
Dimerization of Transmembrane Proteins in Cancer Immunotherapy

1
College of Chemistry, Fuzhou University, Fuzhou 350108, China
2
College of Biological Science and Engineering, Fuzhou University, Fuzhou 350108, China
*
Author to whom correspondence should be addressed.
Membranes 2023, 13(4), 393; https://doi.org/10.3390/membranes13040393
Submission received: 2 February 2023
/
Revised: 24 March 2023
/
Accepted: 26 March 2023
/
Published: 30 March 2023
(This article belongs to the Special Issue Membrane Proteins: Function, Structure, and Dynamic)
Abstract
:Transmembrane proteins (TMEMs) are integrated membrane proteins that span the entire lipid bilayer and are permanently anchored to it. TMEMs participate in various cellular processes. Some TMEMs usually exist and perform their physiological functions as dimers rather than monomers. TMEM dimerization is associated with various physiological functions, such as the regulation of enzyme activity, signal transduction, and cancer immunotherapy. In this review, we focus on the dimerization of transmembrane proteins in cancer immunotherapy. This review is divided into three parts. First, the structures and functions of several TMEMs related to tumor immunity are introduced. Second, the characteristics and functions of several typical TMEM dimerization processes are analyzed. Finally, the application of the regulation of TMEM dimerization in cancer immunotherapy is introduced.
1. Introduction
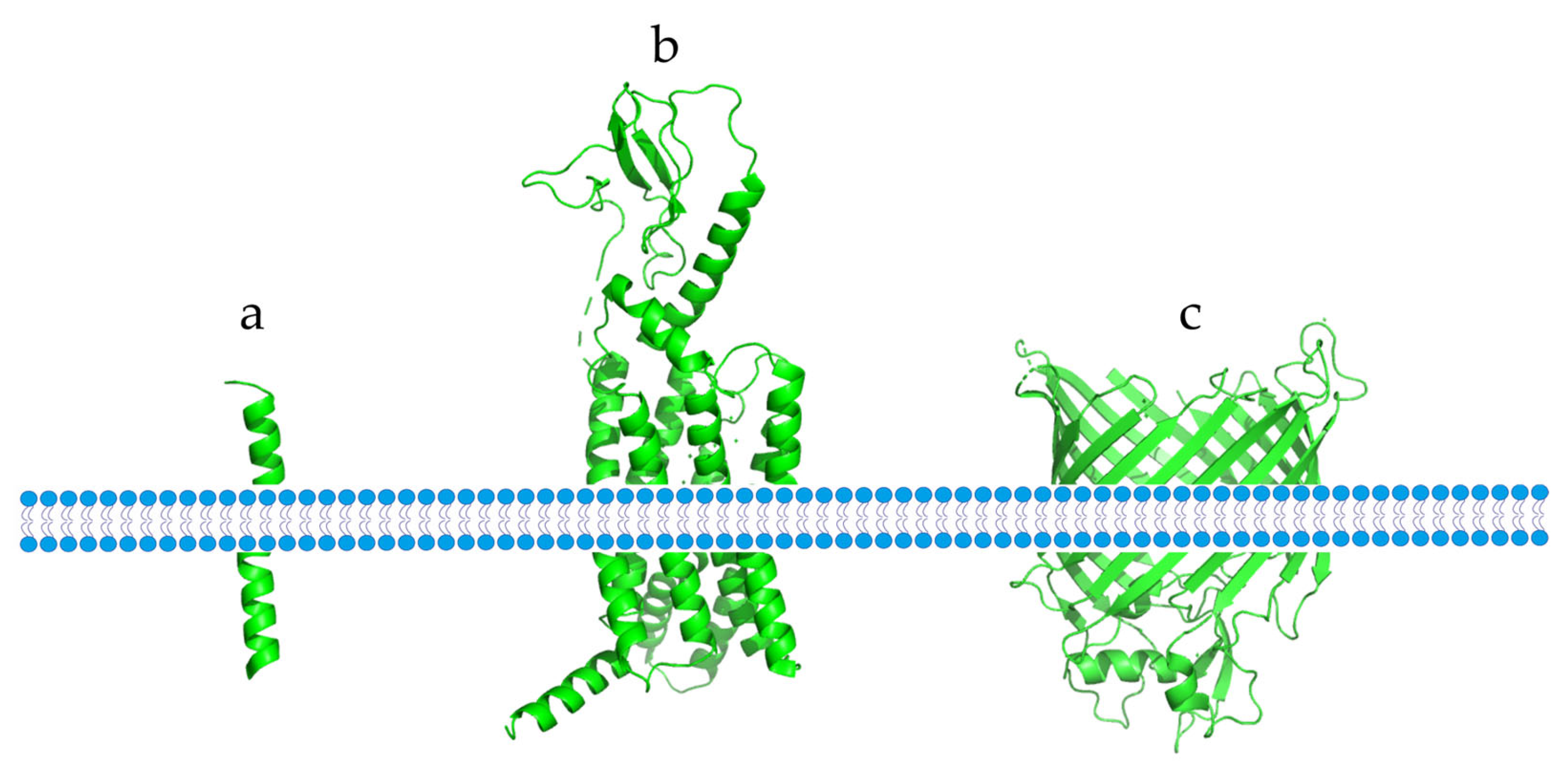
Membrane proteins are essential for the physiological functions of the cell membrane. They can be classified into two types based on their interaction with the membrane: peripheral membrane proteins, which bind to the membrane through non-covalent interactions, and integrated membrane proteins, which more firmly bind to the membrane through hydrophobic interactions [1]. TMEMs are integrated membrane proteins that span the entire lipid bilayer and are permanently anchored in it [2]. Based on their different structures, TMEMs can be divided into two categories: α-helical proteins and β-barrel proteins (Figure 1). These two structures are the dominant structures of all transmembrane proteins, with α-helical proteins being mostly found in the cytoplasm and subcellular septum, while β-barrel proteins are mostly found in chloroplasts, bacteria, and mitochondrial membranes [3].
A variety of TMEMs play a regulatory role in cancer immunotherapy. Within the TMEM family, immune checkpoint proteins are called immune system regulators; they can regulate the activity of various immune cells and play important roles in the process of maintaining the body’s immune balance [4]. Cancer cells can use inhibitory immune checkpoint proteins to generate tumor immune escape [5]. Traditional immune checkpoint proteins mainly target T cells, with Programmed death ligand 1 (PD-L1) and Toll-Like Receptor-4 (TLR-4) being well-known immune checkpoint proteins related to T-cell activation [5,6]. The dimerization of PD-L1 or TLR-4 can cause changes in T-cell activity, making them a potential target for cancer immunotherapy. For example, the PD-L1/Programmed death 1 (PD-1) signaling pathway can inhibit T-cell activity, allowing cancer cells to escape the killing action performed by immune cells [7]. BMS-8, a compound by Bristol-Myers Squibb (BMS), binds to PD-L1 and causes it to form a homodimer, ultimately blocking the interaction between PD-L1 and PD-1, and leading to T-cell activation [8]. Rhamnogalacturonan II (RG-II) could effectively induce the dimerization of TLR-4, activate myeloid differentiation factor 88 (MyD88)-independent and MyD88-dependent pathways, promote the maturation and differentiation of dendritic cells (DCs), produce a series of cytokines to regulate inflammatory responses, and affect the activity of T cells [9].
Recent studies have demonstrated the potential of immune checkpoint proteins in natural killer (NK) cells for cancer immunotherapy [10]. Current immune checkpoint proteins associated with NK-cell activity include natural killer Group 2 member A (NKG2A), leukocyte immunoglobulin-like receptors (LIRS), human leukocyte antigen G (HLA-G), and Cluster of differentiation 96 (CD96) [10,11,12,13,14]. HLA-G is a major histocompatibility complex molecule that binds to inhibitory receptors on white blood cells and protects the fetus from attack by the mother’s immune cells [15]. Because of its immunosuppressive effects, HLA-G has been extensively studied for its role in cancer immunotherapy. For example, after homodimer formation, HLA-G can bind to its ligand, Ig-like transcript 2 (ILT2), with a higher affinity to regulate the cytotoxic activity of T cells and NK cells [16]. NKG2A is one of the key immune checkpoint proteins of NK cells and is involved in the regulation of NK-cell activity [17]. NKG2A can form a heterodimer with Cluster of differentiation 94 (CD94), which results in the inhibition of the activity of NK cells [17]. Therefore, blocking the interaction between NKG2A and CD94 can activate NK-cell activity, further inhibiting the immune escape of cancer cells.
Four immune checkpoint proteins, PD-L1, TLR4, HLA-G, and NKG2A, belong to the TMEM family and can form homodimers or heterodimers to regulate cancer immunotherapy. Therefore, this review focuses on these four immune checkpoint proteins. Firstly, we introduce the structures and functions of these proteins; then, we analyze the binding properties and functions of these immune checkpoint proteins and their receptors. Finally, we discuss the regulation of the dimerization of these immune checkpoint proteins and their potential as targets for cancer immunotherapy through dimerization regulation. Understanding the mechanisms of TMEM dimerization regulation can provide insights into developing novel anti-tumor drugs.
2. Structures and Functions of Immune Checkpoint Proteins in Cancer Immunotherapy
The structures and physiological functions of immune checkpoint proteins are crucial to the development of new drugs for tumor immunotherapy. This section mainly introduces the structures and physiological functions of several immune checkpoint proteins, including PD-L1, TLR4, HLA-G, and NKG2A.
2.1. PD-L1
PD-L1 is a transmembrane protein expressed in macrophages, activated B cells, T cells, and many solid tumor cells at higher levels than in normal tissues [18]. Encoded by the PD-L gene, PD-L1 belongs to the cluster of differentiation 28 (CD28)/B7/cytotoxic T lymphocyte antigen-4 (CTLA-4) family [19,20]. The gene encoding the PD-L1 protein contains seven exons, and the PD-L1 protein contains 290 amino acids and has a molecular mass of 40 kDa [21]. PD-L1 consists of an intracellular domain (30 amino acids), a transmembrane domain (hydrophobic), and two extracellular Ig-like domains (IgC- and IgV-like domains) [22,23,24].
PD-L1 plays a crucial role in tumor immunotherapy, as it inhibits T cells and NK cells when combined with PD-1 [25,26]. The PD-L1/PD-1 complex produces a signal that suppresses cytotoxic T cells, leading to T-cell depletion, which protects local tissues from immune-cell-induced inflammation [27]. The PD-1/PD-L1 signaling pathway is also involved in immune tolerance, which refers to the inability of immune cells to carry out a normal immune response under specific antigen stimulation [28]. Inhibiting the PD-1/PD-L1 signaling pathway enhances the immune response in two ways: it promotes the maturation and differentiation of immune cells, and it enhances the activity of immune cells [29,30]. However, the activation of immune cells can result in skin disorders, such as vitiligo and lichenoid dermatitis associated with renal metabolism, as well as the uncommon but difficult-to-treat psoriasis [29,30]. Therefore, understanding the structure and physiological function of PD-L1 is crucial to developing new drugs for tumor immunotherapy.
