J.; Peng, J.; Guo, L. miR-7 modulates chemoresistance of small cell lung cancer by repressing MRP 1/ABCC 1. Int. J. Exp. Pathol. 2015, 96, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Miglionico, R.; Gerbino, A.; Ostuni, A.; Armentano, M.F.; Monné, M.; Carmosino, M.; Bisaccia, F. New insights into the roles of the N-terminal region of the ABCC6 transporter. J. Bioenerg. Biomembr. 2016, 48, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.; Sparks, K.E.; Fraser, K.; Loe, D.W.; Grant, C.E.; Wilson, G.M.; Deeley, R.G. Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 1994, 54, 5902–5910. [Google Scholar]
- Slot, A.J.; Molinski, S.V.; Cole, S.P. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 2011, 50, 179–207. [Google Scholar]
- Cui, Q.; Yang, Y.; Ji, N.; Wang, J.Q.; Ren, L.; Yang, D.H.; Chen, Z.S. Gaseous signaling molecules and their application in resistant cancer treatment: From invisible to visible. Future Med. Chem. 2019, 11, 323–336. [Google Scholar] [CrossRef]
- Bakos, E.; Evers, R.; Szakács, G.; Tusnády, G.E.; Welker, E.; Szabó, K.; de Haas, M.; van Deemter, L.; Borst, P.; Váradi, A.; et al. Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J. Biol. Chem. 1998, 273, 32167–32175. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.x.; Cui, L.; Riordan, J.R.; Chang, X.b. Allosteric interactions between the two non-equivalent nucleotide binding domains of multidrug resistance protein MRP1. J. Biol. Chem. 2000, 275, 20280–20287. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Cui, H.R.; Loe, D.W.; Grant, C.E.; Almquist, K.C.; Cole, S.P.; Deeley, R.G. Comparison of the functional characteristics of the nucleotide binding domains of multidrug resistance protein 1. J. Biol. Chem. 2000, 275, 13098–13108. [Google Scholar] [CrossRef] [Green Version]
- Moody, J.E.; Millen, L.; Binns, D.; Hunt, J.F.; Thomas, P.J. Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J. Biol. Chem. 2002, 277, 21111–21114. [Google Scholar] [CrossRef] [Green Version]
- Nagata, K.; Nishitani, M.; Matsuo, M.; Kioka, N.; Amachi, T.; Ueda, K. Nonequivalent nucleotide trapping in the two nucleotide binding folds of the human multidrug resistance protein MRP1. J. Biol. Chem. 2000, 275, 17626–17630. [Google Scholar] [CrossRef] [Green Version]
- Payen, L.; Gao, M.; Westlake, C.; Theis, A.; Cole, S.P.; Deeley, R.G. Functional interactions between nucleotide binding domains and leukotriene C4 binding sites of multidrug resistance protein 1 (ABCC1). Mol. Pharmacol. 2005, 67, 1944–1953. [Google Scholar] [CrossRef]
- Tóth, Á.; Janaszkiewicz, A.; Crespi, V.; Di Meo, F. On the interplay between lipids and asymmetric dynamics of an NBS degenerate ABC transporter. Commun. Biol. 2023, 6, 149. [Google Scholar] [CrossRef]
- Arana, M.R.; Altenberg, G.A. ATP-binding Cassette Exporters: Structure and Mechanism with a Focus on P-glycoprotein and MRP1. Curr. Med. Chem. 2019, 26, 1062–1078. [Google Scholar] [CrossRef]
- Conseil, G.; Arama-Chayoth, M.; Tsfadia, Y.; Cole, S.P. Structure-guided probing of the leukotriene C4 binding site in human multidrug resistance protein 1 (MRP1; ABCC1). FASEB J. 2019, 33, 10692–10704. [Google Scholar] [CrossRef] [Green Version]
- Raichaudhuri, A. Arabidopsis thaliana MRP1 (AtABCC1) nucleotide binding domain contributes to arsenic stress tolerance with serine triad phosphorylation. Plant Physiol. Biochem. 2016, 108, 109–120. [Google Scholar]
- Cole, S.P. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [Green Version]
- He, S.M.; Li, R.; R Kanwar, J.; Zhou, S.F. Structural and functional properties of human multidrug resistance protein 1 (MRP1/ABCC1). Curr. Med. Chem. 2011, 18, 439–481. [Google Scholar] [CrossRef]
- Ghanem, C.I.; Manautou, J.E. Modulation of hepatic MRP3/ABCC3 by xenobiotics and pathophysiological conditions: Role in drug pharmacokinetics. Curr. Med. Chem. 2019, 26, 1185–1223. [Google Scholar]
