
Recent Advances in Molecular Dynamics Simulations of Tau Fibrils and Oligomers

{kind=link}
{kind=link}
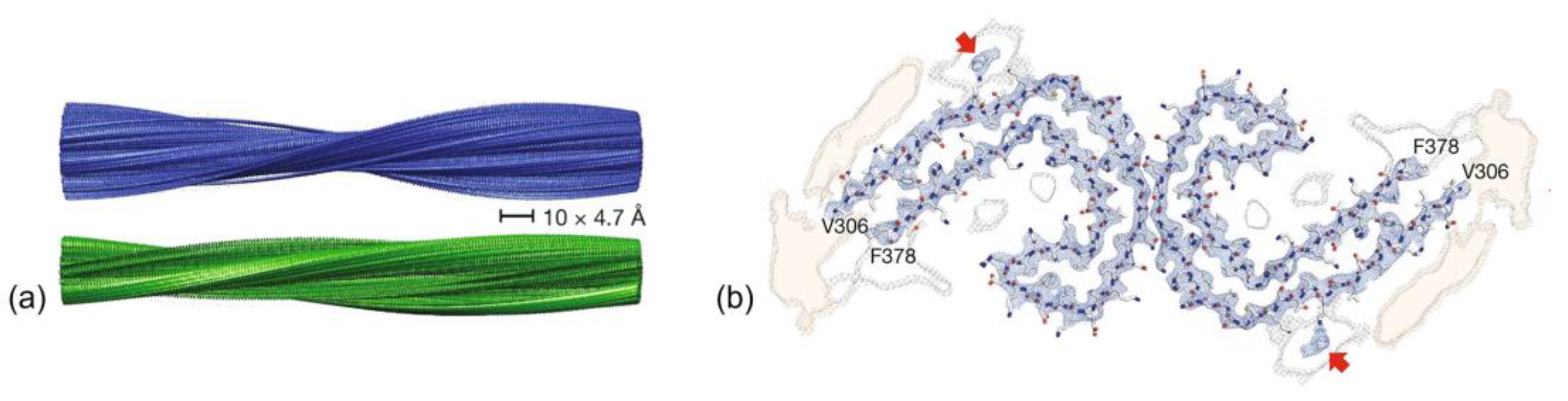{kind=link}
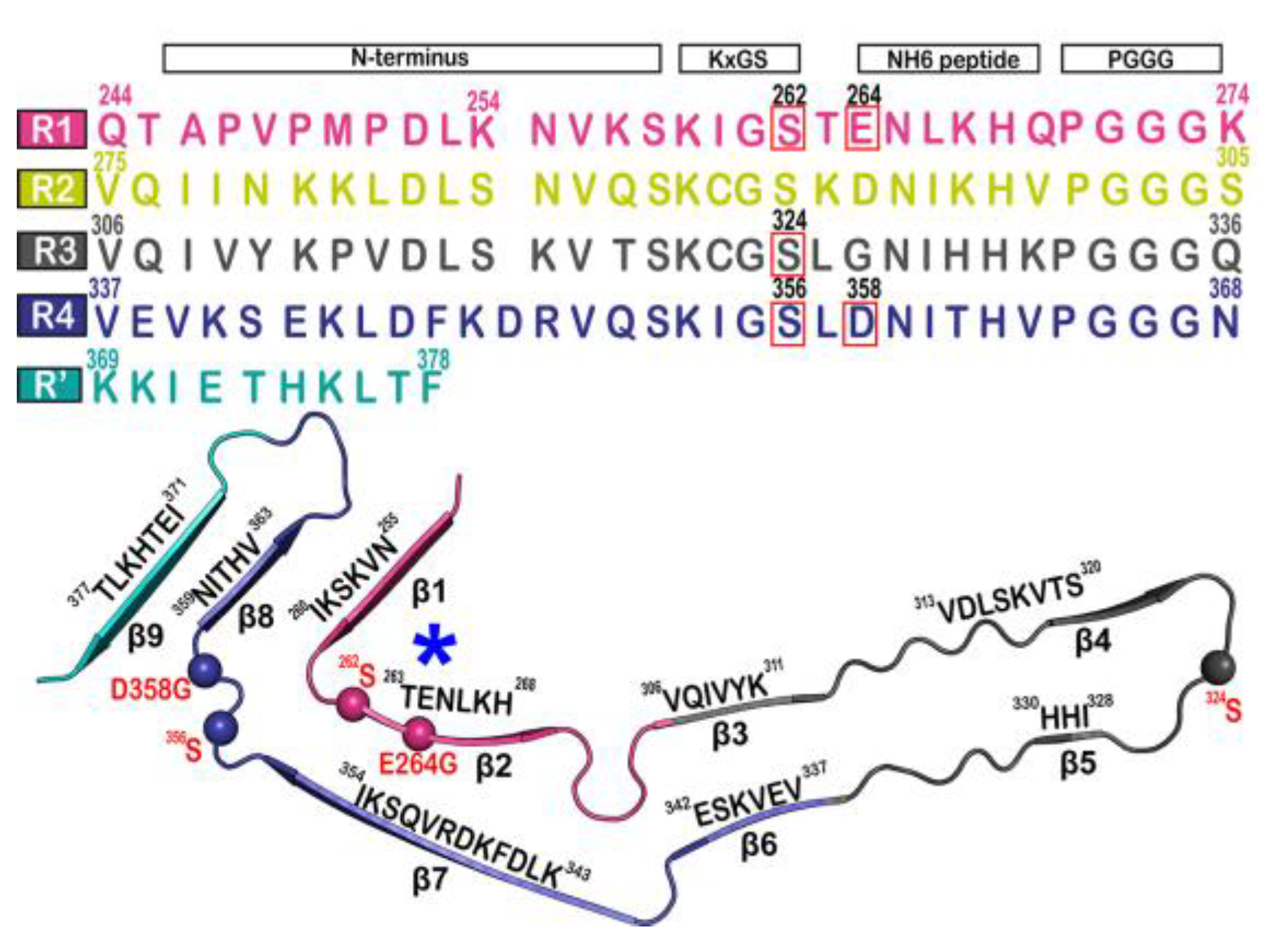{kind=link}
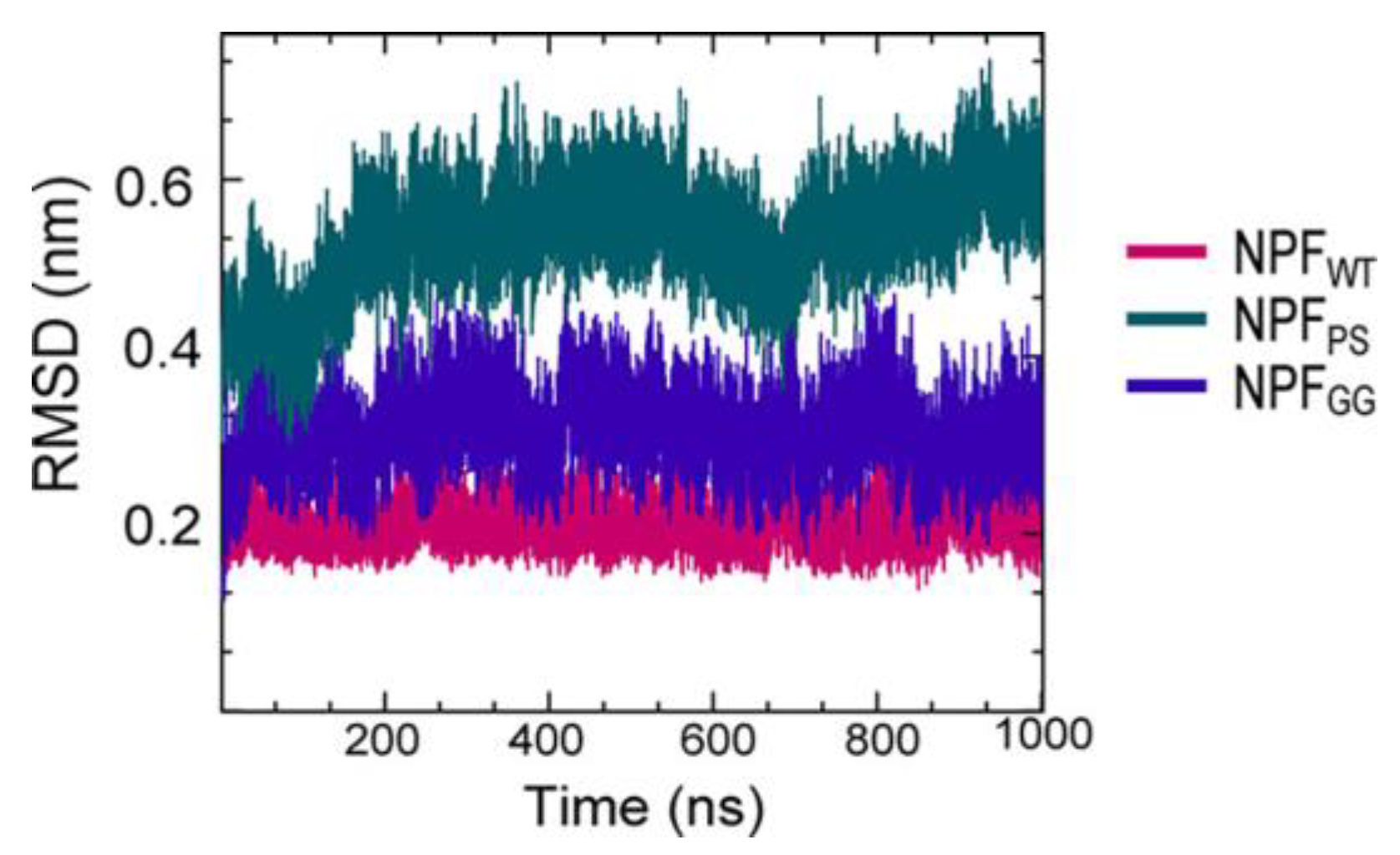{kind=link}
{kind=link}
{kind=link}
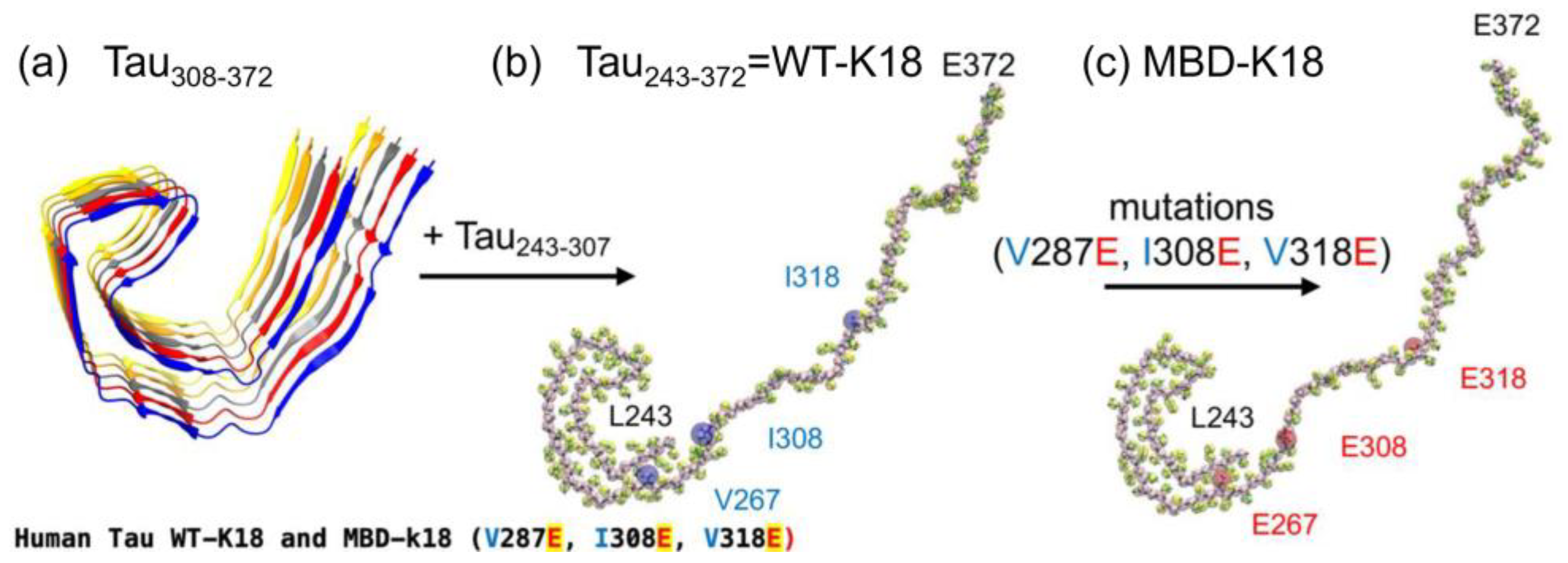{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Multiscale Molecular Dynamics Simulations of Tau Filaments (NPF and PHF/SF) in a Solution and in the Surface of a Lipid Bilayer
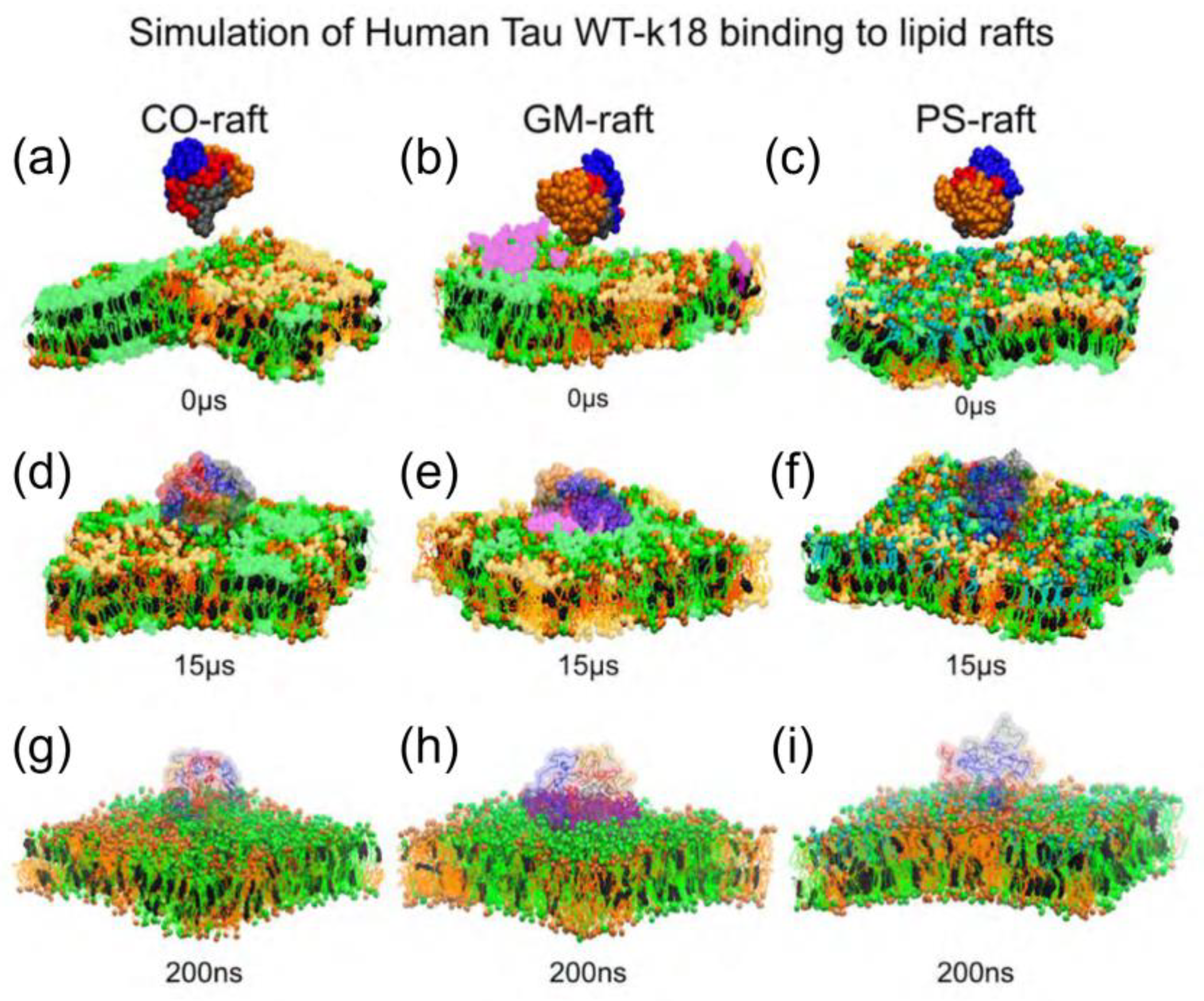
3. Multiscale Molecular Dynamics Simulations of Tau K-18 and R3-R4 Oligomers in Lipid Membrane Systems
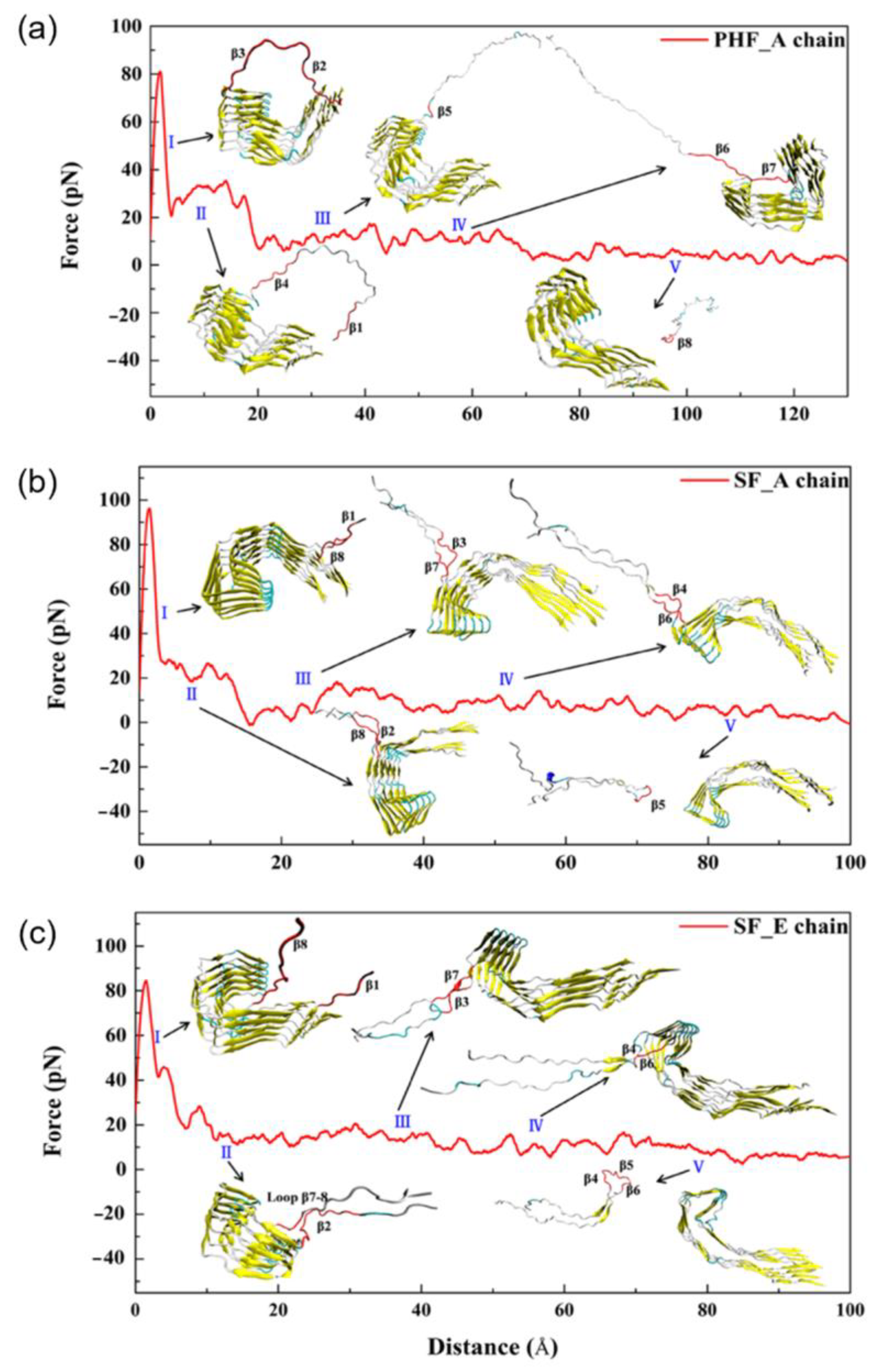
4. Dissociation of Tau Fibrils Studied through MD
5. Challenges and Future Opportunities
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gustke, N.; Trinczek, B.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Domains of tau protein and interactions with microtubules. Biochemistry 1994, 33, 9511–9522. [Google Scholar] [CrossRef] [PubMed]
- Limorenko, G.; Lashuel, H.A. Revisiting the grammar of Tau aggregation and pathology formation: How new insights from brain pathology are shaping how we study and target Tauopathies. Chem. Soc. Rev. 2022, 51, 513–565. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Characterization of tau fibrillization in vitro. Alzheimers. Dement. 