2.2. TLR-4
TLR-4 is a member of the Toll-like receptor (TLR) family that plays a critical role in regulating the balance of the immune system [31,32]. It is predominantly located on the cell membrane of adipose cells, as well as on the outer membrane of natural killer (NK) cells, macrophages, and monocytes [33]. Encoded by the TLR-4 gene, TLR-4 has a molecular mass of 69 KDa in humans. The full-length TLR-4 protein comprises three parts: an extracellular domain containing 608 amino acids, an intracellular domain containing 187 amino acids, and a transmembrane domain [34]. The extracellular domain is further divided into three sections based on its amino acid sequence: an N-terminal domain, a central domain, and a C-terminal domain [35].
TLR-4 plays an essential role in regulating the response to tissue damage and inflammation, which is crucial to maintaining tissue homeostasis [36]. For instance, TLR-4 binds to ligands such as a cluster of differentiation 14 (CD14) or myeloid differentiation protein 2 (MD2) to activate downstream signaling pathways that regulate inflammatory responses via two distinct pathways (myD88-dependent and myD88-independent pathways) [37]. These inflammatory signals link appetite with the mesolimbic dopamine (DA) system and the hypothalamus, significantly affecting appetite [38]. The link between chronic inflammation and cancer development was first discovered in the 19th century [39,40]. Until recently, it was thought that TLR-4-induced inflammation might have two distinct effects on tumor therapy [41]. While chronic inflammation promotes tumor cell growth, inducing acute inflammation can effectively kill tumor cells [41]. By inducing TLR-4 dimerization with small-molecule agonists, downstream signaling pathways can be activated to generate a strong pro-inflammatory response to kill tumor cells, which may have important implications in anti-tumor immunity [41]. Thus, TLR-4 agonists have been widely studied as potential agents for cancer immunotherapy.
2.3. HLA-G
HLA-G is a non-classical major histocompatibility complex-I (MHC-I) molecule that plays a crucial role in fetal and maternal immune tolerance [42]. While primarily expressed in the placental trophoblast, it is also expressed in various cancer cells, suggesting its involvement in regulating cancer development [43,44,45,46]. The gene responsible for HLA-G has eight exons and seven introns, with the extracellular domains being encoded by exons 2, 3, and 4; the transmembrane and cytoplasmic domains being encoded by exons 5 and 6; and exon 1 encoding signal peptides [47]. HLA-G has a molecular mass of 37–39 kDa and consists of seven isomers (HLA-G1 to -G7) that differ in the heavy-chain α1, α2, and α3 domains of the extracellular domain; β2 microglobulin; and transmembrane and cytoplasmic domains [43]. HLA-G1 and HLA-G5, both of which contain complete α1-α2-α3 domains, peptides, and β2m in their extracellular domains, are the most highly expressed members among all isomers [48,49,50]. Other isomers, such as HLA-G2, HLA-G3, and HLA-G4, lack one or both α-domains and are expressed in a membrane-anchored form containing a single α1 domain, α1-α3 domains, and α1-α2 domains, respectively. The remaining isomers are expressed in a non-membrane-anchored form [48,49,50].
HLA-G has been well-studied due to its role in maternal and infant immune tolerance [51,52]. During pregnancy, HLA-G mediates immune tolerance and supports fetal growth by binding to immune cells and exerting its immunosuppressive effects [53]. As an immunosuppressive molecule, HLA-G regulates immune cell activity by inhibiting immune cell maturation and cytotoxicity, inducing immune cell apoptosis, and activating downstream signaling pathways [54]. HLA-G binds to inhibitory receptors located on T cells, resulting in changes in T-cell function and the inhibition of T-cell maturation and proliferation, as well as changes in the cellular activity of CD8+T cells and CD4+T cells [55]. HLA-G also has an inhibitory effect on DCs maturation and differentiation when binding to Ig-like transcript 4 (ILT4), a ligand expressed on DCs, inhibiting antigen presentation and impeding signal transmission between other immune cells and DCs [56,57].
2.4. NKG2A
Killer-cell immunoglobulin-like receptors (KIRs) can be classified as stimulatory or inhibitory receptors based on their function [58]. NKG2A, an inhibitory receptor in the KIR family, is commonly expressed in NK cells and CD8+T cells [59,60]. Located in the NK complex, the NKG2A gene is composed of seven exons [61]. Recombinant human NKG2A, made up of 159 amino acids, has a molecular mass of 26 KDa and comprises three components: an extracellular lectin-like domain, a transmembrane domain, and an intracellular domain [62,63]. The intracellular domain of NKG2A contains two immunoreceptor tyrosine-based inhibitory motifs (ITIMs), which are associated with Src homology region 2 domain-containing phosphatase-1 (SHP-1) and can bind to ligands to cause ITIM tyrosine phosphorylation [64]. The SHP-1 tyrosine phosphatase is then recruited intracellularly, and tyrosine residues on the activated cascade signaling molecules are dephosphorylated, thus transmitting the inhibitory signal [65].
NKG2A plays a critical role in regulating immune cell function and is commonly expressed in NK cells in human peripheral blood [66]. After heterodimerization with CD94, NKG2A can bind to the human leukocyte antigen E (HLA-E) ligand to activate downstream signaling pathways, thereby inhibiting NK-cell function; when the binding of NKG2A to HLA-E is blocked, NK cells can resume cytotoxic activity [67,68,69]. Additionally, NKG2A is expressed in some T cells, such as CD8+T cells, but unlike its expression in NK cells, it is only expressed in CD8+T cells in patients [70,71]. For example, the expression level of NKG2A is increased in cancer patients and patients with chronic viral infections, indirectly indicating the potential of NKG2A as a cancer therapeutic target [70,71]. NKG2A is highly expressed in lymphocytes in different tumor microenvironments [72,73]. For instance, NKG2A is overexpressed in human cervical cancer cells, and this mechanism is dependent on interleukin -15 (IL-15) to upregulate its expression in CD8+T cells, thus inhibiting the cytotoxicity of lymphocytes and rendering them ineffective in killing tumors [74]. Blocking NKG2A-mediated signaling pathways can improve NK-cell dysfunction. Monalizumab, a novel anti-NKG2A antibody drug, has therapeutic effects on chronic lymphocytic leukemia (CLL) when used [75].
3. Binding Characteristics and Function of Transmembrane Protein-Receptor Dimers
Immune checkpoint proteins perform various physiological functions by interacting with receptors. Understanding the dimer structures and functions of these proteins, as well as the ways in which they interact with ligands, is important in the development of small-molecule drugs that regulate immune checkpoint protein dimerization.
3.1. PD-L1 Dimerization
Zak et al. reported the crystal structure diffraction data of the PD-L1 dimer (Figure 2a) [76]. The human PD-L1 dimer consists of two asymmetric PD-L1 molecules with a rotation angle of 30° between them, and the force between the two PD-L1 molecules is not strong, with a contact surface of 814.8 Å [77]. Both PD-1 and PD-L1 have an extracellular IgV-like domain through which they interact and activate downstream signaling pathways, ultimately inhibiting immune T-cell activity [78]. When PD-L1 dimerization occurs, the inhibitory signal of the downstream pathway is blocked, and the inhibition of T cells is removed [79].
PD-1 is an inhibitory receptor on T cells and a member of the CD28 family. It is generally expressed in regulatory T cells (Tregs, CD4+, and Foxp3+) and NK cells. PD-1 is also expressed in other immune cells, such as macrophages and B cells [80]. The full-length PD-1 protein consists of three parts containing a total of 268 amino acids: a cytoplasmic part (94 amino acids), a transmembrane structure (27 amino acids), and an extracellular part (147 amino acids) [81]. The cytoplasmic tail of PD-1 contains an immunoreceptor tyrosine-based switch motif (ITSM) and an ITIM [82]. After the interaction between PD-L1 and the extracellular IgV-like domain of PD-1, the tyrosine residues of these two domains are phosphorylated [81]. The activation of the downstream signaling pathway leads to the activation of SHP-1/-2 and the dephosphorylation of CD28. Various co-inhibitory receptors on T cells inhibit signal transduction, thereby inducing apoptosis and inhibiting cytokine secretion and cell differentiation [82,83,84,85].
A cluster of differentiation 80 (CD80) belongs to the transmembrane protein family, and full-length CD80 is composed of 254 amino acids [86]. Similar to PD-L1, CD80 also has an extracellular IgV-like domain, enabling it to bind to the IgV-like domain of PD-L1, thereby blocking the interaction between PD-1 and PD-L1 [86]. The interaction between CD80 and CTLA-4 can produce signals that inhibit the activity of T cells, and the heterodimerization of PD-L1/CD80 can effectively reduce the affinity of CTLA-4/CD80 interaction. Therefore, inducing the heterodimerization of PD-L1 and CD80 is an effective method to activate T cells [87]. Additionally, both CD80 and PD-1 act by binding to the IgV-like domain of PD-L1, so the heterodimerization of CD80/PD-L1 can effectively reduce the binding affinity between PD-L1 and PD-1, thereby removing the inhibitory effect on T cells [88].