- Zhang, Y.; Yang, S.H.; Guo, X.L. New insights into Vinca alkaloids resistance mechanism and circumvention in lung cancer. Biomed. Pharmacother. 2017, 96, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Conseil, G.; Cole, S.P. Conserved amino acids in the region connecting membrane spanning domain 1 to nucleotide binding domain 1 are essential for expression of the MRP1 (ABCC1) transporter. PLoS ONE 2021, 16, e0246727. [Google Scholar] [CrossRef]
- Cort, A.; Ozben, T.; Saso, L.; De Luca, C.; Korkina, L. Redox control of multidrug resistance and its possible modulation by antioxidants. Oxidative Med. Cell. Longev. 2016, 2016, 4251912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Chen, B. Detection approaches for multidrug resistance genes of leukemia. Drug Des. Dev. Ther. 2017, 11, 1255–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Chen, W.M.; Wang, Z.H.; Wei, T.N.; Chen, Z.Z.; Wu, W.B. CircPAN3 mediates drug resistance in acute myeloid leukemia through the miR-153-5p/miR-183-5p–XIAP axis. Exp. Hematol. 2019, 70, 42–54. [Google Scholar] [CrossRef]
- Feldman, R.E.; Lam, A.C.; Sadow, P.M.; Bleier, B.S. P-glycoprotein is a marker of tissue eosinophilia and radiographic inflammation in chronic rhinosinusitis without nasal polyps. In International Forum of Allergy & Rhinology; Wiley Online Library: Hoboken, NJ, USA, 2013; Volume 3, pp. 684–687. [Google Scholar]
- Westlake, C.J.; Qian, Y.M.; Gao, M.; Vasa, M.; Cole, S.P.; Deeley, R.G. Identification of the structural and functional boundaries of the multidrug resistance protein 1 cytoplasmic loop 3. Biochemistry 2003, 42, 14099–14113. [Google Scholar] [CrossRef]
- Hanssen, K.M.; Wheatley, M.S.; Yu, D.M.; Conseil, G.; Norris, M.D.; Haber, M.; Cole, S.P.; Fletcher, J.I. GSH facilitates the binding and inhibitory activity of novel multidrug resistance protein 1 (MRP1) modulators. FEBS J. 2022, 289, 3854–3875. [Google Scholar] [CrossRef]
- Zhao, Z.J.; Gao, X.Y.; Zeng, J.C.; Zhang, S.L.; Meng, X.M.; Shen, Y.J.; Sheng, X.H. Theoretical insights into the cotransport mechanism of GSH with anticancer drugs by MRP1. J. Phys. Chem. B 2020, 124, 9803–9811. [Google Scholar] [CrossRef]
- Cole, S.P. Targeting multidrug resistance protein 1 (MRP1, ABCC1): Past, present, and future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef]
- DeGorter, M.K.; Conseil, G.; Deeley, R.G.; Campbell, R.L.; Cole, S.P. Molecular modeling of the human multidrug resistance protein 1 (MRP1/ABCC1). Biochem. Biophys. Res. Commun. 2008, 365, 29–34. [Google Scholar] [CrossRef]
- Amram, S.; Ganoth, A.; Tichon, O.; Peer, D.; Nachliel, E.; Gutman, M.; Tsfadia, Y. Structural characterization of the drug translocation path of MRP1/ABCC1. Israel J. Chem. 2014, 54, 1382–1393. [Google Scholar] [CrossRef]
- Weigl, K.E.; Conseil, G.; Rothnie, A.J.; Arama, M.; Tsfadia, Y.; Cole, S.P. An outward-facing aromatic amino acid is crucial for signaling between the membrane-spanning and nucleotide-binding domains of multidrug resistance protein 1 (MRP1; ABCC1). Mol. Pharmacol. 2018, 94, 1069–1078. [Google Scholar] [CrossRef]
- Jagodinsky, J.C.; Akgun, U. Characterizing the binding interactions between P-glycoprotein and eight known cardiovascular transport substrates. Pharmacol. Res. Perspect. 2015, 3, e00114. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.J.U.; dos Santos, D.J.V.A. Insights on P-Glycoprotein’s Efflux Mechanism Obtained by Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 1853–1864. [Google Scholar] [CrossRef]
- Dastvan, R.; Mishra, S.; Peskova, Y.B.; Nakamoto, R.K.; Mchaourab, H.S. Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors. Science 2019, 364, 689–692. [Google Scholar] [CrossRef]
- Domicevica, L.; Biggin, P. Homology modelling of human P-glycoprotein. Biochem. Soc. Trans. 2015, 43, 952–958. [Google Scholar] [CrossRef]
- Prajapati, R.; Sangamwar, A.T. Translocation mechanism of P-glycoprotein and conformational changes occurring at drug-binding site: Insights from multi-targeted molecular dynamics. Biochim. Biophys. Acta (BBA)—Biomembr. 2014, 1838, 2882–2898. [Google Scholar] [CrossRef] [Green Version]