2010, 6, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.M.; Binder, L.I. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am. J. Pathol. 1997, 150, 2181–2195. [Google Scholar]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef]
- von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Mutations of Tau Protein in Frontotemporal Dementia Promote Aggregation of Paired Helical Filaments by Enhancing Local β-Structure. J. Biol. Chem. 2001, 276, 48165–48174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait-Bouziad, N.; Lv, G.; Mahul-Mellier, A.-L.; Xiao, S.; Zorludemir, G.; Eliezer, D.; Walz, T.; Lashuel, H.A. Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun. 2017, 8, 1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; McAninch, E.A.; Wilson, R.S.; Evans, D.A. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimers. Dement. 2021, 17, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An english translation of alzheimer’s 1907 paper, “über eine eigenartige erkankung der hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s Disease. In Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease; Harris, J.R., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 329–352. [Google Scholar]
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J. Cell Biol. 1986, 103, 2739–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef]
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch. Neurol. 1995, 52, 81–88. [Google Scholar] [CrossRef]
- Buchhave, P.; Minthon, L.; Zetterberg, H.; Wallin, A.K.; Blennow, K.; Hansson, O. Cerebrospinal fluid levels of β-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 2012, 69, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Giacobini, E.; Gold, G. Alzheimer disease therapy--moving from amyloid-β to tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Selkoe, D.J. Treatments for Alzheimer’s disease emerge. Science 2021, 373, 624–626. [Google Scholar] [CrossRef]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Cryst. D Biol. Cryst. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Liu, Y.; Sun, Y.; Ding, F. Misfolding and Self-Assembly Dynamics of Microtubule-Binding Repeats of the Alzheimer-Related Protein Tau. J. Chem. Inf. Model. 2021, 61, 2916–2925. [Google Scholar] [CrossRef]
- Chung, D.-E.C.; Roemer, S.; Petrucelli, L.; Dickson, D.W. Cellular and pathological heterogeneity of primary tauopathies. Mol. Neurodegener. 2021, 16, 57. [Google Scholar] [CrossRef]
- Sahayaraj, A.E.; Viswanathan, R.; Pinhero, F.; Abdul Vahid, A.; Vijayan, V. Sequence-Dependent Conformational Properties of PGGG Motif in Tau Repeats: Insights from Molecular Dynamics Simulations of Narrow Pick Filament. ACS Chem. Neurosci. 