
Simulation Study of the Effect of Antimicrobial Peptide Associations on the Mechanism of Action with Bacterial and Eukaryotic Membranes


Abstract
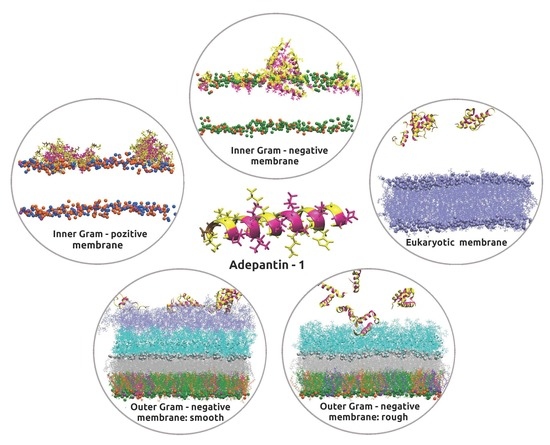
:
1. Introduction
2. Materials and Methods
2.1. Peptide Design and Biological Activity
2.2. Simulation Details
3. Results
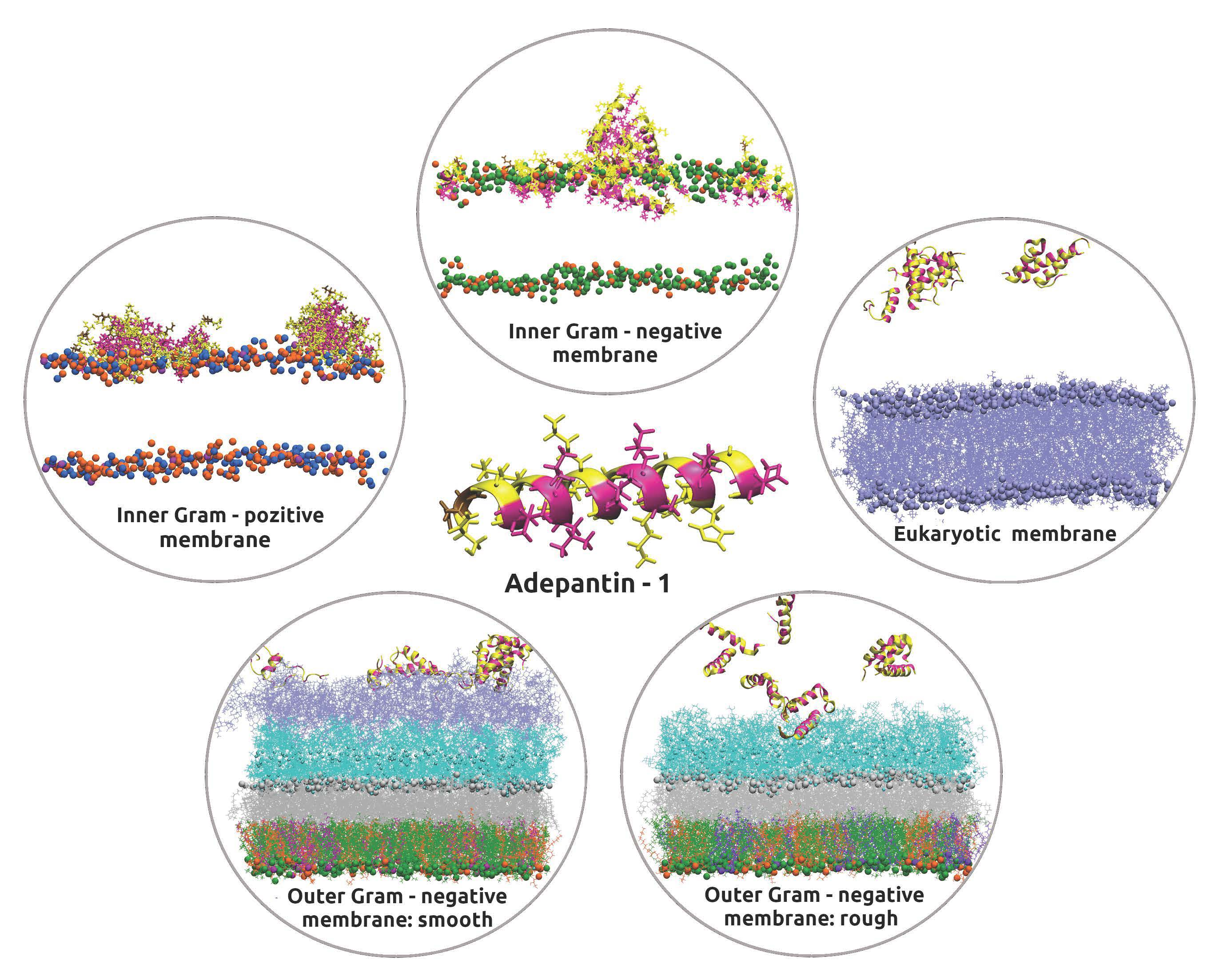
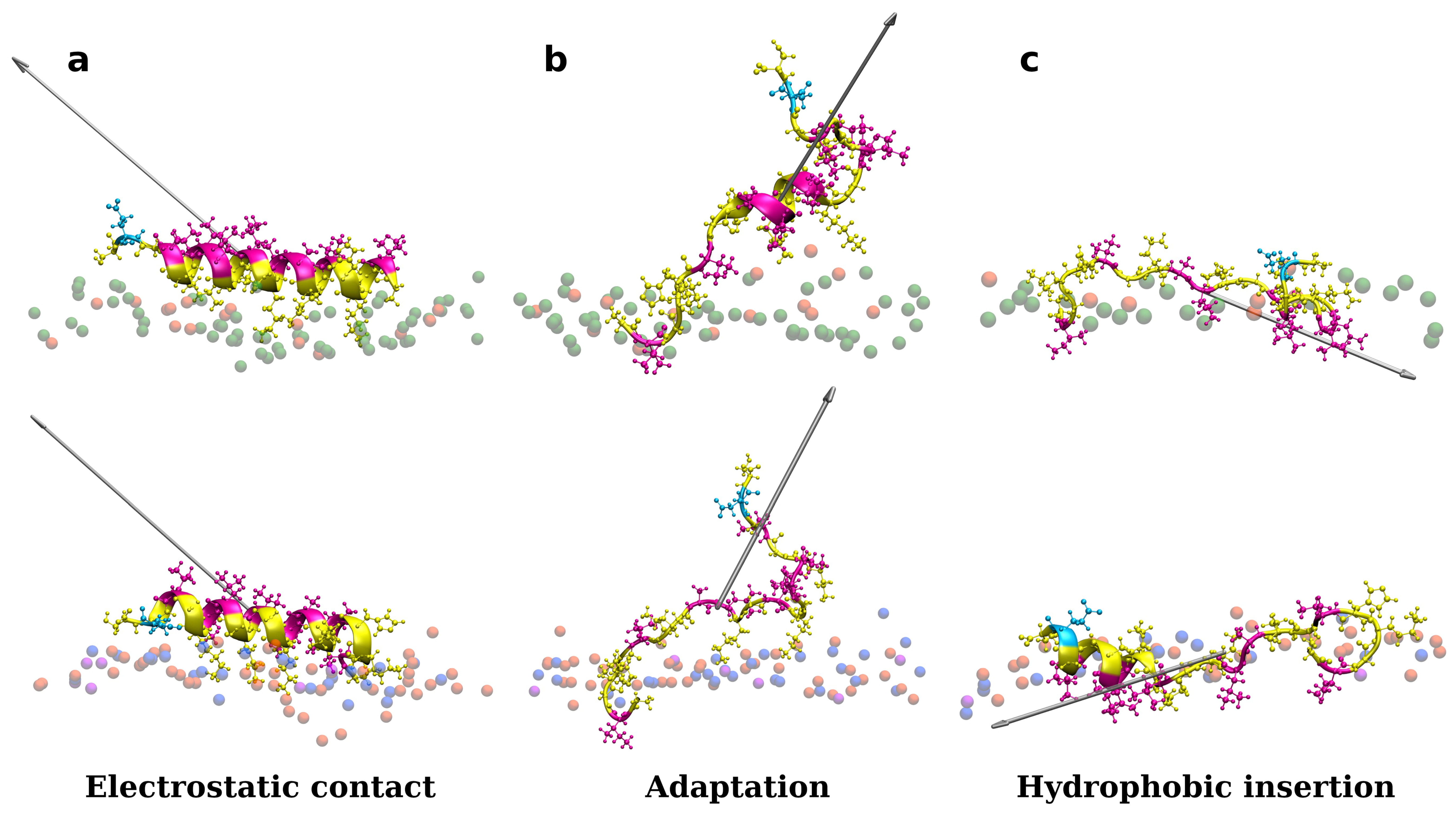
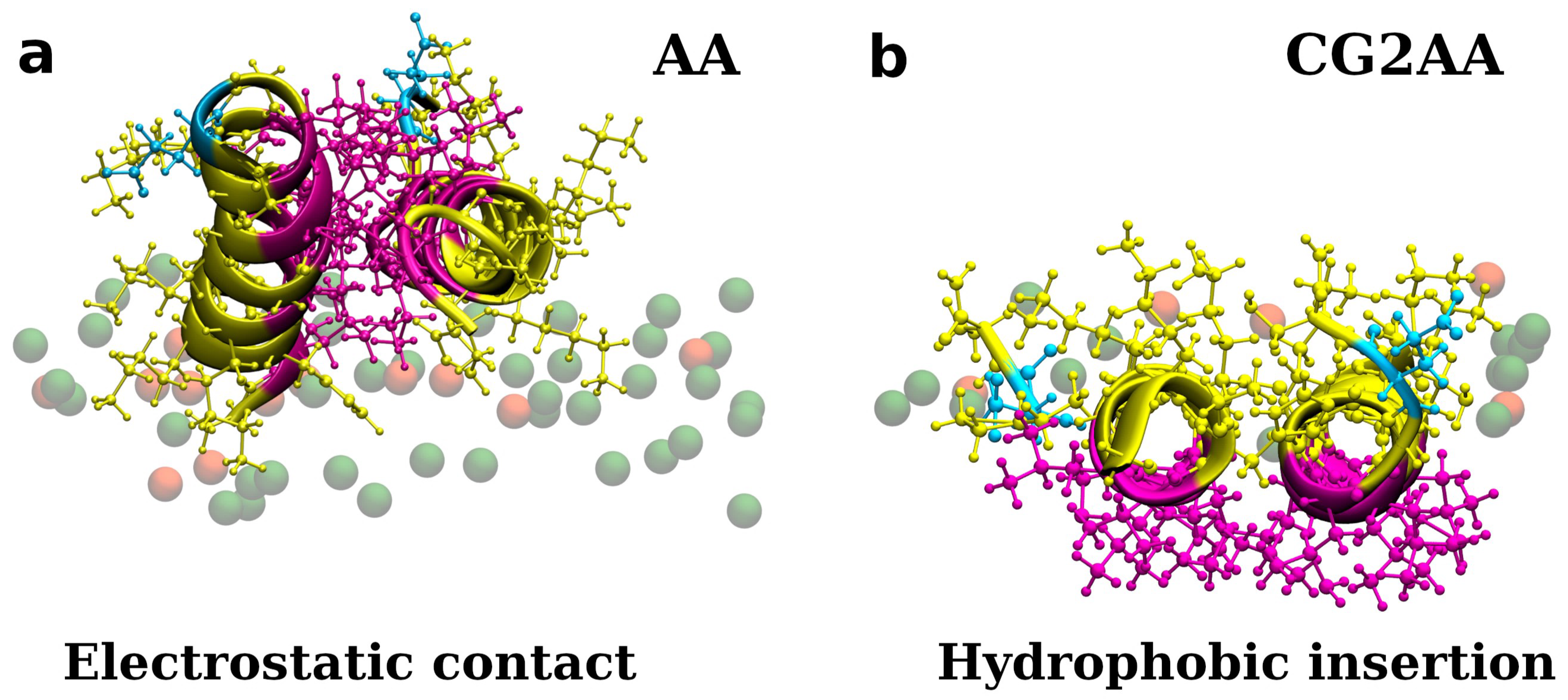
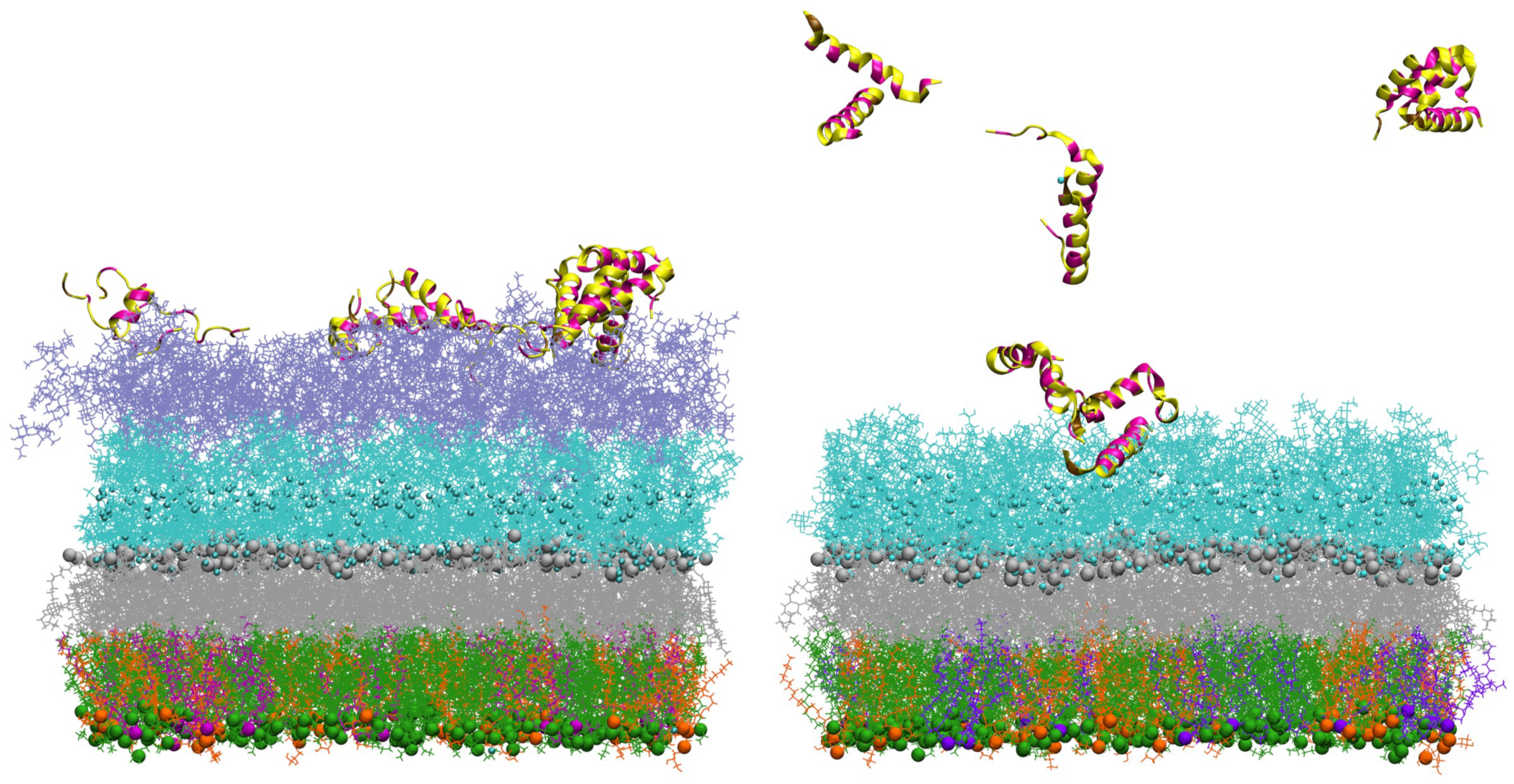
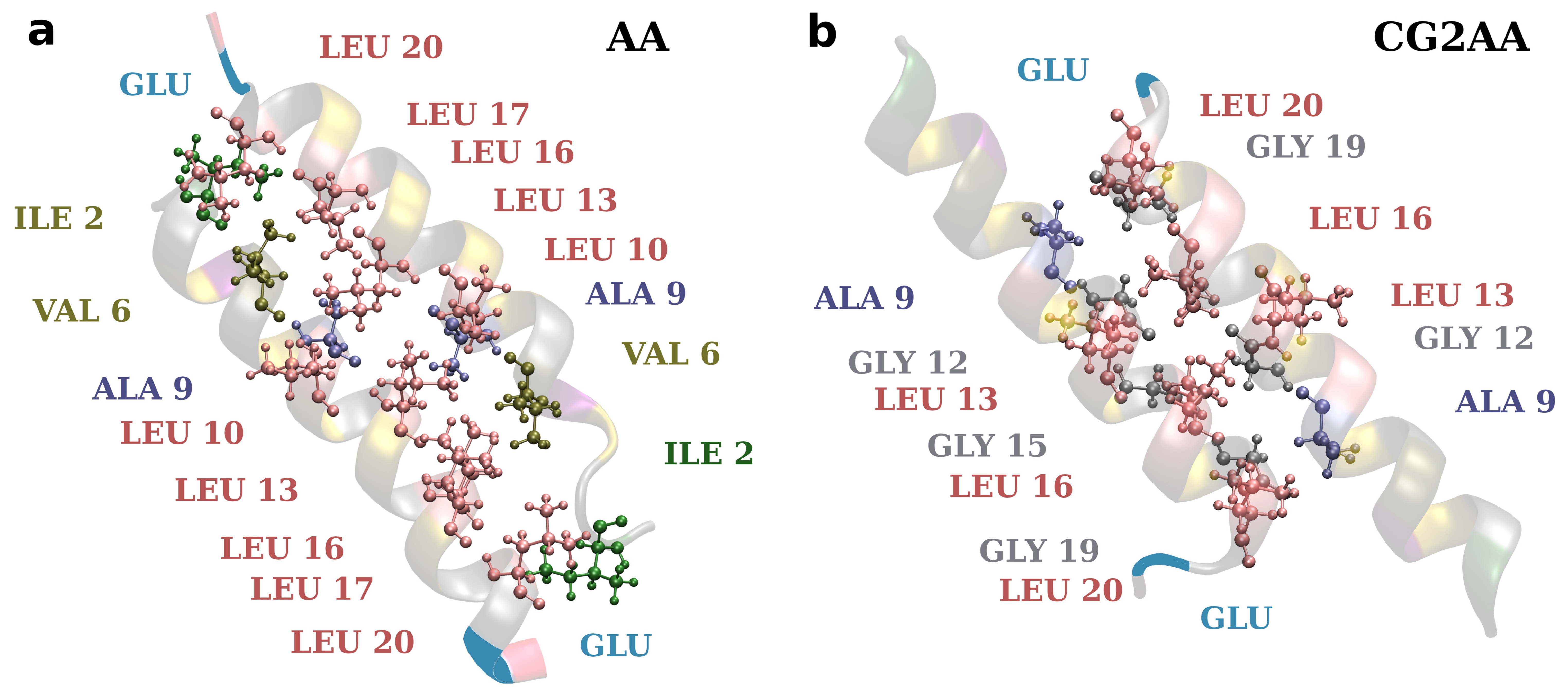
3.1. Accessing Peptide(s) Binding to Various Membrane with AA Modeling
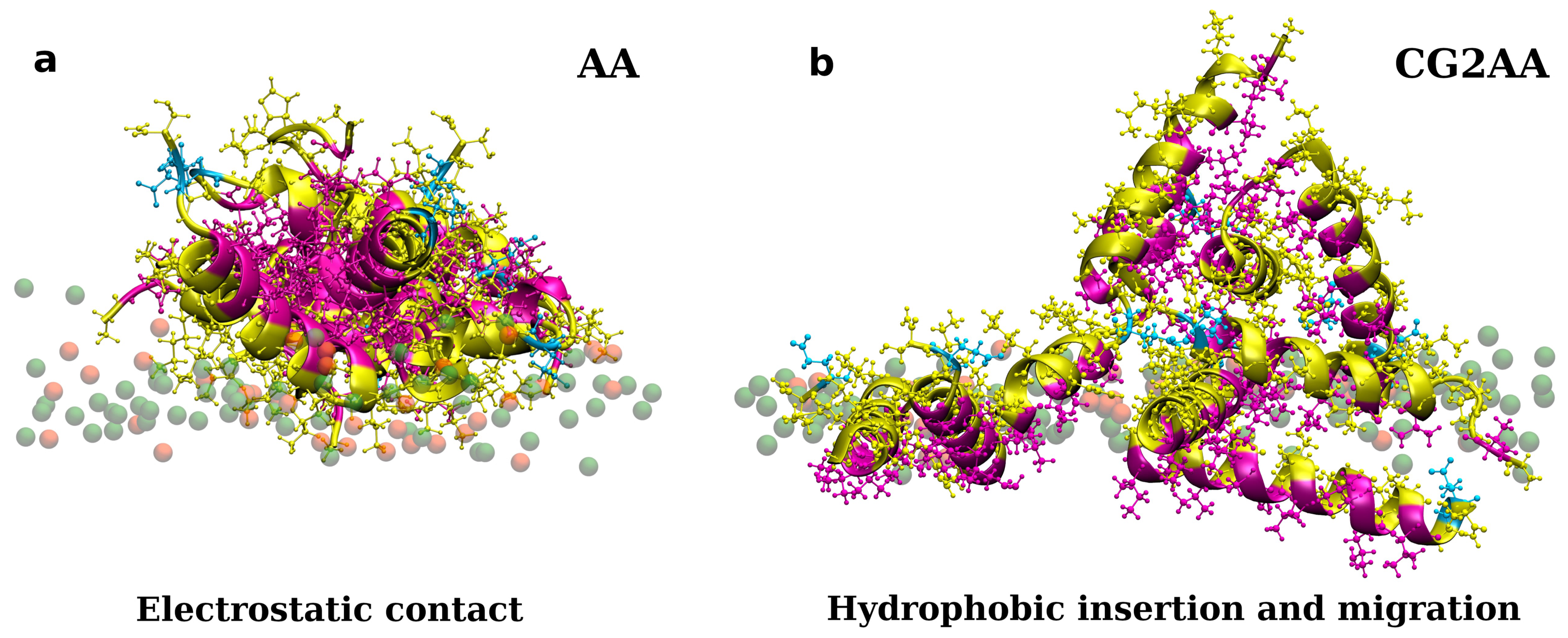
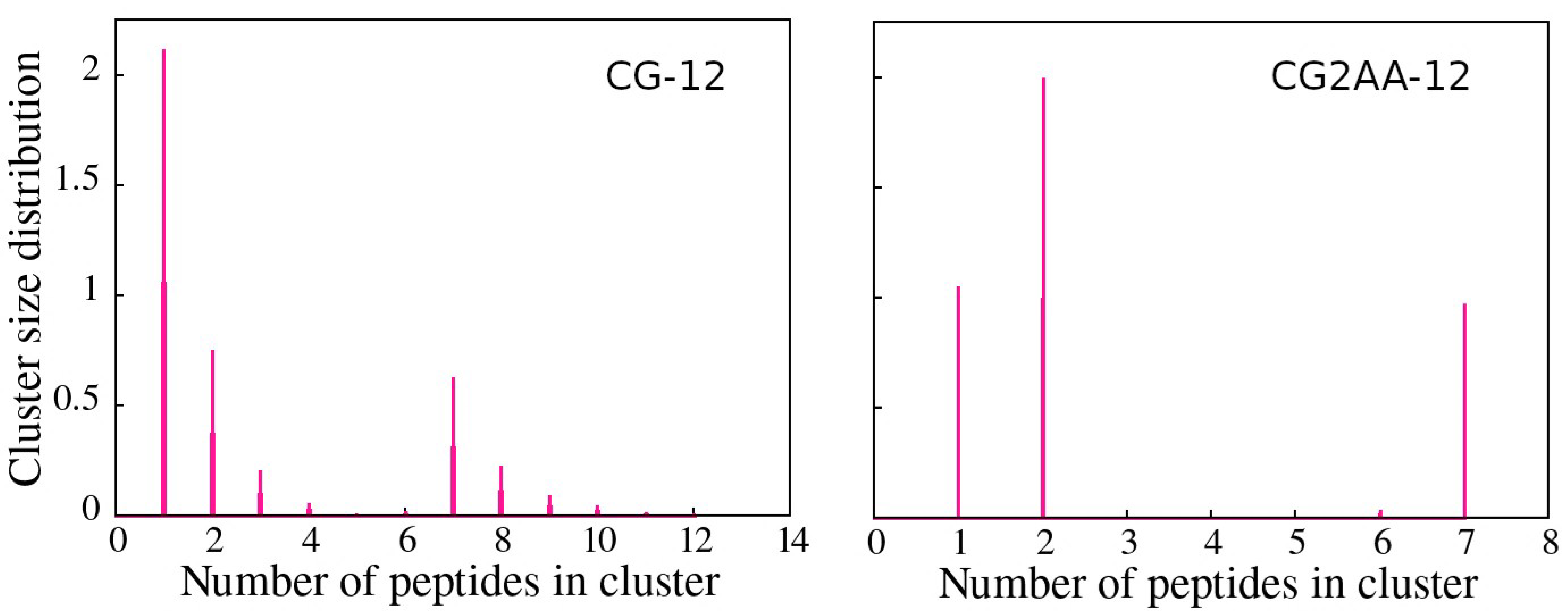
3.2. Accessing Peptide(s) Binding to Gram-Negative Membrane with CG and CG2AA Modeling
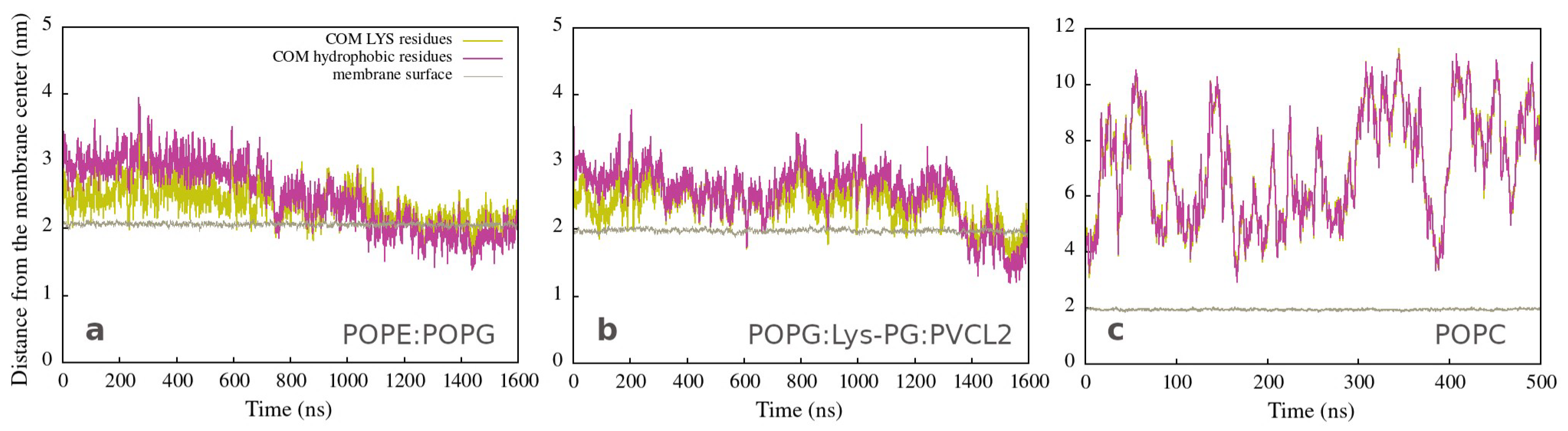
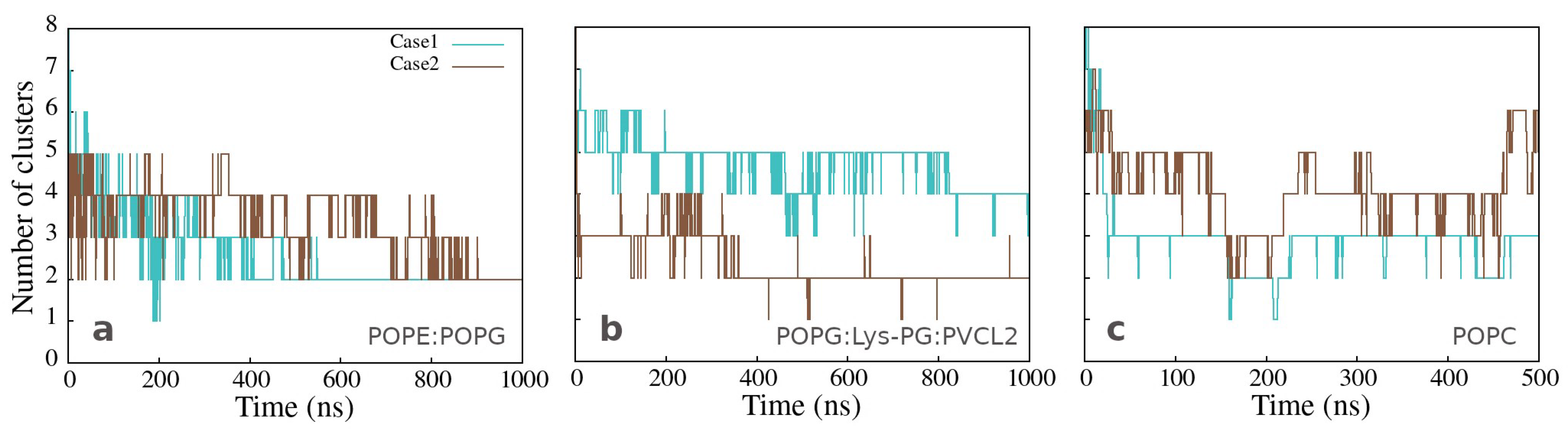
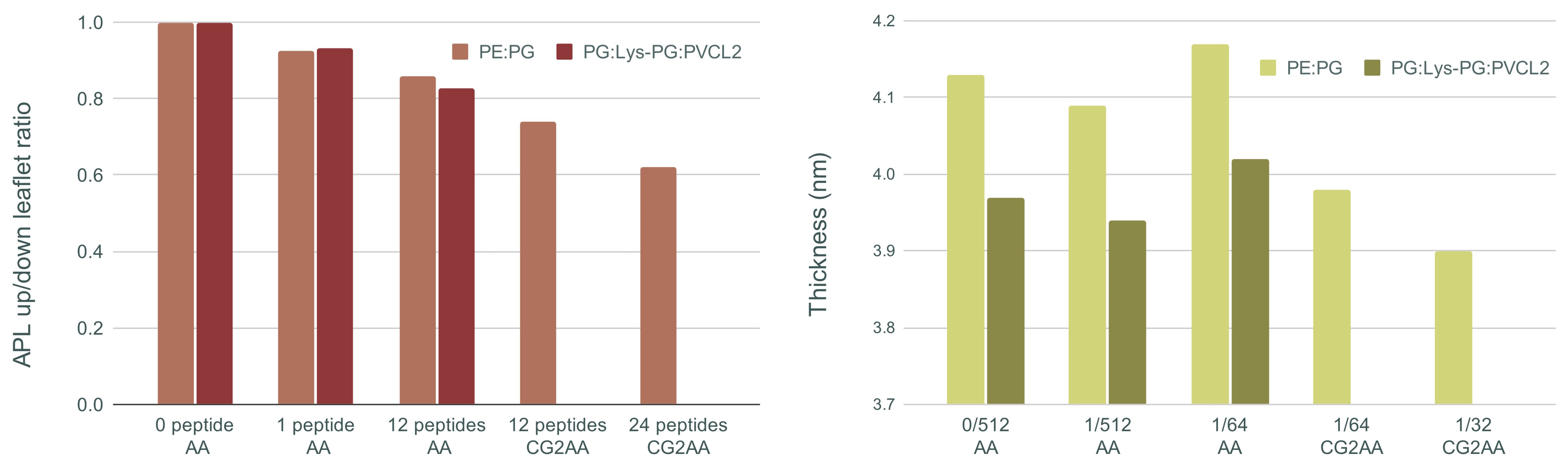
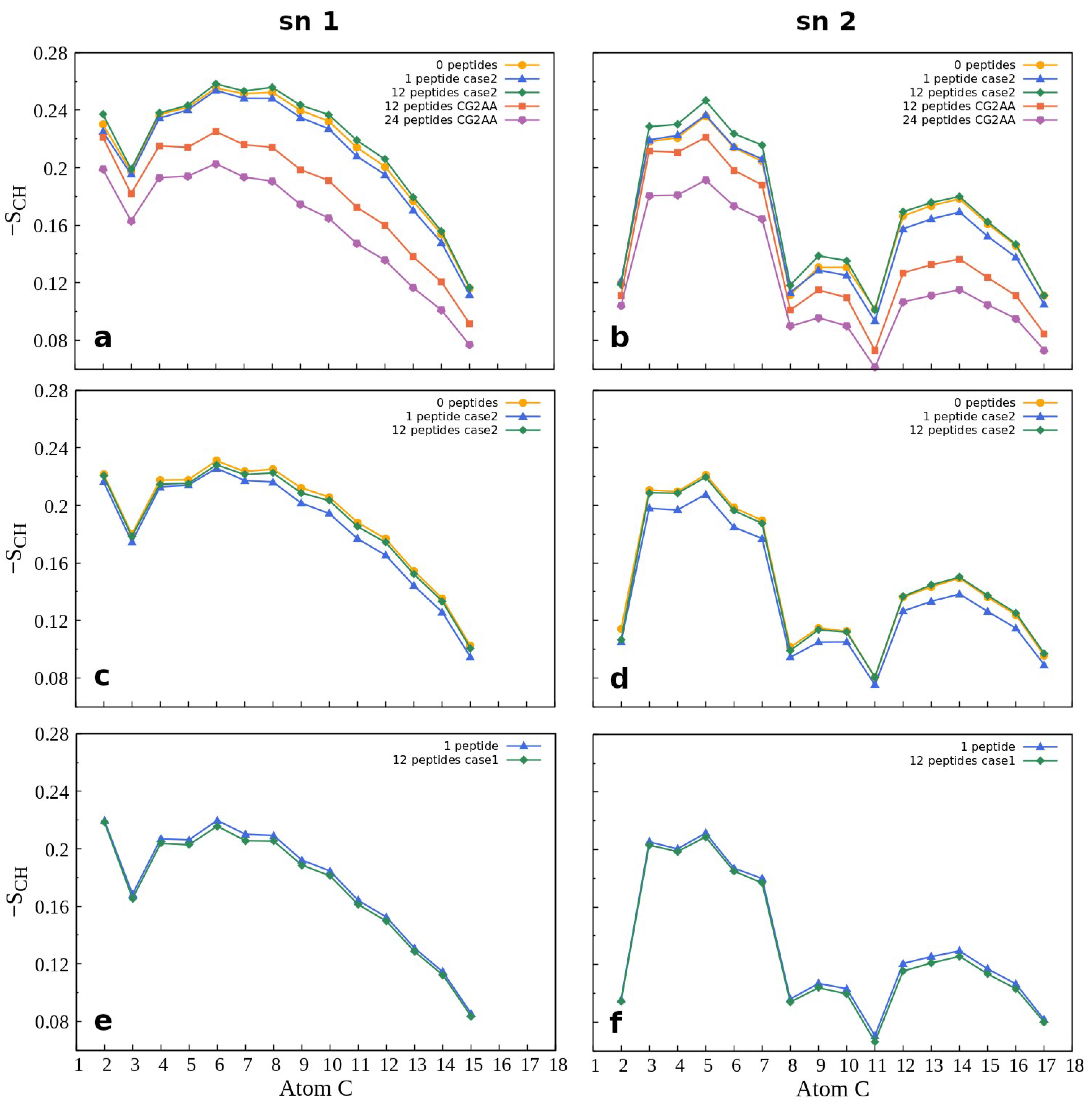
3.3. Changes in Membranes Induced by Peptide Interaction