- Manoharan, J.P.; Karunakaran, K.N.; Vidyalakshmi, S.; Dhananjayan, K. Computational binding affinity and molecular dynamic characterization of annonaceous acetogenins at nucleotide binding domain (NBD) of multi-drug resistance ATP-binding cassette sub-family B member 1 (ABCB1). J. Biomol. Struct. Dyn. 2023, 41, 821–832. [Google Scholar] [CrossRef]
- Esposito, C.; Wang, S.; Lange, U.E.W.; Oellien, F.; Riniker, S. Combining Machine Learning and Molecular Dynamics to Predict P-Glycoprotein Substrates. J. Chem. Inf. Model. 2020, 60, 4730–4749. [Google Scholar] [CrossRef]
- Prajapati, R.; Singh, U.; Patil, A.; Khomane, K.S.; Bagul, P.; Bansal, A.K.; Sangamwar, A.T. In silico model for P-glycoprotein substrate prediction: Insights from molecular dynamics and in vitro studies. J. Comput.-Aided Mol. Des. 2013, 27, 347–363. [Google Scholar] [CrossRef]
- Karthika, C.; Sureshkumar, R.; Zehravi, M.; Akter, R.; Ali, F.; Ramproshad, S.; Mondal, B.; Tagde, P.; Ahmed, Z.; Khan, F.S.; et al. Multidrug Resistance of Cancer Cells and the Vital Role of P-Glycoprotein. Life 2022, 12, 897. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.L.; Duong, V.A.; Maeng, H.J. Pharmaceutical Formulations with P-Glycoprotein Inhibitory Effect as Promising Approaches for Enhancing Oral Drug Absorption and Bioavailability. Pharmaceutics 2021, 13, 1103. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; Liu, F.F.; Dong, X.Y.; Sun, Y. Molecular insight into conformational transmission of human P-glycoprotein. J. Chem. Phys. 2013, 139, 225102. [Google Scholar] [CrossRef]
- Wen, P.C.; Verhalen, B.; Wilkens, S.; Mchaourab, H.S.; Tajkhorshid, E. On the Origin of Large Flexibility of P-glycoprotein in the Inward-facing State. J. Biol. Chem. 2013, 288, 19211–19220. [Google Scholar] [CrossRef] [Green Version]
- Hulyer, A.R.C.; Briggs, D.A.; O’Mara, M.L.; Kerr, I.D.; Harmer, J.R.; Callaghan, R. Cross-linking, DEER-spectroscopy and molecular dynamics confirm the inward facing state of P-glycoprotein in a lipid membrane. J. Struct. Biol. 2020, 211, 107513. [Google Scholar] [CrossRef]
- Zhang, Y.; Gong, W.; Wang, Y.; Liu, Y.; Li, C. Exploring movement and energy in human P-glycoprotein conformational rearrangement. J. Biomol. Struct. Dyn. 2019, 37, 1104–1119. [Google Scholar] [CrossRef]
- Al-Shawi, M.K.; Polar, M.K.; Omote, H.; Figler, R.A. Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem. 2003, 278, 52629–52640. [Google Scholar] [CrossRef] [Green Version]
- Frank, G.A.; Shukla, S.; Rao, P.; Borgnia, M.J.; Bartesaghi, A.; Merk, A.; Mobin, A.; Esser, L.; Earl, L.A.; Gottesman, M.M.; et al. Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle. Mol. Pharmacol. 2016, 90, 35–41. [Google Scholar]
- Rosenberg, M.F.; Kamis, A.B.; Callaghan, R.; Higgins, C.F.; Ford, R.C. Three-dimensional structures of the mammalian multidrug resistance P-glycoprotein demonstrate major conformational changes in the transmembrane domains upon nucleotide binding. J. Biol. Chem. 2003, 278, 8294–8299. [Google Scholar]
- St-Pierre, J.F.; Bunker, A.; Róg, T.; Karttunen, M.; Mousseau, N. Molecular Dynamics Simulations of the Bacterial ABC Transporter SAV1866 in the Closed Form. J. Phys. Chem. B 2012, 116, 2934–2942. [Google Scholar] [CrossRef]
- Taggi, V.; Riera Romo, M.; Piquette-Miller, M.; zu Schwabedissen, H.E.; Neuhoff, S. Transporter Regulation in Critical Protective Barriers: Focus on Brain and Placenta. Pharmaceutics 2022, 14, 1376. [Google Scholar] [CrossRef]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef] [Green Version]
- Zibell, G.; Unkrüer, B.; Pekcec, A.; Hartz, A.M.; Bauer, B.; Miller, D.S.; Potschka, H. Prevention of seizure-induced up-regulation of endothelial P-glycoprotein by COX-2 inhibition. Neuropharmacology 2009, 56, 849–855. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.; Pekcec, A.; Toellner, K.; Miller, D.S.; Potschka, H. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Mol. Pharmacol. 2008, 73, 1444–1453. [Google Scholar] [CrossRef] [Green Version]