2023, 14, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks III, C.L.; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kalé, L.V.; Skeel, R.D.; Schulten, K. NAMD: A Parallel, Object-Oriented Molecular Dynamics Program. Int. J. Supercomput. Appl. High Perform. Comput. 1996, 10, 251–268. [Google Scholar] [CrossRef]
- Sinha, S.; Tam, B.; Wang, S.M. Applications of Molecular Dynamics Simulation in Protein Study. Membranes 2022, 12, 844. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [PubMed] [Green Version]
- Lee, C.A.; Gasster, S.D.; Plaza, A.; Chang, C.I.; Huang, B. Recent Developments in High Performance Computing for Remote Sensing: A Review. IEEE J. Sel. Top. Appl. Earth Obs. Remote Sens. 2011, 4, 508–527. [Google Scholar] [CrossRef]
- Kohnke, B.; Kutzner, C.; Grubmüller, H. A GPU-Accelerated Fast Multipole Method for GROMACS: Performance and Accuracy. J. Chem. Theory Comput. 2020, 16, 6938–6949. [Google Scholar] [CrossRef]
- Li, X.; Dong, X.; Wei, G.; Margittai, M.; Nussinov, R.; Ma, B. The distinct structural preferences of tau protein repeat domains. Chem. Commun. 2018, 54, 5700–5703. [Google Scholar] [CrossRef]
- Chowdhury, U.D.; Paul, A.; Bhargava, B.L. The effect of lipid composition on the dynamics of tau fibrils. Proteins 2022, 90, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.-E.; Dokholyan, N.V.; De Simone, A.; et al. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, B.; Nussinov, R.; Wei, G. Structural Insight into Tau Protein’s Paradox of Intrinsically Disordered Behavior, Self-Acetylation Activity, and Aggregation. J. Phys. Chem. Lett. 2014, 5, 3026–3031. [Google Scholar] [CrossRef]