3.2. TLR-4 Dimerization
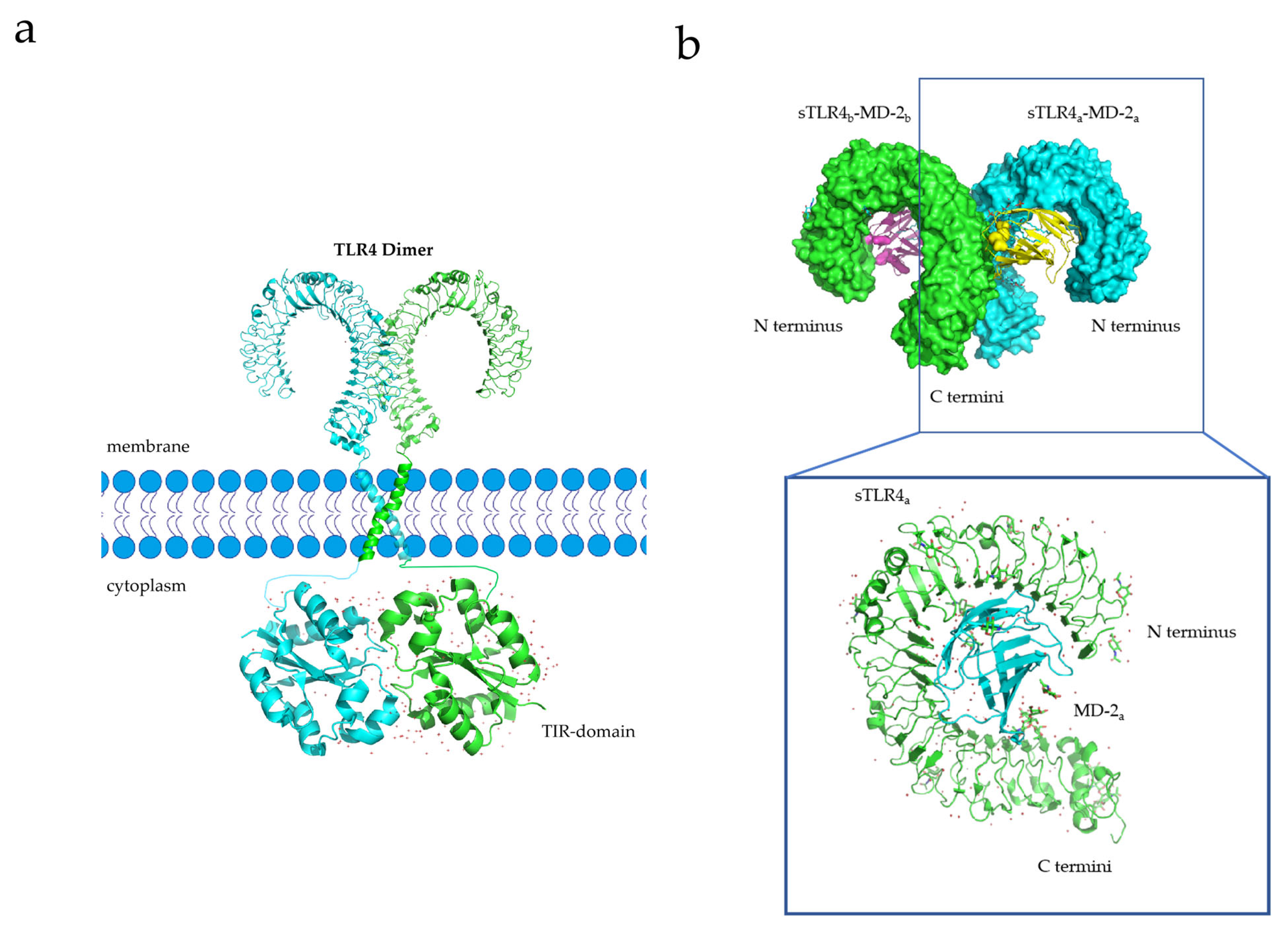
X-ray crystallography and experimental studies have demonstrated that TLRs are capable of forming homodimer signaling complexes, and small-molecule agonists can induce the homodimerization of TLR-4 to initiate downstream signaling pathways (Figure 3a) [90,91]. For example, Vladimir et al. designed a library of cell-permeating decoy peptides, each of which represents a nonfragmented patch of the TLR4 TIR surface [91]. They tested these peptides for the ability to inhibit early cytokine mRNA expression and mitogen-activated protein kinase (MAPK) activation in lipopolysaccharide (LPS)-stimulated primary murine macrophages. Five peptides-4R1, 4R3, 4BB, 4R9, and 4aE-potently inhibited all manifestations of TLR4, but not TLR2 signaling. These findings suggest that the area between the BB loop of TLR4 and its fifth helical region mediates TLR4 TIR dimerization [91]. The transmembrane domain of TLR4 plays a key role in its dimerization process, and the sequences that constitute the transmembrane domain, as well as their secondary and tertiary structures formed by the insertion into the membrane, affect dimerization [92]. For instance, 25% of the amino acids in the TLR-4 transmembrane domain (632TIIGVSVLSVLVVSVVAVLVY652) constitute polar residues. The 637SxxS640 motif (x = any amino acid) among these polar residues is believed to contribute to the stability of homologous dimer complexes [93]. Once dimerization occurs, TLR-4 activates various downstream signaling pathways, inducing inflammatory responses, and activating monocytes (macrophages and DCs) and neutrophils, thereby enhancing the immune response and promoting tumor killing [41,94,95].
Lipopolysaccharide (LPS), an immune system agonist, activates monocytes and neutrophils via the TLR-4 signaling pathway, ultimately activating the body’s innate immune system [41]. The process whereby LPS activates the TLR-4 signaling pathway is as follows: First, LPS forms a complex with its binding protein, LBP (LPS-binding protein). This complex then binds to a cluster of differentiation 14 (CD14) and transfers LPS to myeloid differential protein-2 (MD2) [96]. Upon interaction with LPS, the structure of MD2 changes, and the TLR-4 receptor binds to the MD2/TLR-4 complex to form a homodimer (Figure 3b) [96]. Activated TLR-4-MD2-LPS homodimer complexes perform downstream signal transduction to initiate innate immune responses [34].
High Mobility Group Box 1 (HMGB1) is typically expressed in the nucleus, where it can enhance transcription after binding to DNA [97,98,99,100]. HMGB1 is also released into the cytoplasm during apoptosis, injury, or death, and it is secreted in cancer cells and immune cells as a response mechanism to external stimuli [97,98,99,100]. The full-length HMGB1 protein contains 215 amino acids, and its structure is highly conserved [101]. It is mainly composed of three parts: the first part, called Box A, comprises amino acid residues 9–79; the second part, Box B, contains amino acid residues 95–163; and the last part is a C-terminal tail containing amino acid residues 186–215 [101]. The B box has been identified as a functional domain recognized by TLR-4, and amino acid residues 89–108 are the site of interaction with TLR-4 [101]. Upon interaction with TLR-4, downstream signaling pathways can be activated to regulate the inflammatory response and innate immunity [102,103]. For example, TLR-4-deficient animals are protected from ischemia–reperfusion injury in the liver, kidney, and heart, indicating that TLR-4 plays a critical role in aseptic inflammation [104].
Fibronectin (FN), typically expressed in the extracellular matrix, is a protein with multiple domains that is overexpressed in many types of cancer cells and plays a crucial role in tumor growth and metastasis [105,106]. FN molecules contain two distinct chains with molecular weights of 220 and 250 kDa, which are connected at their C ends by two disulfide bonds to form FN molecules [107,108]. The two chains can be divided into three parts according to the amino acid sequence: first, there are 12 modules composed of 40 amino acid residues; then, 2 modules contain 50 amino acid residues; finally, there are 15 to 17 modules made up of 90-to-100 amino acid residues [107,108]. It is worth mentioning that FNIII EDA (extra domains A) and FNIII EDB are the key structures for FN to play its physiological functions, and FNIII EDA is the site where FN interacts with TLR-4. After FN combines with TLR-4, it can activate the downstream (MyD88-dependent) signaling pathway and, finally, activate the innate immune system of the body [109].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.3. HLA-G Dimerization
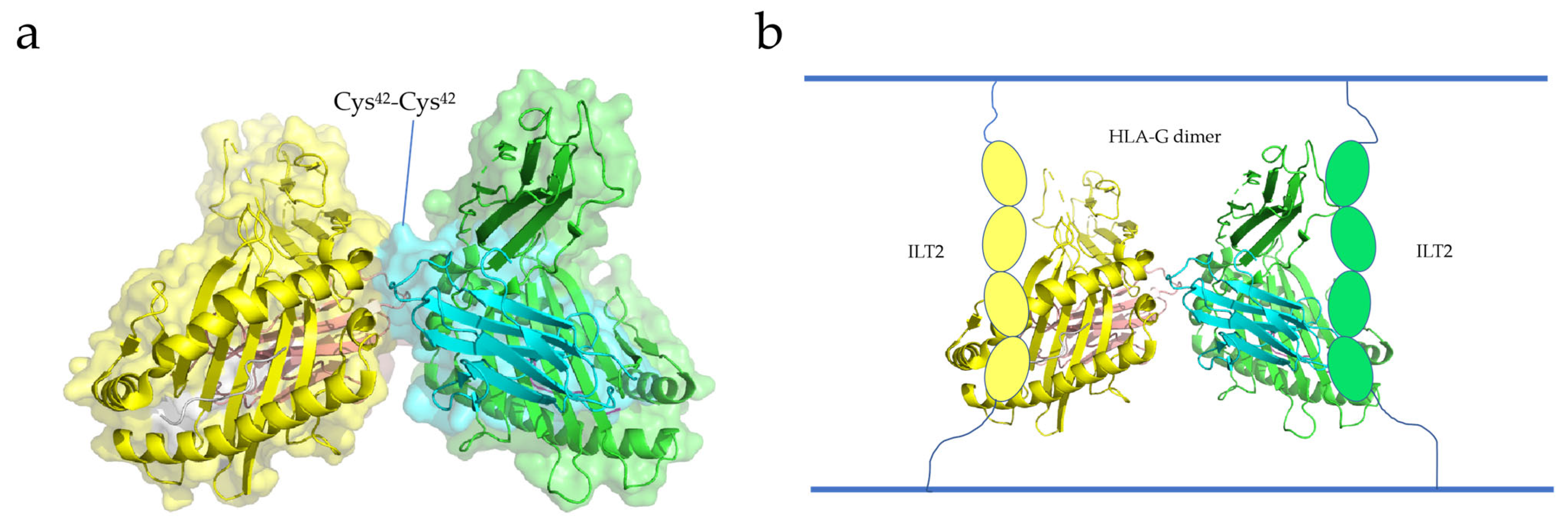
Boyson and Shiroishi et al. reported the crystal structure of the HLA-G dimer, showing that the soluble form of HLA-G can form dimers with intermolecular Cys42-Cys42 disulfide bonds (Figure 4a) [111,112]. The Cys42 residue, located in the center of the α1 helical structure, contributes to the stability of the HLA-G dimer by interacting with the Cys42 residue of the other HLA-G protein [111]. The two exposed binding sites above the HLA-G dimer bind to CD8 and LILRB-1/-2, respectively. Shiroishi et al. proposed a complex model of HLA-G binding to its receptor, ILT2 (HLA-G dimer: receptor = 1:2) (Figure 4b). These findings suggest that the stability of the HLA-G dimer is dependent on its structural orientation [111]. Interestingly, HLA-G dimers are associated with T-cell activation and disease occurrence [113]. Therefore, HLA-G dimerization has great potential in cancer immunotherapy and may become a new target for cancer therapy.
Ig-like transcripts (ILTs) are members of the immunomodulatory receptor family and are commonly expressed on various immune cells [114]. Ig-like transcripts 4 (ILT4) and 2 (ILT2), which belong to the immunoglobulin-like receptor (LILR) family, are expressed on leukocytes [115]. ILT2 is frequently expressed on dendritic cells, T cells, and NK cells, while ILT4 is mainly expressed on DCs and monocytes [115]. The extracellular domains of ILT2 and ILT4 consist of four parts (immunoglobulin domains D1–D4), as well as a cytoplasmic tail and transmembrane domain [116]. The binding sites of HLA-G are the D1 and D2 domains located outside the cell (Figure 4b) [117,118,119]. The binding of ILT to HLA-G can inhibit the cytokine secretion, cell differentiation, and cell proliferation of immune cells, and induce cytotoxicity and apoptosis [115]. For example, after ILT4 binds to HLA-G, it can recruit SHP-1/-2 and activate the nuclear factor-κ-gene binding (NF-κB) pathway, leading to the increase in Interleukin-6 (IL-6) level and then the activation of the STAT3 pathway, which affects the function and maturation of DCs and leads to impaired innate immunity [120].
KIR2DL4 is a member of the killer-cell immunoglobulin-like receptor (KIR) and is commonly expressed on killer cells; it consists of two domains outside the cell, a positively charged transmembrane arginine residue, and an intracellular ITIM tail [121]. KIR2DL4 contains two distinct signaling domains (activation and inhibition), so Attia et al. hypothesized that it has both functions [57]. To demonstrate this, they created a chimeric receptor with both activation and inhibition domains, and the results showed that both KIR2DL4 signaling domains were active, suggesting that under different conditions, KIR2DL4 can inhibit or activate the activity of NK cells [57]. The interaction between KIR2DL4 and HLA-G can activate the downstream signaling pathway, thus regulating the activity of NK cells [122]. The structure of KIR2DL4 is unique, and its role in cancer will likely be the focus of future research.