- Chai, A.B.; Callaghan, R.; Gelissen, I.C. Regulation of P-Glycoprotein in the Brain. Int. J. Mol. Sci. 2022, 23, 14667. [Google Scholar] [CrossRef]
- Wise, J.G. Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry 2012, 51, 5125–5141. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Huang, S.; Heng, Y.; Mei, H.; Pan, X. Computational Insights into Allosteric Conformational Modulation of P-Glycoprotein by Substrate and Inhibitor Binding. Molecules 2020, 25, 6006. [Google Scholar] [CrossRef]
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef] [Green Version]
- Klepsch, F.; Ecker, G.F. Impact of the recent mouse P-glycoprotein structure for structure-based ligand design. Mol. Inform. 2010, 29, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.F.; Velarde, G.; Ford, R.C.; Martin, C.; Berridge, G.; Kerr, I.D.; Callaghan, R.; Schmidlin, A.; Wooding, C.; Linton, K.J.; et al. Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 2001, 20, 5615–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrier, I.; Gros, P. Investigating the role of the invariant carboxylate residues E552 and E1197 in the catalytic activity of Abcb1a (mouse Mdr3). FEBS J. 2008, 275, 3312–3324. [Google Scholar] [CrossRef]
- Futamata, R.; Ogasawara, F.; Ichikawa, T.; Kodan, A.; Kimura, Y.; Kioka, N.; Ueda, K. In vivo FRET analyses reveal a role of ATP hydrolysis–associated conformational changes in human P-glycoprotein. J. Biol. Chem. 2020, 295, 5002–5011. [Google Scholar] [CrossRef] [PubMed]
- Immadisetty, K.; Hettige, J.; Moradi, M. Lipid-Dependent Alternating Access Mechanism of a Bacterial Multidrug ABC Exporter. ACS Cent. Sci. 2019, 5, 43–56. [Google Scholar] [CrossRef]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Sakiyama, K.; Hipolito, C.J.; Fujioka, A.; Hirokane, R.; Ikeguchi, K.; Watanabe, B.; Hiratake, J.; et al. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. USA 2014, 111, 4049–4054. [Google Scholar] [CrossRef] [Green Version]
- Bársony, O.; Szalóki, G.; Türk, D.; Tarapcsák, S.; Gutay-Tóth, Z.; Bacsó, Z.; Holb, I.J.; Székvölgyi, L.; Szabó, G.; Csanády, L.; et al. A single active catalytic site is sufficient to promote transport in P-glycoprotein. Sci. Rep. 2016, 6, 24810. [Google Scholar] [CrossRef] [Green Version]
- Sharom, F.J.; Lugo, M.R.; Eckford, P.D. New insights into the drug binding, transport and lipid flippase activities of the p-glycoprotein multidrug transporter. J. Bioenerg. Biomembr. 2005, 37, 481–487. [Google Scholar] [CrossRef]
- Raub, T.J. P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol. Pharm. 2006, 3, 3–25. [Google Scholar] [CrossRef]
- Mora Lagares, L.; Pérez-Castillo, Y.; Minovski, N.; Novič, M. Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2022, 23, 362. [Google Scholar] [CrossRef]
- Swartz, D.J.; Weber, J.; Urbatsch, I.L. P-glycoprotein is fully active after multiple tryptophan substitutions. Biochim. Biophys. Acta (BBA)—Biomembr. 2013, 1828, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Aller, S.G. Equilibrated Atomic Models of Outward-Facing P-glycoprotein and Effect of ATP Binding on Structural Dynamics. Sci. Rep. 2015, 5, 7880. [Google Scholar] [CrossRef] [Green Version]
- Gil-Martins, E.; Barbosa, D.J.; Silva, V.; Remião, F.; Silva, R. Dysfunction of ABC transporters at the blood-brain barrier: Role in neurological disorders. Pharmacol. Ther. 2020, 213, 107554. [Google Scholar] [CrossRef]
- Ledwitch, K.V.; Roberts, A.G. Cardiovascular ion channel inhibitor drug-drug interactions with P-glycoprotein. AAPS J. 2017, 19, 409–420. [Google Scholar] [CrossRef]
- Miller, D.S. Regulation of ABC transporters blood–brain barrier: The good, the bad, and the ugly. Adv. Cancer Res. 2015, 125, 43–70. [Google Scholar]
- Al-Shawi, M.K.; Omote, H. The Remarkable Transport Mechanism of P-Glycoprotein: A Multidrug Transporter. J. Bioenerg. Biomembr. 2005, 37, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Seelig, A. P-Glycoprotein: One Mechanism, many tasks and the Consequences for Pharmacotherapy of Cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef]