- Cheng, K.H.; Graf, A.; Lewis, A.; Pham, T.; Acharya, A. Exploring Membrane Binding Targets of Disordered Human Tau Aggregates on Lipid Rafts Using Multiscale Molecular Dynamics Simulations. Membranes 2022, 12, 1098. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; Pluhackova, K.; Böckmann, R.A.; Marrink, S.J.; Tieleman, D.P. Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput. 2014, 10, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumer, M.; Harris, S.; Li, M.; Martinez, L.; Untereiner, M.; Saeta, P.N.; Carpenter, T.S.; Ingólfsson, H.I.; Bennett, W.F.D. Simulations of Asymmetric Membranes Illustrate Cooperative Leaflet Coupling and Lipid Adaptability. Front. Cell Dev. Biol. 2020, 8, 575. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Derreumaux, P. Molecular Dynamics Simulations of the Tau R3–R4 Domain Monomer in the Bulk Solution and at the Surface of a Lipid Bilayer Model. J. Phys. Chem. B 2022, 126, 3431–3438. [Google Scholar] [CrossRef] [PubMed]
- Pastor, R.W.; MacKerell, A.D., Jr. Development of the CHARMM Force Field for Lipids. J. Phys. Chem. Lett. 2011, 2, 1526–1532. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Derreumaux, P. Molecular Dynamics Simulations of the Tau Amyloid Fibril Core Dimer at the Surface of a Lipid Bilayer Model: I. In Alzheimer’s Disease. J. Phys. Chem. B 2022, 126, 4849–4856. [Google Scholar] [CrossRef]