3.4. Gram-Negative Outer Membrane Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kabelka, I.; Vácha, R. Advances in Molecular Understanding of α-Helical Membrane-Active Peptides. Acc. Chem. Res. 2021, 54, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Stillwell, W. An Introduction to Biological Membranes: Composition, Structure and Function, 2nd ed.; Elsevier Science: Amsterdam, The Netherlands, 2016; pp. 1–597. [Google Scholar] [CrossRef]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E.W. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharmacol. Ther. 2015, 40, 277–283. [Google Scholar]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.D.F.; Hancock, R.E.W. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti-Infect. Ther. 2007, 5, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, A.B.; Jiang, X.; Bergen, P.J.; Zhu, Y. Antimicrobial peptides: An update on classifications and databases. Int. J. Mol. Sci. 2021, 22, 11691. [Google Scholar] [CrossRef]
- Baltzer, S.A.; Brown, M.H. Antimicrobial peptides-promising alternatives to conventional antibiotics. J. Mol. Microbiol. Biotechnol. 2011, 20, 228–235. [Google Scholar] [CrossRef]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.-J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta BBA Biomembr. 2008, 1778, 2308–2317. [Google Scholar] [CrossRef] [Green Version]
- Rončević, T.; Puizina, J.; Tossi, A. Antimicrobial peptides as anti-infective agents in pre-post-antibiotic era? Int. J. Mol. Sci. 2019, 20, 5713. [Google Scholar] [CrossRef]
- Guha, S.; Ghimire, J.; Wu, E.; Wimley, W.C. Mechanistic Landscape of Membrane-Permeabilizing Peptides. Chem. Rev. 2019, 119, 6040–6085. [Google Scholar] [CrossRef]
- Park, S.-C.; Park, Y.; Hahm, K.-S. The Role of Antimicrobial Peptides in Preventing Multidrug-Resistant Bacterial Infections and Biofilm Formation. Int. J. Mol. Sci. 2011, 12, 5971–5992. [Google Scholar] [CrossRef]