- van Wonderen, J.H.; McMahon, R.M.; O’Mara, M.L.; McDevitt, C.A.; Thomson, A.J.; Kerr, I.D.; MacMillan, F.; Callaghan, R. The central cavity of ABCB 1 undergoes alternating access during ATP hydrolysis. FEBS J. 2014, 281, 2190–2201. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.; Berridge, G.; Mistry, P.; Higgins, C.; Charlton, P.; Callaghan, R. Drug binding sites on P-glycoprotein are altered by ATP binding prior to nucleotide hydrolysis. Biochemistry 2000, 39, 11901–11906. [Google Scholar] [CrossRef]
- Urgaonkar, S.; Nosol, K.; Said, A.M.; Nasief, N.N.; Bu, Y.; Locher, K.P.; Lau, J.Y.N.; Smolinski, M.P. Discovery and Characterization of Potent Dual P-Glycoprotein and CYP3A4 Inhibitors: Design, Synthesis, Cryo-EM Analysis, and Biological Evaluations. J. Med. Chem. 2022, 65, 191–216. [Google Scholar] [CrossRef]
- Thonghin, N.; Collins, R.F.; Barbieri, A.; Shafi, T.; Siebert, A.; Ford, R.C. Novel features in the structure of P-glycoprotein (ABCB1) in the post-hydrolytic state as determined at 7.9 Å resolution. BMC Struct. Biol. 2018, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Matsuoka, K.; Kimura, Y.; Ueda, K.; Kato, H. Inward- and outward-facing X-ray crystal structures of homodimeric P-glycoprotein CmABCB1. Nat. Commun. 2019, 10, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, K.; Pant, S.; Tajkhorshid, E. Active participation of membrane lipids in inhibition of multidrug transporter P-glycoprotein. Chem. Sci. 2021, 12, 6293–6306. [Google Scholar] [CrossRef] [PubMed]
- Govind Kumar, V.; Ogden, D.S.; Isu, U.H.; Polasa, A.; Losey, J.; Moradi, M. Prefusion spike protein conformational changes are slower in SARS-CoV-2 than in SARS-CoV-1. J. Biol. Chem. 2022, 298, 101814. [Google Scholar] [CrossRef] [PubMed]
- Polasa, A.; Moradi, M. Deciphering the Inter-domain Decoupling in the Gram-negative Bacterial Membrane Insertase. bioRxiv 2022. [Google Scholar] [CrossRef]
- Domicevica, L.; Koldsø, H.; Biggin, P.C. Multiscale molecular dynamics simulations of lipid interactions with P-glycoprotein in a complex membrane. J. Mol. Graph. Model. 2018, 80, 147–156. [Google Scholar] [CrossRef]
- Isu, U.H.; Badiee, S.A.; Khodadadi, E.; Moradi, M. Cholesterol in Class C GPCRs: Role, Relevance, and Localization. Membranes 2023, 13, 301. [Google Scholar] [CrossRef]
- Wang, L.; O’Mara, M.L. Effect of the Force Field on Molecular Dynamics Simulations of the Multidrug Efflux Protein P-Glycoprotein. J. Chem. Theory Comput. 2021, 17, 6491–6508. [Google Scholar] [CrossRef]
- Behmard, E.; Barzegari, E.; Najafipour, S.; Kouhpayeh, A.; Ghasemi, Y.; Asadi-Pooya, A.A. Efflux dynamics of the antiseizure drug, levetiracetam, through the P-glycoprotein channel revealed by advanced comparative molecular simulations. Sci. Rep. 2022, 12, 13674. [Google Scholar] [CrossRef]
- Li, H.; Gong, W. Study of Allosteric Transitions of Human P-Glycoprotein by Using the Two-State Anisotropic Network Model. Front. Med. 2022, 9, 26. [Google Scholar] [CrossRef]
- Vahedi, S.; Chufan, E.E.; Ambudkar, S.V. Global alteration of the drug-binding pocket of human P-glycoprotein (ABCB1) by substitution of fifteen conserved residues reveals a negative correlation between substrate size and transport efficiency. Biochem. Pharmacol. 2017, 143, 53–64. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. A salt bridge in intracellular loop 2 is essential for folding of human P-glycoprotein. Biochemistry 2013, 52, 3194–3196. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2002, 277, 44332–44338. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Human P-glycoprotein contains a greasy ball-and-socket joint at the second transmission interface. J. Biol. Chem. 2013, 288, 20326–20333. [Google Scholar] [CrossRef] [Green Version]