- Liu, H.; Liu, X.; Zhou, S.; An, X.; Liu, H.; Yao, X. Disclosing the Template-Induced Misfolding Mechanism of Tau Protein by Studying the Dissociation of the Boundary Chain from the Formed Tau Fibril Based on a Steered Molecular Dynamics Simulation. ACS Chem. Neurosci. 2019, 10, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Leonard, C.; Phillips, C.; McCarty, J. Insight Into Seeded Tau Fibril Growth From Molecular Dynamics Simulation of the Alzheimer’s Disease Protofibril Core. Front. Mol. Biosci. 2021, 8, 624302. [Google Scholar] [CrossRef] [PubMed]
- Izrailev, S.; Stepaniants, S.; Isralewitz, B.; Kosztin, D.; Lu, H.; Molnar, F.; Wriggers, W.; Schulten, K. Steered Molecular Dynamics. In Computational Molecular Dynamics: Challenges, Methods, Ideas; Deuflhard, P., Hermans, J., Leimkuhler, B., Mark, A.E., Reich, S., Skeel, R.D., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; pp. 39–65. [Google Scholar]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Lemkul, J.A.; Bevan, D.R. Assessing the Stability of Alzheimer’s Amyloid Protofibrils Using Molecular Dynamics. J. Phys. Chem. B 2010, 114, 1652–1660. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Derreumaux, P. Structures of the intrinsically disordered Aβ, tau and α-synuclein proteins in aqueous solution from computer simulations. Biophys. Chem. 2020, 264, 106421. [Google Scholar] [CrossRef]