3.4. NKG2A Dimerization
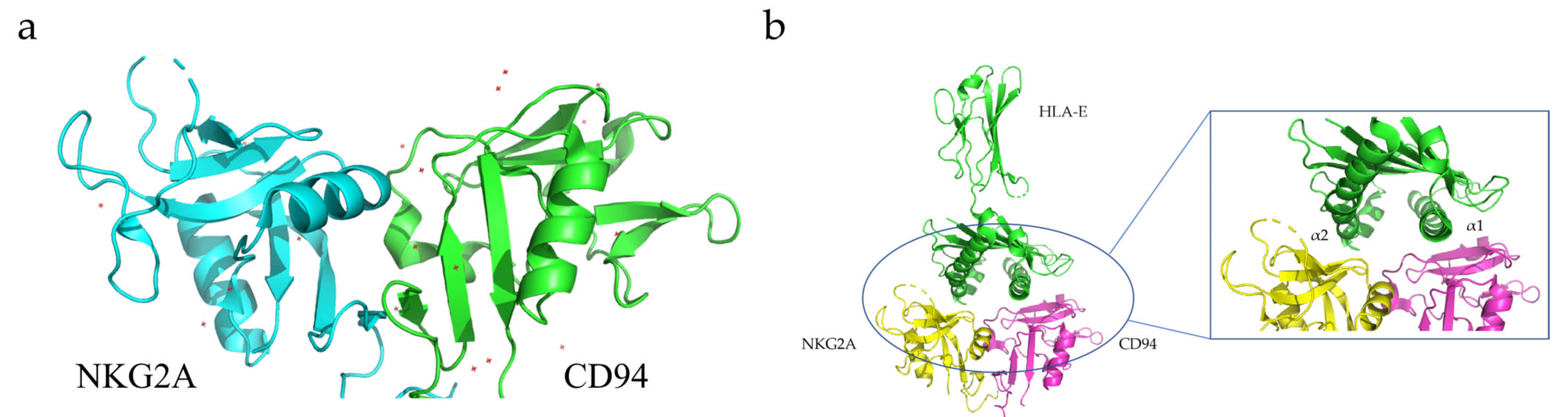
CD94, also known as Kp43, is a transmembrane protein expressed on the surface of most freshly isolated natural killer (NK) cells, as well as some subpopulations of γδT and αβT cells, although at different levels [123,124]. CD94 belongs to the Type II transmembrane protein family, and its carbohydrate recognition domain (CRD) is located outside the cell and contains two glycosylation sites [123,124]. The intracellular domain consists of only seven amino acids. CD94 can form heterodimers with NKG2A to perform its physiological functions [123,124]. The NKG2A/CD94 heterodimer can bind to the HLA-E expressed on immune T and NK cells to inhibit their activity and function [125]. Brett K. Kaiser and co-workers determined the crystal structure of the HLAE/NKG2A/CD94 protein complex using X-ray crystallography (Figure 5) [126].
HLA-E is a major histocompatibility complex (MHC) Class I molecule highly expressed in many solid tumor cells [127]. Generally, HLA-E forms trimers on the outer membrane of the cell, consisting of an α-heavy chain with a molecular weight of 45 kDa and a light chain (beta-2 microglobulin) encoded by chromosome 15 [128,129]. The binding of HLA-E to ligands has high specificity and only peptides with specific amino acid sequences can bind to HLA-E and be stably expressed [128,129]. For example, HLA-E can specifically bind to NKG2A/CD94 receptors on CD8+ T and NK cells and activate downstream signaling pathways that lead to the inhibition of target-cell function and activity [128,129]. When formed in a complex with CD94/NKG2A, HLA-E can generate inhibitory signals, reduce the secretion of cytokines, and directly inhibit the killing effect of immune cells on cancer cells. This signaling pathway is often used by tumor cells to evade immune-cell-mediated killing [75].
4. Application of Regulation of TMEM Dimerization in Anti-Tumor Immunity
Several TMEMs play important physiological roles as dimers. Among them, immune checkpoint proteins are crucial to tumor immunotherapy, and small-molecule drugs that regulate their dimerization have been extensively studied.
PD-L1, which exists in a monomeric form, interacts with PD-1 to produce signals that suppress immune cells, protecting cancer cells from being killed by the immune system [77]. Therefore, inhibiting the interaction between PD-L1 and PD-1 using small-molecule inhibitors is a promising direction in cancer immunotherapy. Two major classes of synthetic small-molecule inhibitors have been reported to date, one that directly blocks the PD-L1 binding site to PD-1 and another that induces PD-L1 dimerization, thus blocking the PD-1 binding site [130]. The drug-induced formation of PD-L1 homodimers can prevent PD-L1/PD-1 binding, thereby blocking downstream signaling pathways [77]. For example, BMS company has reported that the compound with (2-methyl-3-biphenyl) methanol as a scaffold can effectively induce the dimerization of PD-L1 and interact with the hydrophobic tunnel formed by two PD-L1 molecules (Figure 2b) [76,131]. These compounds can form hydrogen bonds with the amino acid residues (such as Ala121 or Tyr123) of PD-L1 (Figure 2c), making the formed PD-L1 dimer more stable and thus preventing the binding of PD-L1 to its ligands (such as PD-1) [132]. (S)-BMS-200, a compound with core scaffolds of 2,3-dihydro-1,4-benzodioxinyl, can stably bind to the PD-L1 homodimer and induce remarkable conformational changes in the key residues on the dimer, thus accelerating compact interactions [133]. Moreover, intermolecular interaction studies revealed that compound N-[2-(aminocarbonyl) phenyl] [1,1’-biphenyl]-4-carboxamide (APBC) can bind to the hydrophobic pocket formed by two PD-L1 monomers, especially anchoring residues Y56, M115, and A121. APBC forms a key hydrogen bond with critical residue D122, thereby stabilizing the structure of the dimer. APBC can bind and block the binding interface of PD-L1, thus blocking the binding of PD-1 [134]. In a mouse tumor model with high PD-L1 expression, the administration of APBC resulted in enhanced infiltration of CD8+T cells and increased cytokine levels in the tumor microenvironment [134]. Compared with the control group, the tumor growth and survival rate of mice in the APBC group were significantly improved, and the tumor growth inhibition rate reached 62.1% after administering 10 mg/kg APBC [134]. In conclusion, small-molecule drugs that induce PD-L1 dimerization generally bind to PD-L1 and induce its dimerization, thus blocking the binding of PD-1 to PD-L1 and achieving the objective of immunotherapy. Therefore, these small-molecule drugs may become potential candidates for new anti-cancer drugs.
The dimerization of TLR-4 is crucial to regulating both innate and adaptive immune systems. Immunomax®, a TLR-4 agonist, binds to TLR-4 and promotes TLR-4 homodimerization, which in turn triggers the formation of intracellular signal transduction complexes and activates downstream signaling pathways [135,136]. In a 4T1 breast cancer mouse model, Immunomax® significantly improved survival; inhibited tumor growth and metastasis; increased the percentage of NK cells, CD4+T cells, and CD8+T cells in the mouse spleen; and significantly reduced the percentage of bone marrow-derived suppressor cells (MDSCs), making it a potential anti-cancer drug candidate [136]. Huang et al. established a sarcoma-containing C57BL/10J mouse model to validate the anti-cancer effects of cationic polymers such as cationic dextran (C-dextran) and polyethylenimine (PEI) [137]. These cationic polymers activated the TLR-4 signaling pathway, resulting in TLR-4 dimerization, acute inflammation, and direct tumor cell killing, thereby prolonging the survival time of mice, and inhibiting tumor growth and metastasis [137]. Park et al. found that rhamnogalacturonan II (RG-II) could activate the downstream signaling pathway of TLR-4. Further studies showed that RG-II could effectively induce the dimerization of TLR-4, activate MyD88-independent and MyD88-dependent pathways, promote the maturation and differentiation of DCs, and produce a series of cytokines to regulate inflammatory responses [9]. The effect of RG-II on mice carrying lymphoma C57BL6 was further verified, and the results showed that the activity of CD8+T cells was significantly enhanced after the administration of RG-II and that the tumor growth and metastasis of mice were effectively alleviated [9]. In conclusion, inducing TLR-4 dimerization is an emerging cancer immunotherapy approach. Therefore, TLR-4 agonists have been widely studied by the scientific community, and investigations are still in progress.
HLA-G is typically expressed on specific immune cells or as a soluble dimer (sHLA-G) in the blood [138]. Upon the formation of sHLA-G dimers, the activity of HLA-G molecules is increased, enhancing their binding affinity for ligand ILT-2/ILT-4, which is expressed by tumor-associated white blood cells [138,139]. The binding of sHLA-G dimers to their ligand activates the downstream signaling pathway, leading to the inhibition of DC function and maturation and weakened activity of NK cells and CD8+T cells, ultimately inhibiting the immune response [138,139]. In a study by Nathalie et al., HLA-G dimers were identified in Fon+ cell lines from melanoma patients, and the formation of HLA-G dimers inhibited NK-cell activity, protecting Fon+ cells from NK-cell-mediated killing and promoting rapid tumor growth and metastasis, potentially leading to patient death [140]. Singer et al. found different levels of HLA-G5 homodimers in malignant effusions of four ovarian cancer patients, and disease severity was positively correlated with the level of dimers [141,142]. These findings suggest that high levels of sHLA-G dimers are associated with advanced disease and a poor prognosis. Currently, there is limited knowledge regarding the regulation of the HLA-G dimer, which is typically regulated at the gene level [143]. For instance, the 3’-UTR of HLA-G can interact with multiple microRNAs (miR-152, miR-133a, and miR-148a) to reduce HLA-G expression and further downregulate the level of sHLA-G dimers in the blood [143]. In addition to microRNAs, Reches et al. identified an RNA-binding protein, HNRNPR (RBP HNRNPR), that interacts with the 3′-UTR of HLA-G and regulates its expression [144]. In summary, reducing the expression level of the HLA-G protein can reduce the formation of HLA-G dimers, thereby reducing the binding affinity of HLA-G and its receptors, and enhancing the immune response. This class of drugs is currently under development and has great potential for cancer immunotherapy.