- Batoni, G.; Maisetta, G.; Esin, S. Antimicrobial peptides and their interaction with biofilms of medically relevant bacteria. Biochim. Biophys. Acta BBA Biomembr. 2016, 1858, 1044–1060. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Yan, Z.-B.; Meng, Y.-M.; Hong, X.-Y.; Shao, G.; Ma, J.-J.; Cheng, X.-R.; Liu, J.; Kang, J.; Fu, C.-Y. Antimicrobial peptides: Mechanism of action, activity and clinical potential. Mil. Med. Res. 2021, 8, 1–25. [Google Scholar] [CrossRef]
- Lee, T.-H.; Hall, K.N.; Aguilar, M.-I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2015, 16, 25–39. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hancock, R.E.W.; Jenssen, H. Computer-Aided Design of Antimicrobial Peptides. Curr. Pharm. Anal. 2010, 6, 66–75. [Google Scholar] [CrossRef]
- Rončević, T.; Vukičević, D.; Ilić, N.; Krce, L.; Gajski, G.; Tonkić, M.; Goić-Barišić, I.; Zoranić, L.; Sonavane, Y.; Benincasa, M.; et al. Antibacterial Activity Affected by the Conformational Flexibility in Glycine-Lysine Based α-Helical Antimicrobial Peptides. J. Med. Chem. 2018, 61, 2924–2936. [Google Scholar] [CrossRef]