- Thangapandian, S.; Kapoor, K.; Tajkhorshid, E. Probing cholesterol binding and translocation in P-glycoprotein. Biochim. Biophys. Acta (BBA)—Biomembr. 2020, 1862, 183090. [Google Scholar] [CrossRef]
- Clouser, A.F.; Alam, Y.H.; Atkins, W.M. Cholesterol Asymmetrically Modulates the Conformational Ensemble of the Nucleotide-Binding Domains of P-Glycoprotein in Lipid Nanodiscs. Biochemistry 2021, 60, 85–94. [Google Scholar] [CrossRef]
- Yang, X.; Ye, W.; Qi, Y.; Ying, Y.; Xia, Z. Overcoming Multidrug Resistance in Bacteria Through Antibiotics Delivery in Surface-Engineered Nano-Cargos: Recent Developments for Future Nano-Antibiotics. Front. Bioeng. Biotechnol. 2021, 9, 696514. [Google Scholar] [CrossRef]
- Chegini, Z.; Khoshbayan, A.; Vesal, S.; Moradabadi, A.; Hashemi, A.; Shariati, A. Bacteriophage therapy for inhibition of multi drug-resistant uropathogenic bacteria: A narrative review. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 30. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug Resistance in Bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [Green Version]
- Sodani, K.; Patel, A.; Kathawala, R.; Chen, Z.S. Multidrug resistance associated proteins in multidrug resistance. Chin. J. Cancer 2011, 31, 58–72. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Cater, R.; Choy, B.; Mancia, F. Structural Insights into Transporter-Mediated Drug Resistance in Infectious Diseases. J. Mol. Biol. 2021, 433, 167005. [Google Scholar] [CrossRef] [PubMed]
- Velamakanni, S.; Yao, Y.; Gutmann, D.A.P.; van Veen, H.W. Multidrug Transport by the ABC Transporter Sav1866 from Staphylococcus aureus. Biochemistry 2008, 47, 9300–9308. [Google Scholar] [CrossRef] [PubMed]
- Dashtbani-Roozbehani, A.; Brown, M.H. Efflux Pump Mediated Antimicrobial Resistance by Staphylococci in Health-Related Environments: Challenges and the Quest for Inhibition. Antibiotics 2021, 10, 1502. [Google Scholar] [CrossRef] [PubMed]
- Zolnerciks, J.K.; Wooding, C.; Linton, K.J. Evidence for a Sav1866-like architecture for the human multidrug transporter P-glycoprotein. FASEB J. 2007, 21, 3937–3948. [Google Scholar] [CrossRef]
- Göddeke, H.; Schäfer, L.V. Capturing Substrate Translocation in an ABC Exporter at the Atomic Level. J. Am. Chem. Soc. 2020, 142, 12791–12801. [Google Scholar] [CrossRef]
- Stockner, T.; Gradisch, R.; Schmitt, L. The role of the degenerate nucleotide binding site in type I ABC exporters. FEBS Lett. 2020, 594, 3815–3838. [Google Scholar] [CrossRef]
- Dastvan, R.; Rasouli, A.; Dehghani-Ghahnaviyeh, S.; Gies, S.; Tajkhorshid, E. Proton-driven alternating access in a spinster lipid transporter. Nat. Commun. 2022, 13, 5161. [Google Scholar] [CrossRef]
- Mishra, S.; Verhalen, B.; Stein, R.A.; Wen, P.C.; Tajkhorshid, E.; Mchaourab, H.S. Conformational dynamics of the nucleotide binding domains and the power stroke of a heterodimeric ABC transporter. eLife 2014, 3, e02740. [Google Scholar] [CrossRef]
- Göddeke, H.; Timachi, M.H.; Hutter, C.A.J.; Galazzo, L.; Seeger, M.A.; Karttunen, M.; Bordignon, E.; Schäfer, L.V. Atomistic Mechanism of Large-Scale Conformational Transition in a Heterodimeric ABC Exporter. J. Am. Chem. Soc. 2018, 140, 4543–4551. [Google Scholar] [CrossRef]
- Oliveira, A.S.; Baptista, A.M.; Soares, C.M. Conformational changes induced by ATP-hydrolysis in an ABC transporter: A molecular dynamics study of the Sav1866 exporter. Proteins Struct. Funct. Bioinform. 2011, 79, 1977–1990. [Google Scholar] [CrossRef]
- Nöll, A.; Thomas, C.; Herbring, V.; Zollmann, T.; Barth, K.; Mehdipour, A.R.; Tomasiak, T.M.; Brüchert, S.; Joseph, B.; Abele, R.; et al. Crystal structure and mechanistic basis of a functional homolog of the antigen transporter TAP. Proc. Natl. Acad. Sci. USA 2017, 114, E438–E447. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.P.; Van Bambeke, F.; Tulkens, P.M.; Prevost, M. Dynamics and Structural Changes Induced by ATP Binding in SAV1866, a Bacterial ABC Exporter. J. Phys. Chem. B 2010, 114, 15948–15957. [Google Scholar] [CrossRef] [Green Version]