NKG2A forms a heterodimer with CD94 in the majority of NK and CD8+T cells; this heterodimer binds to its ligand, HLA-E, to suppress NK- and CD8+T-cell activity. Tumor cells exploit this mechanism to evade the immune response [145,146], making it essential to block the interaction of HLA-E and NKG2A to enhance anti-tumor immune responses [86]. NKG2A and CD94 can only perform their functions after forming heterodimers. For instance, in klrd1-knockout mice, NKG2A could not form a heterodimer with CD94 due to the lack of the gene encoding CD94, resulting in the absence of inhibition of NK-cell maturation and development, as well as no reduction in the number and activity of NK cells [147]. Likewise, in DBA/2J mice with spontaneous mutations in the Klrd1 gene, CD94 expression was prevented, yet normal NK-cell development was maintained following the induction of NKG2A expression, without significant immunosuppression [148]. CD94 was expressed in both CD8+T cells and CD4+T cells, whereas NKG2A was only expressed in CD8+T cells. Thus, inducing CD94/NKG2A expression in mixed-culture lymphocyte populations resulted in impaired CD8+T-cell activity, which was restored upon the addition of anti-CD94 antibodies [149]. These findings suggest that blocking the formation of NKG2A/CD94 heterodimers can prevent downstream signaling pathways, thereby eliminating the inhibitory effect on immune cells [75]. In conclusion, inhibiting the formation of NKG2A/CD94 heterodimers can block the immunosuppressive signal of NKG2A, which has significant implications for the development of anti-tumor drugs.
5. Summary and Outlook
Cancer has long been one of the leading causes of death in humans, and the introduction of immune checkpoint inhibitors has opened new opportunities for cancer therapy. However, a significant proportion of patients do not respond to immune checkpoint inhibitors, and some solid tumors have primary resistance to immune checkpoint inhibitors [150]. Therefore, the search for more effective immunotherapy is urgent. A variety of TMEMs play important roles in cancer immunotherapy, and activating their downstream signaling pathways can regulate the activity of immune cells and play a role in killing tumor cells. The dimerization of TMEMs is often postulated to be the initial step in controlling their downstream signaling pathways [151].
Drugs (e.g., monoclonal antibodies and small-molecule immune checkpoint inhibitors) that regulate the dimerization of several TMEMs are under development, and some of these compounds show good biological activity [77]. A breakthrough has been made in the research of antibody immune checkpoint inhibitors; the U.S. Food and Drug Administration (FDA) has approved seven types of monoclonal antibodies, but these drugs directly act on immune checkpoint proteins and their ligands, which can cause multiple, nonspecific toxic effects and side effects in some patients, which are expensive to treat [152,153]. In contrast, research on small-molecule immune checkpoint inhibitors is still in its infancy. Small-molecule inhibitors have the advantages of weak immune-related adverse reactions, good penetration in tumor cells, and high bioavailability [154,155]. Despite many exciting advantages, the regulation of the dimerization of transmembrane proteins using small-molecule inhibitors still faces several challenges. First, although protein dimerization is thought to act as an on–off switch for cascade signaling, small-molecule drug-regulatory protein dimerization is not yet universally applicable, and most current research targets are immune checkpoint proteins. Secondly, current research on these small-molecule drugs is still in the initial stage, and the mechanism and characteristics of the drugs have not been fully understood, so the development cost is high; the yield is low; and the time span is long. Third, because these small-molecule inhibitors inhibit signaling pathways through indirect action, their effects are generally weaker than those of antibodies, and there are challenges to increasing their efficacy to antibody levels.
Indeed, cancer immune escape and immune tolerance might overpower drug monotherapy; then, based on biomarker analysis, targeted combinational treatments might be needed to achieve a meaningful immune response. Several promising novel immunomodulatory therapies and their optimal combinations are currently under clinical investigation. However, severe immune-related side effects may be the result of disinhibiting the brakes of the immune system, and their management must be contemplated. In conclusion, the patient-specific configuration of immune-system–cancer-cell interactions and the specific immune escape mechanism will need to be understood to guide personalized treatment options for cancer immunotherapy.
Author Contributions
L.L. conceived and designed the review; J.L. contributed new ideas; L.L. and J.L. wrote the draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Key R&D Program of China (2020YFA0210800), National Natural Science Foundation of China (Nos. 21974022 and 22027805), Major Project of Science and Technology of Fujian Province (2020HZ06006).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
There are no conflicts of interest to declare. The funders had no role in the design or implementation of the research.
References
- Smith, S.M. Strategies for the purification of membrane proteins. In Protein Chromatography; Springer: Berlin/Heidelberg, Germany, 2011; pp. 485–496. [Google Scholar] [CrossRef] [Green Version]
- Schmit, K.; Michiels, C. TMEM proteins in cancer: A review. Front. Pharmacol. 2018, 9, 1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinothkumar, K.R.; Henderson, R. Structures of membrane proteins. Q. Rev. Biophys. 2010, 43, 65–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Liang, D.; Liu, J.; Zeng, J.; Zeng, Y. The Breakthroughs in Cancer Immune Checkpoint Based Therapy: A Review of Development in Immune Checkpoint Study and its Application. Comb. Chem. High Throughput Screen. 2017, 20, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhang, C.; Jin, S.; Gao, Z.; Cao, J.; Wang, A.; Li, D.; Wang, Q.; Sun, X.; Bai, D. Progress of immune checkpoint therapy in the clinic (Review). Oncol. Rep. 2019, 41, 3–14. [Google Scholar] [CrossRef]
- Litak, J.; Grochowski, C.; Litak, J.; Osuchowska, I.; Gosik, K.; Radzikowska, E.; Kamieniak, P.; Rolinski, J. TLR-4 Signaling vs. Immune Checkpoints, miRNAs Molecules, Cancer Stem Cells, and Wingless-Signaling Interplay in Glioblastoma Multiforme-Future Perspectives. Int. J. Mol. Sci. 2020, 21, 3114. [Google Scholar] [CrossRef]
- Ying, H.; Zhang, X.; Duan, Y.; Lao, M.; Xu, J.; Yang, H.; Liang, T.; Bai, X. Non-cytomembrane PD-L1: An atypical target for cancer. Pharmacol. Res. 2021, 170, 105741. [Google Scholar] [CrossRef]
- Sugiura, D.; Maruhashi, T.; Okazaki, I.M.; Shimizu, K.; Maeda, T.K.; Takemoto, T.; Okazaki, T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 2019, 364, 558–566. [Google Scholar] [CrossRef]
- Park, S.N.; Noh, K.T.; Jeong, Y.I.; Jung, I.D.; Kang, H.K.; Cha, G.S.; Lee, S.J.; Seo, J.K.; Kang, D.H.; Hwang, T.H.; et al. Rhamnogalacturonan II is a Toll-like receptor 4 agonist that inhibits tumor growth by activating dendritic cell-mediated CD8+ T cells. Exp. Mol. Med. 2013, 45, e8. [Google Scholar] [CrossRef]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Godal, R.; Bachanova, V.; Gleason, M.; McCullar, V.; Yun, G.H.; Cooley, S.; Verneris, M.R.; McGlave, P.B.; Miller, J.S. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin-like receptor-negative natural killer cells after NKG2A and LIR-1 blockade. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2010, 16, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Biassoni, R.; Mingari, M.C.; Moretta, L. Receptors for HLA class-I molecules in human natural killer cells. Annu. Rev. Immunol. 1996, 14, 619–648. [Google Scholar] [CrossRef] [PubMed]
- Moretta, L.; Mingari, M.C.; Pende, D.; Bottino, C.; Biassoni, R.; Moretta, A. The molecular basis of natural killer (NK) cell recognition and function. J. Clin. Immunol. 1996, 16, 243–253. [Google Scholar] [CrossRef]
- Wagtmann, N.; Rajagopalan, S.; Winter, C.C.; Peruzzi, M.; Long, E.O. Killer cell inhibitory receptors specific for HLA-C and HLA-B identified by direct binding and by functional transfer. Immunity 1995, 3, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Clements, C.S.; Kjer-Nielsen, L.; McCluskey, J.; Rossjohn, J. Structural studies on HLA-G: Implications for ligand and receptor binding. Hum. Immunol. 2007, 68, 220–226. [Google Scholar] [CrossRef]
- Hò, G.T.; Celik, A.A.; Huyton, T.; Hiemisch, W.; Blasczyk, R.; Simper, G.S.; Bade-Doeding, C. NKG2A/CD94 Is a New Immune Receptor for HLA-G and Distinguishes Amino Acid Differences in the HLA-G Heavy Chain. Int. J. Mol. Sci. 2020, 21, 4362. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiong, H.; Ning, Z. Implications of NKG2A in immunity and immune-mediated diseases. Front. Immunol. 2022, 13, 960852. [Google Scholar] [CrossRef]
- Brown, J.A.; Dorfman, D.M.; Ma, F.R.; Sullivan, E.L.; Munoz, O.; Wood, C.R.; Greenfield, E.A.; Freeman, G.J. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J. Immunol. 2003, 170, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Honjo, T. PD-1: An inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 2001, 22, 265–268. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Lin, D.Y.-W.; Tanaka, Y.; Iwasaki, M.; Gittis, A.G.; Su, H.-P.; Mikami, B.; Okazaki, T.; Honjo, T.; Minato, N.; Garboczi, D.N. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 3011–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, D.; Zhou, S.; Liu, X.; Zhao, C.; Liu, H.; Yao, X. Understanding the structural and energetic basis of PD-1 and monoclonal antibodies bound to PD-L1: A molecular modeling perspective. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A.J.S. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.; Simonetta, F.; Baker, J.; Morrison, A.R.; Wenokur, A.S.; Pierini, A.; Berraondo, P.; Negrin, R.S. Indirect impact of PD-1/PD-L1 blockade on a murine model of NK cell exhaustion. Front. Immunol. 2020, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M.J.N.R.I. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Swaika, A.; Hammond, W.A.; Joseph, R.W. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol. Immunol. 2015, 67, 4–17. [Google Scholar] [CrossRef]