- Man, V.H.; He, X.; Gao, J.; Wang, J. Phosphorylation of Tau R2 Repeat Destabilizes Its Binding to Microtubules: A Molecular Dynamics Simulation Study. ACS Chem. Neurosci. 2023, 14, 458–467. [Google Scholar] [CrossRef]
- Xu, L.; Zheng, J.; Margittai, M.; Nussinov, R.; Ma, B. How Does Hyperphopsphorylation Promote Tau Aggregation and Modulate Filament Structure and Stability? ACS Chem. Neurosci. 2016, 7, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Dinkel, P.; Yu, X.; Margittai, M.; Zheng, J.; Nussinov, R.; Wei, G.; Ma, B. Molecular insights into the reversible formation of tau protein fibrils. Chem. Commun. 2013, 49, 3582–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barredo, P.A.; Balanay, M.P. Recent Advances in Molecular Dynamics Simulations of Tau Fibrils and Oligomers. Membranes 2023, 13, 277. https://doi.org/10.3390/membranes13030277
Barredo PA, Balanay MP. Recent Advances in Molecular Dynamics Simulations of Tau Fibrils and Oligomers. Membranes. 2023; 13(3):277. https://doi.org/10.3390/membranes13030277
Chicago/Turabian StyleBarredo, Prechiel A., and Mannix P. Balanay. 2023. "Recent Advances in Molecular Dynamics Simulations of Tau Fibrils and Oligomers" Membranes 13, no. 3: 277. https://doi.org/10.3390/membranes13030277