- Pollock, N.L.; Lloyd, J.; Montinaro, C.; Rai, M.; Dafforn, T.R. Conformational trapping of an ABC transporter in polymer lipid nanoparticles. Biochem. J. 2022, 479, 145–159. [Google Scholar] [CrossRef]
- Aittoniemi, J.; de Wet, H.; Ashcroft, F.M.; Sansom, M.S. Asymmetric switching in a homodimeric ABC transporter: A simulation study. PLoS Comput. Biol. 2010, 6, e1000762. [Google Scholar] [CrossRef] [Green Version]
- Zaitseva, J.; Oswald, C.; Jumpertz, T.; Jenewein, S.; Wiedenmann, A.; Holland, I.B.; Schmitt, L. A structural analysis of asymmetry required for catalytic activity of an ABC-ATPase domain dimer. EMBO J. 2006, 25, 3432–3443. [Google Scholar] [CrossRef]
- Roberts, A.G. The Structure and Mechanism of Drug Transporters BT—Enzyme Kinetics in Drug Metabolism: Fundamentals and Applications; Springer: New York, NY, USA, 2021; pp. 193–234. [Google Scholar] [CrossRef]
- Zeng, Y.; Charkowski, A.O. The role of ATP-binding cassette transporters in bacterial phytopathogenesis. Phytopathology 2021, 111, 600–610. [Google Scholar] [CrossRef]
- Doerrler, W.T.; Gibbons, H.S.; Raetz, C.R. MsbA-dependent translocation of lipids across the inner membrane of Escherichia coli. J. Biol. Chem. 2004, 279, 45102–45109. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; White, K.A.; Polissi, A.; Georgopoulos, C.; Raetz, C.R. Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem. 1998, 273, 12466–12475. [Google Scholar] [CrossRef] [Green Version]
- Zgurskaya, H.I.; Lopez, C.A.; Gnanakaran, S. Permeability barrier of Gram-negative cell envelopes and approaches to bypass it. ACS Infect. Dis. 2015, 1, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Bertani, B.; Ruiz, N. Function and biogenesis of lipopolysaccharides. EcoSal Plus 2018, 8, 1–19. [Google Scholar] [CrossRef]
- Lyu, J.; Liu, C.; Zhang, T.; Schrecke, S.; Elam, N.P.; Packianathan, C.; Hochberg, G.K.; Russell, D.; Zhao, M.; Laganowsky, A. Structural basis for lipid and copper regulation of the ABC transporter MsbA. Nat. Commun. 2022, 13, 7291. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Lakatos, A.; Spadaccini, R.; Vogel, R.; Hoffmann, C.; Becker-Baldus, J.; Ouari, O.; Tordo, P.; Mchaourab, H.; Glaubitz, C. The ABC exporter MsbA probed by solid state NMR–challenges and opportunities. Biol. Chem. 2015, 396, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T. Structural dynamics of ABC transporters: Molecular simulation studies. Biochem. Soc. Trans. 2021, 49, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Orelle, C.; Mathieu, K.; Jault, J.M. Multidrug ABC transporters in bacteria. Res. Microbiol. 2019, 170, 381–391. [Google Scholar] [CrossRef]
- Thélot, F.; Orlando, B.J.; Li, Y.; Liao, M. High-resolution views of lipopolysaccharide translocation driven by ABC transporters MsbA and LptB2FGC. Curr. Opin. Struct. Biol. 2020, 63, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Moradi, M.; Tajkhorshid, E. Mechanistic picture for conformational transition of a membrane transporter at atomic resolution. Proc. Natl. Acad. Sci. USA 2013, 110, 18916–18921. [Google Scholar] [CrossRef] [Green Version]
- Moradi, M.; Tajkhorshid, E. Computational recipe for efficient description of large-scale conformational changes in biomolecular systems. J. Chem. Theory Comput. 2014, 10, 2866–2880. [Google Scholar] [CrossRef] [Green Version]
- Mittal, A.; Böhm, S.; Grütter, M.G.; Bordignon, E.; Seeger, M.A. Asymmetry in the homodimeric ABC transporter MsbA recognized by a DARPin. J. Biol. Chem. 2012, 287, 20395–20406. [Google Scholar] [CrossRef] [Green Version]
- Siarheyeva, A.; Sharom, F.J. The ABC transporter MsbA interacts with lipid A and amphipathic drugs at different sites. Biochem. J. 2009, 419, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Spadaccini, R.; Kaur, H.; Becker-Baldus, J.; Glaubitz, C. The effect of drug binding on specific sites in transmembrane helices 4 and 6 of the ABC exporter MsbA studied by DNP-enhanced solid-state NMR. Biochim. Biophys. Acta (BBA)—Biomembr. 2018, 1860, 833–840. [Google Scholar] [CrossRef]