- Xing, Y.; Hogquist, K. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Prim. 2020, 6, 38. [Google Scholar] [CrossRef]
- Sibaud, V. Dermatologic reactions to immune checkpoint inhibitors. Am. J. Clin. Dermatol. 2018, 19, 345–361. [Google Scholar] [CrossRef]
- Hua, Z.; Hou, B. TLR signaling in B-cell development and activation. Cell. Mol. Immunol. 2013, 10, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.-J.; Gong, H.-F.; Zhao, Q.-Q.; Liu, X.-S.; Liu, C.; Wang, H.J.T.L. Critical role of toll-like receptor 4 (TLR4) in dextran sulfate sodium (DSS)-Induced intestinal injury and repair. Toxicol. Lett. 2019, 315, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Pahwa, R.; Devaraj, S.; Jialal, I. The effect of the accessory proteins, soluble CD14 and lipopolysaccharide-binding protein on Toll-like receptor 4 activity in human monocytes and adipocytes. Int. J. Obes. 2016, 40, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Pathak, C. Human Toll-Like Receptor 4 (hTLR4): Structural and functional dynamics in cancer. Int. J. Biol. Macromol. 2019, 122, 425–451. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Park, B.S.; Kim, J.-I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.J.C. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Zhang, L.; Li, Z.; Zhang, Q. The roles of toll-like receptors in carcinogenesis and cancer immunotherapy. Chin.-Ger. J. Clin. Oncol. 2010, 9, 118–120. [Google Scholar] [CrossRef]
- Yirmiya, R.; Goshen, I.J.B. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.J.N. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Kluwe, J.; Mencin, A.; Schwabe, R.F. Toll-like receptors, wound healing, and carcinogenesis. J. Mol. Med. 2009, 87, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, K.; Selmani, Z.; Ishii, H.; Yamaguchi, K. Innate immunity and cancer therapy. Int. Immunopharmacol. 2011, 11, 350–357. [Google Scholar] [CrossRef]
- Shetab Boushehri, M.A.; Lamprecht, A. TLR4-based immunotherapeutics in cancer: A review of the achievements and shortcomings. Mol. Pharm. 2018, 15, 4777–4800. [Google Scholar] [CrossRef]
- Foroni, I.; Couto, A.R.; Bettencourt, B.F.; Santos, M.; Lima, M.; Bruges-Armas, J.J.H.; Diseases, A.I. HLA-E, HLA-F and HLA-G—The non-classical side of the MHC cluster. In HLA and Associated Important Diseases; IntechOpen: London, UK, 2014; Volume 3, pp. 61–109. [Google Scholar] [CrossRef] [Green Version]
- Carosella, E.D.; Rouas-Freiss, N.; Tronik-Le Roux, D.; Moreau, P.; LeMaoult, J. HLA-G: An immune checkpoint molecule. Adv. Immunol. 2015, 127, 33–144. [Google Scholar] [CrossRef]
- de Kruijf, E.M.; Sajet, A.; van Nes, J.G.; Natanov, R.; Putter, H.; Smit, V.T.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.; Kuppen, P.J.K. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J. Immunol. 2010, 185, 7452–7459. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Wang, P.; Dai, P.; Jin, B.; Tong, Y.; Lin, H.; Shi, G. Correlation between human leukocyte antigen-G expression and clinical parameters in oral squamous cell carcinoma. Indian J. Cancer 2018, 55, 340. [Google Scholar] [CrossRef] [PubMed]
- Zeestraten, E.; Reimers, M.; Saadatmand, S.; Dekker, J.T.; Liefers, G.; Van Den Elsen, P.; Van De Velde, C.; Kuppen, P.J.K. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br. J. Cancer 2014, 110, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Arnaiz-Villena, A.; Juarez, I.; Suarez-Trujillo, F.; López-Nares, A.; Vaquero, C.; Palacio-Gruber, J.; Martin-Villa, J.M. HLA-G: Function, polymorphisms and pathology. Int. J. Immunogenet. 2021, 48, 172–192. [Google Scholar] [CrossRef] [PubMed]
- Diehl, M.; Münz, C.; Keilholz, W.; Stevanović, S.; Holmes, N.; Loke, Y.W.; Rammensee, H.-G.J.C.B. Nonclassical HLA-G molecules are classical peptide presenters. Curr. Biol. 1996, 6, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Paul, P.; Cabestre, F.A.; Lefebvre, S.; Khalil-Daher, I.; Vazeux, G.; Quiles, R.M.M.; Bermond, F.; Dausset, J.; Carosella, E.D. Identification of HLA-G7 as a new splice variant of the HLA-G mRNA and expression of soluble HLA-G5,-G6, and-G7 transcripts in human transfected cells. Hum. Immunol. 2000, 61, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kuroki, K.; Okabe, Y.; Kasai, Y.; Matsumoto, N.; Yamada, C.; Takai, T.; Ose, T.; Kon, S.; Matsuda, T. The immunosuppressive effect of domain-deleted dimer of HLA-G2 isoform in collagen-induced arthritis mice. Hum. Immunol. 2016, 77, 754–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.-X.; Xie, Y.-M.; Zhao, S.-J.; Liu, C.-Y.; Mor, G.; Liao, A.-H. Human leukocyte antigens: The unique expression in trophoblasts and their crosstalk with local immune cells. Int. J. Biol. Sci. 2022, 18, 4043–4052. [Google Scholar] [CrossRef] [PubMed]
- Moffett, A.; Colucci, F. Co-evolution of NK receptors and HLA ligands in humans is driven by reproduction. Immunol. Rev. 2015, 267, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhou, Y.; Wei, H. Roles of HLA-G in the maternal-fetal immune microenvironment. Front. Immunol. 2020, 11, 592010. [Google Scholar] [CrossRef] [PubMed]
- Saurabh, A.; Chakraborty, S.; Kumar, P.; Mohan, A.; Bhatnagar, A.K.; Rishi, N.; Mitra, D.K.J.T. Inhibiting HLA-G restores IFN-γ and TNF-α producing T cell in pleural tuberculosis. Tuberculosis 2018, 109, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Martín-Villa, J.M.; Vaquero-Yuste, C.; Molina-Alejandre, M.; Juarez, I.; Suárez-Trujillo, F.; López-Nares, A.; Palacio-Gruber, J.; Barrera-Gutiérrez, L.; Fernández-Cruz, E.; Rodríguez-Sainz, C. HLA-G: Too Much or Too Little? Role in Cancer and Autoimmune Disease. Front. Immunol. 2022, 67, 796054. [Google Scholar] [CrossRef] [PubMed]
- Ristich, V.; Liang, S.; Zhang, W.; Wu, J.; Horuzsko, A. Tolerization of dendritic cells by HLA-G. Eur. J. Immunol. 2005, 35, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Attia, J.V.; Dessens, C.E.; van de Water, R.; Houvast, R.D.; Kuppen, P.J.; Krijgsman, D. The molecular and functional characteristics of HLA-G and the interaction with its receptors: Where to intervene for cancer immunotherapy? Int. J. Mol. Sci. 2020, 21, 8678. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Ochoa, M.C.; Cordeiro, L.; Berraondo, P.; de Cerio, A.L.-D.; Cabo, M.; López-Botet, M.; Melero, I. Targeting NK-cell checkpoints for cancer immunotherapy. Curr. Opin. Immunol. 2017, 45, 73–81. [Google Scholar] [CrossRef]
- Li, F.; Wei, H.; Wei, H.; Gao, Y.; Xu, L.; Yin, W.; Sun, R.; Tian, Z. Blocking the Natural Killer Cell Inhibitory Receptor NKG2A Increases Activity of Human Natural Killer Cells and Clears Hepatitis B Virus Infection in Mice. Gastroenterology 2013, 144, 392–401. [Google Scholar] [CrossRef]
- Rapaport, A.S.; Schriewer, J.; Gilfillan, S.; Hembrador, E.; Crump, R.; Plougastel, B.F.; Wang, Y.; Le Friec, G.; Gao, J.; Cella, M.; et al. The Inhibitory Receptor NKG2A Sustains Virus-Specific CD8+ T Cells in Response to a Lethal Poxvirus Infection. Immunity 2015, 43, 1112–1124. [Google Scholar] [CrossRef] [Green Version]
- Plougastel, B.; Jones, T.; Trowsdale, J. Genomic structure, chromosome location, and alternative splicing of the humanNKG2A gene. Immunogenetics 1996, 44, 286–291. [Google Scholar] [CrossRef]
- Iwaszko, M.; Bogunia-Kubik, K. Clinical Significance of the HLA-E and CD94/NKG2 Interaction. Arch. Immunol. et Ther. Exp. 2011, 59, 353–367. [Google Scholar] [CrossRef]
- Mingari, M.C.; Pietra, G.; Moretta, L. Immune Checkpoint Inhibitors: Anti-NKG2A Antibodies on Board. Trends Immunol. 2019, 40, 83–85. [Google Scholar] [CrossRef]
- Tang, L.; Bai, J.; Chung, C.-S.; Lomas-Neira, J.; Chen, Y.; Huang, X.; Ayala, A. Programmed Cell Death Receptor Ligand 1 Modulates the Regulatory T Cells’ Capacity to Repress Shock/Sepsis–Induced Indirect Acute Lung Injury by Recruiting Phosphatase Src Homology Region 2 Domain-Containing Phosphatase 1. Shock 2015, 43, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.-Z.; Ding, K.; Wang, Z.-R.; Ding, C.-H.; Lei, S.-J.; Liu, J.-P.; Yin, C.; Hu, P.-F.; Ding, J.; Chen, W.-S.; et al. SHP-1 Acts as a Tumor Suppressor in Hepatocarcinogenesis and HCC Progression. Cancer Res. 2018, 78, 4680–4691. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, S.; Mace, E.M.; Minard, C.G.; Forbes, L.R.; Vargas-Hernandez, A.; Duryea, T.K.; Makedonas, G.; Banerjee, P.P.; Shearer, W.; Orange, J.S. High-resolution phenotyping identifies NK cell subsets that distinguish healthy children from adults. PLoS ONE 2017, 12, e0181134. [Google Scholar] [CrossRef]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cui, Y.; Luo, G.; Wang, Q.; Hu, J.; He, W.; Yuan, J.; Zhou, J.; Wu, Y.; Sun, X.; et al. Activated mouse CD4+Foxp3− T cells facilitate melanoma metastasis via Qa-1-dependent suppression of NK-cell cytotoxicity. Cell Res. 2012, 22, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, X.-M.; Li, S.-R.; Twelkmeyer, T.; Wang, W.-H.; Zhang, S.-Y.; Wang, S.-F.; Chen, J.-Z.; Jin, X.; Wu, Y.-Z.; et al. NKG2A is a NK cell exhaustion checkpoint for HCV persistence. Nat. Commun. 2019, 10, 1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A–HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef] [PubMed]
- Van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; Van Der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Li, Q.; Cai, S.; Li, M.; Zhou, X.; Wu, G.; Kang, K.; Yuan, J.; Wang, R.; Huyan, T.; Zhang, W. Natural killer cell exhaustion in lung cancer. Int. Immunopharmacol. 2021, 96, 107764. [Google Scholar] [CrossRef]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; André, P.; et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018, 175, 1744–1755.e15. [Google Scholar] [CrossRef] [Green Version]
- Sheu, B.-C.; Chiou, S.-H.; Lin, H.-H.; Chow, S.-N.; Huang, S.-C.; Ho, H.-N.; Hsu, S.-M. Up-regulation of Inhibitory Natural Killer Receptors CD94/NKG2A with Suppressed Intracellular Perforin Expression of Tumor-Infiltrating CD8+ T Lymphocytes in Human Cervical Carcinoma. Cancer Res. 2005, 65, 2921–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, E.M.; Mele, J.M.; Cheney, C.; Timmerman, E.A.; Fiazuddin, F.; Strattan, E.J.; Mo, X.; Byrd, J.C.; Muthusamy, N.; Awan, F.T. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. OncoImmunology 2016, 5, e1226720. [Google Scholar] [CrossRef] [Green Version]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [Green Version]
- Bailly, C.; Vergoten, G. Protein homodimer sequestration with small molecules: Focus on PD-L1. Biochem. Pharmacol. 2020, 174, 113821. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Grudnik, P.; Magiera-Mularz, K.; Dömling, A.; Dubin, G.; Holak, T.A. Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2. Structure 2017, 25, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.; Mills, J.J.; Zhao, B.; Wang, F.; Sun, Q.; Christov, P.P.; Tarr, J.C.; Rietz, T.A.; Olejniczak, E.T.; Lee, T.; et al. Fragment-based screening of programmed death ligand 1 (PD-L1). Bioorganic Med. Chem. Lett. 2019, 29, 786–790. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Schöniger, S.; Jasani, B. The PD-1/PD-L1 Pathway: A Perspective on Comparative Immuno-Oncology. Animals 2022, 12, 2661. [Google Scholar] [CrossRef]
- Wang, Q.; Bardhan, K.; Boussiotis, V.A.; Patsoukis, N. The PD-1 Interactome. Adv. Biol. 2021, 5, e2100758. [Google Scholar] [CrossRef]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.-A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.Y.-C.; Wang, Y.; Jhatakia, A.D.; Deng, A.X.; Bee, C.; Deshpande, S.; Rangan, V.S.; Bezman, N.; Gudmundsson, O.; Chen, G. Higher-Order Structure Characterization of NKG2A/CD94 Protein Complex and Anti-NKG2A Antibody Binding Epitopes by Mass Spectrometry-Based Protein Footprinting Strategies. J. Am. Soc. Mass Spectrom. 2021, 32, 1567–1574. [Google Scholar] [CrossRef]
- Kang-Pettinger, T.; Walker, K.; Brown, R.; Cowan, R.; Wright, H.; Baravalle, R.; Waters, L.C.; Muskett, F.W.; Bowler, M.W.; Sawmynaden, K.; et al. Identification, binding, and structural characterization of single domain anti-PD-L1 antibodies inhibitory of immune regulatory proteins PD-1 and CD80. J. Biol. Chem. 2022, 299, 102769. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, C.K.; Lin, C.-H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.-F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, S.; Cheng, Y.; Zhang, W.; Wang, M.; Sun, H.; Guo, B.; Li, Z.; Xiao, Y.; Jiang, S. Discovery of Small-Molecule Inhibitors of the PD-1/PD-L1 Axis That Promote PD-L1 Internalization and Degradation. J. Med. Chem. 2022, 65, 3879–3893. [Google Scholar] [CrossRef]
- Mineev, K.S.; Goncharuk, S.A.; Goncharuk, M.V.; Volynsky, P.E.; Novikova, E.V.; Aresinev, A.S. Spatial structure of TLR4 transmembrane domain in bicelles provides the insight into the receptor activation mechanism. Sci. Rep. 2017, 7, 6864. [Google Scholar] [CrossRef] [Green Version]
- Toshchakov, V.Y.; Szmacinski, H.; Couture, L.A.; Lakowicz, J.R.; Vogel, S.N. Targeting TLR4 Signaling by TLR4 Toll/IL-1 Receptor Domain-Derived Decoy Peptides: Identification of the TLR4 Toll/IL-1 Receptor Domain Dimerization Interface. J. Immunol. 2011, 186, 4819–4827. [Google Scholar] [CrossRef] [Green Version]
- Reuven, E.M.; Fink, A.; Shai, Y. Regulation of innate immune responses by transmembrane interactions: Lessons from the TLR family. Biochim. et Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1586–1593. [Google Scholar] [CrossRef] [Green Version]
- Kargas, V.; Marzinek, J.K.; Holdbrook, D.A.; Yin, H.; Ford, R.C.; Bond, P.J. A polar SxxS motif drives assembly of the transmembrane domains of Toll-like receptor 4. Biochim. et Biophys. Acta (BBA)-Biomembr. 2017, 1859, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Kashani, B.; Zandi, Z.; Pourbagheri-Sigaroodi, A.; Bashash, D.; Ghaffari, S.H. The role of toll-like receptor 4 (TLR4) in cancer progression: A possible therapeutic target? J. Cell. Physiol. 2020, 236, 4121–4137. [Google Scholar] [CrossRef] [PubMed]
- Krüger, C.L.; Zeuner, M.-T.; Cottrell, G.S.; Widera, D.; Heilemann, M. Quantitative single-molecule imaging of TLR4 reveals ligand-specific receptor dimerization. Sci. Signal. 2017, 10, eaan1308. [Google Scholar] [CrossRef] [Green Version]
- Soares, J.-B.; Pimentel-Nunes, P.; Jr, R.R.-A.; Leite-Moreira, A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol. Int. 2010, 4, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.-J.; Cheng, J.; Feng, X.; Yu, Y.; Tian, L.; Huang, Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget 2017, 8, 64534–64550. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in Cancer: Good, Bad, or Both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef] [Green Version]
- Krysko, O.; Løve Aaes, T.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Cui, X.; Liu, Q. Emerging role of HMGB1 in lung diseases: Friend or foe. J. Cell. Mol. Med. 2016, 21, 1046–1057. [Google Scholar] [CrossRef]
- Lee, C.-H.; Yoon, S.-J.; Lee, S.-M. Chlorogenic Acid Attenuates High Mobility Group Box 1 (HMGB1) and Enhances Host Defense Mechanisms in Murine Sepsis. Mol. Med. 2012, 18, 1437–1448. [Google Scholar] [CrossRef]
- Tadie, J.-M.; Bae, H.-B.; Deshane, J.S.; Bell, C.P.; Lazarowski, E.R.; Chaplin, D.D.; Thannickal, V.J.; Abraham, E.; Zmijewski, J.W. Toll-Like Receptor 4 Engagement Inhibits Adenosine 5′-Monophosphate–Activated Protein Kinase Activation through a High Mobility Group Box 1 Protein–Dependent Mechanism. Mol. Med. 2012, 18, 659–668. [Google Scholar] [CrossRef]
- Poulain, L.; Richard, V.; Lévy, P.; Dematteis, M.; Arnaud, C. Toll-Like Receptor-4 Mediated Inflammation Is Involved in the Cardiometabolic Alterations Induced by Intermittent Hypoxia. Mediat. Inflamm. 2015, 2015, 620258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insua-Rodríguez, J.; Oskarsson, T. The extracellular matrix in breast cancer. Adv. Drug Deliv. Rev. 2016, 97, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Tivari, S.; Lu, H.; Dasgupta, T.; De Lorenzo, M.S.; Wieder, R. Reawakening of dormant estrogen-dependent human breast cancer cells by bone marrow stroma secretory senescence. Cell Commun. Signal. 2018, 16, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [Green Version]
- Wu, C. Roles of integrins in fibronectin matrix assembly. Histol. Histopathol. 1997, 12, 233–240. [Google Scholar] [PubMed]
- Julier, Z.; Martino, M.M.; de Titta, A.; Jeanbart, L.; Hubbell, J.A. The TLR4 Agonist Fibronectin Extra Domain A is Cryptic, Exposed by Elastase-2; use in a fibrin matrix cancer vaccine. Sci. Rep. 2015, 5, 8569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.-I.; Hong, M.; Wilson, I.A. An unusual dimeric structure and assembly for TLR4 regulator RP105–MD-1. Nat. Struct. Mol. Biol. 2011, 18, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Shiroishi, M.; Kuroki, K.; Ose, T.; Rasubala, L.; Shiratori, I.; Arase, H.; Tsumoto, K.; Kumagai, I.; Kohda, D.; Maenaka, K. Efficient Leukocyte Ig-like Receptor Signaling and Crystal Structure of Disulfide-linked HLA-G Dimer. J. Biol. Chem. 2006, 281, 10439–10447. [Google Scholar] [CrossRef] [Green Version]
- Boyson, J.E.; Erskine, R.; Whitman, M.C.; Chiu, M.; Lau, J.M.; Koopman, L.A.; Valter, M.M.; Angelisova, P.; Horejsí, V.; Strominger, J.L. Disulfide bond-mediated dimerization of HLA-G on the cell surface. Proc. Natl. Acad. Sci. USA 2002, 99, 16180–16185. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.; Powis, S.; Arosa, F.A. Misfolding of Major Histocompatibility Complex Class I Molecules in Activated T Cells Allows cis-Interactions with Receptors and Signaling Molecules and Is Associated with Tyrosine Phosphorylation. J. Biol. Chem. 2004, 279, 53062–53070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, X.; Kim, J.; Deng, M.; John, S.; Chen, H.; Wu, G.; Phan, H.; Zhang, C.C. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 2016, 15, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krijgsman, D.; Roelands, J.; Hendrickx, W.; Bedognetti, D.; Kuppen, P.J.K. HLA-G: A New Immune Checkpoint in Cancer? Int. J. Mol. Sci. 2020, 21, 4528. [Google Scholar] [CrossRef] [PubMed]
- Hirayasu, K.; Arase, H. Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. J. Hum. Genet. 2015, 60, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, K.; Matsubara, H.; Kanda, R.; Miyashita, N.; Shiroishi, M.; Fukunaga, Y.; Kamishikiryo, J.; Fukunaga, A.; Fukuhara, H.; Hirose, K.; et al. Structural and Functional Basis for LILRB Immune Checkpoint Receptor Recognition of HLA-G Isoforms. J. Immunol. 2019, 203, 3386–3394. [Google Scholar] [CrossRef] [PubMed]
- Shiroishi, M.; Kuroki, K.; Rasubala, L.; Tsumoto, K.; Kumagai, I.; Kurimoto, E.; Kato, K.; Kohda, D.; Maenaka, K. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc. Natl. Acad. Sci. USA 2006, 103, 16412–16417. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Song, H.; Cheng, H.; Qi, J.; Nam, G.; Tan, S.; Wang, J.; Fang, M.; Shi, Y.; Tian, Z.; et al. Structures of the four Ig-like domain LILRB2 and the four-domain LILRB1 and HLA-G1 complex. Cell. Mol. Immunol. 2019, 17, 966–975. [Google Scholar] [CrossRef]
- Mandel, I.; Ziv, D.H.; Goldshtein, I.; Peretz, T.; Alishekevitz, D.; Dror, A.F.; Hakim, M.; Hashmueli, S.; Friedman, I.; Sapir, Y.; et al. BND-22, a first-in-class humanized ILT2-blocking antibody, promotes antitumor immunity and tumor regression. J. Immunother. Cancer 2022, 10, e004859. [Google Scholar] [CrossRef]
- Lin, A.; Yan, W.-H. Heterogeneity of HLA-G Expression in Cancers: Facing the Challenges. Front. Immunol. 2018, 9, 2164. [Google Scholar] [CrossRef]
- Zheng, G.; Jia, L.; Yang, A.-G. Roles of HLA-G/KIR2DL4 in Breast Cancer Immune Microenvironment. Front. Immunol. 2022, 13, 791975. [Google Scholar] [CrossRef]