- Woebking, B.; Velamakanni, S.; Federici, L.; Seeger, M.A.; Murakami, S.; van Veen, H.W. Functional role of transmembrane helix 6 in drug binding and transport by the ABC transporter MsbA. Biochemistry 2008, 47, 10904–10914. [Google Scholar] [CrossRef]
- Furuta, T.; Yamaguchi, T.; Kato, H.; Sakurai, M. Analysis of the structural and functional roles of coupling helices in the ATP-binding cassette transporter MsbA through enzyme assays and molecular dynamics simulations. Biochemistry 2014, 53, 4261–4272. [Google Scholar] [CrossRef]
- Doshi, R.; Ali, A.; Shi, W.; Freeman, E.V.; Fagg, L.A.; van Veen, H.W. Molecular disruption of the power stroke in the ATP-binding cassette transport protein MsbA. J. Biol. Chem. 2013, 288, 6801–6813. [Google Scholar] [CrossRef] [Green Version]
- Westfahl, K.M.; Merten, J.A.; Buchaklian, A.H.; Klug, C.S. Functionally important ATP binding and hydrolysis sites in Escherichia coli MsbA. Biochemistry 2008, 47, 13878–13886. [Google Scholar] [CrossRef] [Green Version]
- Randak, C.O.; Dong, Q.; Ver Heul, A.R.; Elcock, A.H.; Welsh, M.J. ATP and AMP mutually influence their interaction with the ATP-binding cassette (ABC) adenylate kinase cystic fibrosis transmembrane conductance regulator (CFTR) at separate binding sites. J. Biol. Chem. 2013, 288, 27692–27701. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.S.; Tainer, J.A. Learning our ABCs: Rad50 directs MRN repair functions via adenylate kinase activity from the conserved ATP binding cassette. Mol. Cell 2007, 25, 789–791. [Google Scholar] [CrossRef]
- Yaginuma, H.; Kawai, S.; Tabata, K.V.; Tomiyama, K.; Kakizuka, A.; Komatsuzaki, T.; Noji, H.; Imamura, H. Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci. Rep. 2014, 4, 6522. [Google Scholar] [CrossRef] [Green Version]
- Kaur, H.; Lakatos-Karoly, A.; Vogel, R.; Nöll, A.; Tampé, R.; Glaubitz, C. Coupled ATPase-adenylate kinase activity in ABC transporters. Nat. Commun. 2016, 7, 13864. [Google Scholar] [CrossRef]
- Kaur, H.; Abreu, B.; Akhmetzyanov, D.; Lakatos-Karoly, A.; Soares, C.M.; Prisner, T.; Glaubitz, C. Unexplored nucleotide binding modes for the ABC exporter MsbA. J. Am. Chem. Soc. 2018, 140, 14112–14125. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | PDB ID * | Resolution | Method ** |
|---|---|---|---|
| MRP1 | 6BHU [54] | 3.14 Å | Cryo-EM |
| 6UY0 [55] | 3.23 Å | Cryo-EM | |
| 8F4B [56] | 3.27 Å | Cryo-EM | |
| 5UJA [57] | 3.34 Å | Cryo-EM | |
| 5UJ9 [57] | 3.49 Å | Cryo-EM | |
| 7M68 [58] | 4.04 Å | Cryo-EM | |
| Pgp | 5KO2 [59] | 3.30 Å | X-ray |
| 5KPD [60] | 3.35 Å | X-ray | |
| 4Q9H [61] | 3.40 Å | X-ray | |
| 6C0V [62] | 3.40 Å | Cryo–EM | |
| 5KPJ [59] | 3.50 Å | X-ray | |
| 4XWK [63] | 3.50 Å | X-ray | |
| 4Q9L [61] | 3.80 Å | X-ray | |
| 4M1M [64] | 3.80 Å | X-ray | |
| 5KOI [59] | 3.85 Å | X-ray | |
| 6UJN [65] | 3.98 Å | X-ray | |
| 5KPI [59] | 4.01 Å | X-ray | |
| Sav1866 | 2HYD [66] | 3.00 Å | X-ray |
| 2ONJ [67] | 3.40 Å | X-ray | |
| MsbA | 6BPL [68] | 2.70 Å | X-ray |
| 6BPP [68] | 2.92 Å | X-ray | |
| 6BL6 [69] | 2.80 Å | X-ray | |
| 3B60 [70] | 3.70 Å | X-ray | |
| 5TV4 [71] | 4.20 Å | Cryo-EM | |
| 6UZL [72] | 4.40 Å | Cryo-EM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badiee, S.A.; Isu, U.H.; Khodadadi, E.; Moradi, M. The Alternating Access Mechanism in Mammalian Multidrug Resistance Transporters and Their Bacterial Homologs. Membranes 2023, 13, 568. https://doi.org/10.3390/membranes13060568
Badiee SA, Isu UH, Khodadadi E, Moradi M. The Alternating Access Mechanism in Mammalian Multidrug Resistance Transporters and Their Bacterial Homologs. Membranes. 2023; 13(6):568. https://doi.org/10.3390/membranes13060568
Chicago/Turabian StyleBadiee, Shadi A, Ugochi H. Isu, Ehsaneh Khodadadi, and Mahmoud Moradi. 2023. "The Alternating Access Mechanism in Mammalian Multidrug Resistance Transporters and Their Bacterial Homologs" Membranes 13, no. 6: 568. https://doi.org/10.3390/membranes13060568