- Van Beneden, K.; De Creus, A.; Stevenaert, F.; Debacker, V.; Plum, J.; Leclercq, G. Expression of Inhibitory Receptors Ly49E and CD94/NKG2 on Fetal Thymic and Adult Epidermal TCR Vγ3 Lymphocytes. J. Immunol. 2002, 168, 3295–3302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.S.; McCullar, V. Human natural killer cells with polyclonal lectin and immunoglobulinlike receptors develop from single hematopoietic stem cells with preferential expression of NKG2A and KIR2DL2/L3/S2. Blood 2001, 98, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Eugène, J.; Jouand, N.; Ducoin, K.; Dansette, D.; Oger, R.; Deleine, C.; Leveque, E.; Meurette, G.; Podevin, J.; Matysiak, T.; et al. The inhibitory receptor CD94/NKG2A on CD8+ tumor-infiltrating lymphocytes in colorectal cancer: A promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod. Pathol. 2020, 33, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Pizarro, J.C.; Kerns, J.; Strong, R.K. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc. Natl. Acad. Sci. USA 2008, 105, 6696–6701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veuillen, C.; Aurran-Schleinitz, T.; Castellano, R.; Rey, J.; Mallet, F.; Orlanducci, F.; Pouyet, L.; Just-Landi, S.; Coso, D.; Ivanov, V.; et al. Primary B-CLL Resistance to NK Cell Cytotoxicity can be Overcome In Vitro and In Vivo by Priming NK Cells and Monoclonal Antibody Therapy. J. Clin. Immunol. 2012, 32, 632–646. [Google Scholar] [CrossRef]
- Prašnikar, E.; Perdih, A.; Borišek, J. What a Difference an Amino Acid Makes: An All-Atom Simulation Study of Nonameric Peptides in Inhibitory HLA-E/NKG2A/CD94 Immune Complexes. Front. Pharmacol. 2022, 13, 925427. [Google Scholar] [CrossRef]
- Ruibal, P.; Franken, K.L.M.C.; van Meijgaarden, K.E.; van Loon, J.J.F.; van der Steen, D.; Heemskerk, M.H.M.; Ottenhoff, T.H.M.; Joosten, S.A. Peptide Binding to HLA-E Molecules in Humans, Nonhuman Primates, and Mice Reveals Unique Binding Peptides but Remarkably Conserved Anchor Residues. J. Immunol. 2020, 205, 2861–2872. [Google Scholar] [CrossRef]
- Chai, I.; Kornyeyev, D.; Hsieh, E.; Magombedze, G.; Stapleton, L.; Hung, M.; Kwon, H.J.; Stefanutti, E.; Belzile, J.; Czerwieniec, G.; et al. Effects of small molecule-induced dimerization on the programmed death ligand 1 protein life cycle. Sci. Rep. 2022, 12, 21286. [Google Scholar] [CrossRef]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Dömling, A.; Dubin, G.; Holak, T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Liu, C.; Seeram, N.P.; Ma, H. Small molecule inhibitors against PD-1/PD-L1 immune checkpoints and current methodologies for their development: A review. Cancer Cell Int. 2021, 21, 239. [Google Scholar] [CrossRef]
- Guo, Y.; Jin, Y.; Wang, B.; Liu, B. Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization. Int. J. Mol. Sci. 2021, 22, 4766. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ye, W.; Wang, S.; He, Y.; Zhong, H.; Wang, Y.; Zhu, Y.; Han, J.; Bing, Z.; Ji, S.; et al. Discovery of a new inhibitor targeting PD-L1 for cancer immunotherapy. Neoplasia 2021, 23, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Ghochikyan, A.; Pichugin, A.; Bagaev, A.; Davtyan, A.; Hovakimyan, A.; Tukhvatulin, A.; Davtyan, H.; Shcheblyakov, D.; Logunov, D.; Chulkina, M.; et al. Targeting TLR-4 with a novel pharmaceutical grade plant derived agonist, Immunomax®, as a therapeutic strategy for metastatic breast cancer. J. Transl. Med. 2014, 12, 322. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Yang, Y.; Jiang, Y.; Shao, J.; Sun, X.; Chen, J.; Dong, L.; Zhang, J. Anti-tumor immune responses of tumor-associated macrophages via toll-like receptor 4 triggered by cationic polymers. Biomaterials 2013, 34, 746–755. [Google Scholar] [CrossRef]
- Ajith, A.; Portik-Dobos, V.; Nguyen-Lefebvre, A.T.; Callaway, C.; Horuzsko, D.D.; Kapoor, R.; Zayas, C.; Maenaka, K.; Mulloy, L.L.; Horuzsko, A. HLA-G dimer targets Granzyme B pathway to prolong human renal allograft survival. FASEB J. 2019, 33, 5220–5236. [Google Scholar] [CrossRef]
- Zilberman, S.; Schenowitz, C.; Agaugué, S.; Favier, B.; Riteau, B.; Rouzier, R.; Carosella, E.D.; Rouas-Freiss, N.; Menier, C. HLA-G1 and HLA-G5 active dimers are present in malignant cells and effusions: The influence of the tumor microenvironment. Eur. J. Immunol. 2012, 42, 1599–1608. [Google Scholar] [CrossRef] [Green Version]
- Rouas-Freiss, N.; Bruel, S.; Menier, C.; Marcou, C.; Moreau, P.; Carosella, E.D. Switch ofHLA-G alternative splicing in a melanoma cell line causes loss of HLA-G1 expression and sensitivity to NK lysis. Int. J. Cancer 2005, 117, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Elstrand, M.B.; McMaster, M.T.; Berner, A.; Kurman, R.J.; Risberg, B.; Trope, C.G.; Shih, I.-M. HLA-G expression in effusions is a possible marker of tumor susceptibility to chemotherapy in ovarian carcinoma. Gynecol. Oncol. 2005, 96, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Singer, G.; Rebmann, V.; Chen, Y.-C.; Liu, H.-T.; Ali, S.Z.; Reinsberg, J.; McMaster, M.T.; Pfeiffer, K.; Chan, D.W.; Wardelmann, E.; et al. HLA-G is a potential tumor marker in malignant ascites. Clin. Cancer Res. 2003, 9, 4460–4464. [Google Scholar]
- Schwich, E.; Rebmann, V.; Michita, R.T.; Rohn, H.; Voncken, J.W.; Horn, P.A.; Kimmig, R.; Kasimir-Bauer, S.; Buderath, P. HLA-G 3′ untranslated region variants +3187G/G, +3196G/G and +3035T define diametrical clinical status and disease outcome in epithelial ovarian cancer. Sci. Rep. 2019, 9, 5407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reches, A.; Nachmani, D.; Berhani, O.; Duev-Cohen, A.; Shreibman, D.; Ophir, Y.; Seliger, B.; Mandelboim, O. HNRNPR Regulates the Expression of Classical and Nonclassical MHC Class I Proteins. J. Immunol. 2016, 196, 4967–4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducoin, K.; Oger, R.; Mutala, L.B.; Deleine, C.; Jouand, N.; Desfrançois, J.; Podevin, J.; Duchalais, E.; Cruard, J.; Benlalam, H.; et al. Targeting NKG2A to boost anti-tumor CD8 T-cell responses in human colorectal cancer. Oncoimmunology 2022, 11, 2046931. [Google Scholar] [CrossRef] [PubMed]
- Ram, D.R.; Lucar, O.; Hueber, B.; Reeves, R.K. Simian Immunodeficiency Virus Infection Modulates CD94 + (KLRD1+) NK Cells in Rhesus Macaques. J. Virol. 2019, 93, e00731-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, M.T.; Wu, J.; Fang, M.; Sigal, L.J.; Spee, P.; Egebjerg, T.; Dissen, E.; Fossum, S.; Phillips, J.H.; Lanier, L.L. Development and Function of CD94-Deficient Natural Killer Cells. PLoS ONE 2010, 5, e15184. [Google Scholar] [CrossRef] [Green Version]
- Vance, R.E.; Jamieson, A.M.; Cado, D.; Raulet, D.H. Implications of CD94 deficiency and monoallelic NKG2A expression for natural killer cell development and repertoire formation. Proc. Natl. Acad. Sci. USA 2002, 99, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Mingari, M.C.; Ponte, M.; Bertone, S.; Schiavetti, F.; Vitale, C.; Bellomo, R.; Moretta, A.; Moretta, L. HLA class I-specific inhibitory receptors in human T lymphocytes: Interleukin 15-induced expression of CD94/NKG2A in superantigen- or alloantigen-activated CD8 + T cells. Proc. Natl. Acad. Sci. USA 1998, 95, 1172–1177. [Google Scholar] [CrossRef] [Green Version]
- Starzer, A.M.; Preusser, M.; Berghoff, A.S. Immune escape mechanisms and therapeutic approaches in cancer: The cancer-immunity cycle. Ther. Adv. Med Oncol. 2022, 14, 17588359221096219. [Google Scholar] [CrossRef]
- Wang, L.; Liang, H.; Sun, J.; Liu, Y.; Li, J.; Li, J.; Li, J.; Yang, H. Bispecific Aptamer Induced Artificial Protein-Pairing: A Strategy for Selective Inhibition of Receptor Function. J. Am. Chem. Soc. 2019, 141, 12673–12681. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Geng, Q.; Jin, P.; Su, G.; Teng, H.; Dong, J.; Yan, B. Small Molecules as PD-1/PD-L1 Pathway Modulators for Cancer Immunotherapy. Curr. Pharm. Des. 2019, 24, 4911–4920. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, S.; Huizinga, H.P.S.; Butera, R.; Kouchi, A.; Guzik, K.; Magieramularz, K.; Holak, T.A.; Domling, A. A patent review on PD-1/PD-L1 antagonists: Small molecules, peptides, and macrocycles (2015–2018). Expert Opin. Ther. Pat. 2018, 28, 665–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Different types of TMEMs found in the cell membrane. (a) A single transmembrane α-helix protein. (b) An α-helix protein that crosses the membrane multiple times. (c) A transmembrane β-barrel protein.
Figure 1.
Different types of TMEMs found in the cell membrane. (a) A single transmembrane α-helix protein. (b) An α-helix protein that crosses the membrane multiple times. (c) A transmembrane β-barrel protein.

Figure 4.
(a) Crystal structure of a dimer of a disulfide-bonded HLA-G-peptide complex. HLA-G heavy chains and β2m are shown in cartoon models, wrapped in translucent surfaces (yellow and green) [111]. (b) ILT2 and HLA-G dimer binding model. The HLA-G dimer is presented in a cartoon model, and the four Ig domains of ILT2 are circular (PDB entry 2D31) [15].
Figure 4.
(a) Crystal structure of a dimer of a disulfide-bonded HLA-G-peptide complex. HLA-G heavy chains and β2m are shown in cartoon models, wrapped in translucent surfaces (yellow and green) [111]. (b) ILT2 and HLA-G dimer binding model. The HLA-G dimer is presented in a cartoon model, and the four Ig domains of ILT2 are circular (PDB entry 2D31) [15].

Figure 5.
(a) Crystal structure of NKG2A/CD94 complex (PDB entry 3BDW). (b) Co-crystal structure of human HLA-E/NKG2A/CD94 (PDB entry 3CDG) and amplified structure of HLA-E binding interface with NKG2A/CD94 [86].
Figure 5.
(a) Crystal structure of NKG2A/CD94 complex (PDB entry 3BDW). (b) Co-crystal structure of human HLA-E/NKG2A/CD94 (PDB entry 3CDG) and amplified structure of HLA-E binding interface with NKG2A/CD94 [86].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, L.; Li, J. Dimerization of Transmembrane Proteins in Cancer Immunotherapy. Membranes 2023, 13, 393. https://doi.org/10.3390/membranes13040393
AMA Style
Li L, Li J. Dimerization of Transmembrane Proteins in Cancer Immunotherapy. Membranes. 2023; 13(4):393. https://doi.org/10.3390/membranes13040393
Chicago/Turabian StyleLi, Lei, and Jingying Li. 2023. "Dimerization of Transmembrane Proteins in Cancer Immunotherapy" Membranes 13, no. 4: 393. https://doi.org/10.3390/membranes13040393
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.