- Bopp, P.A.; Hawlicka, E.; Fritzsche, S. The Hitchhiker’s guide to molecular dynamics: A lecture companion, mostly for master’s and PhD students interested in using molecular dynamics simulations. ChemTexts 2018, 4, 2. [Google Scholar] [CrossRef]
- Braun, E.; Gilmer, J.; Mayes, H.B.; Mobley, D.L.; Monroe, J.I.; Prasad, S.; Zuckerman, D.M. Best Practices for Foundations in Molecular Simulations [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5957. [Google Scholar] [CrossRef] [Green Version]
- Sani, M.-A.; Separovic, F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem Res. 2016, 49, 1130–1138. [Google Scholar] [CrossRef]
- Marrink, S.J.; Corradi, V.; Souza, P.C.T.; Ingólfsson, H.I.; Tieleman, D.P.; Sansom, M.S.P. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef]
- Carey, A.B.; Ashenden, A.; Köper, I. Model architectures for bacterial membranes. Biophys. Rev. 2022, 14, 111–143. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Fernandez, D.I.; Le Brun, A.P.; Whitwell, T.C.; Sani, M.-A.; James, M.; Separovic, F. The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 2012, 14, 15739–15751. [Google Scholar] [CrossRef]
- Pieta, P.; Mirza, J.; Lipkowski, J. Direct visualization of the alamethicin pore formed in a planar phospholipid matrix. Proc. Natl. Acad. Sci. USA 2012, 109, 21223–21227. [Google Scholar] [CrossRef]
- Allende, D.; Simon, S.A.; McIntosh, T.J. Melittin-induced bilayer leakage depends on lipid material properties: Evidence for toroidal pores. Biophys. J. 2005, 88, 1828–1837. [Google Scholar] [CrossRef]
- Yoneyama, F.; Imura, Y.; Ohno, K.; Zendo, T.; Nakayama, J.; Matsuzaki, K.; Sonomoto, K. Peptide-lipid huge toroidal pore, a new antimicrobial mechanism mediated by a lactococcal bacteriocin, lacticin Q. Antimicrob. Agents Chemother. 2009, 53, 3211–3217. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Juretić, D.; Vukičević, D.; Ilić, N.; Antcheva, N.; Tossi, A. Computational design of highly selective antimicrobial peptides. J. Chem. Inf. Model. 2009, 49, 2873–2882. [Google Scholar] [CrossRef]
- Ilić, N.; Novkovic, M.; Guida, F.; Xhindoli, D.; Benincasa, M.; Tossi, A.; Juretić, D. Selective antimicrobial activity and mode of action of adepantins, glycine-rich peptide antibiotics based on anuran antimicrobial peptide sequences. Biochim. Biophys. Acta BBA Biomembr. 2013, 1828, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Zelezetsky, I.; Pacor, S.; Pag, U.; Papo, N.; Shai, Y.; Sahl, H.-G.; Tossi, A. Controlled alteration of the shape and conformational stability of alpha-helical cell-lytic peptides: Effect on mode of action and cell specificity. Biochem. J. 2005, 390, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Tytler, E.M.; Anantharamaiah, G.M.; Walker, D.E.; Mishra, V.K.; Palgunachari, M.N.; Segrest, J.P. Molecular basis for prokaryotic specificity of magainin-induced lysis. Biochemistry 1995, 34, 4393–4401. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Murzyn, K.; Róg, T.; Pasenkiewicz-Gierula, M. Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophys. J. 2005, 88, 1091–1103. [Google Scholar] [CrossRef]
- Malanovic, N.; Lohner, K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochim. Biophys. Acta BBA Biomembr. 2016, 1858, 936–946. [Google Scholar] [CrossRef]
- Yethon, J.A.; Heinrichs, D.E.; Monteiro, M.A.; Perry, M.B.; Whitfield, C. Involvement of waaY, waaQ, and waaP in the modification of Escherichia coli lipopolysaccharide and their role in the formation of a stable outer membrane. J. Biol. Chem. 1998, 273, 26310–26316. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; Pluhackova, K.; Böckmann, R.A.; Marrink, S.J.; Tieleman, D.P. Going backward: A flexible geometric approach to reverse transformation from coarse grained to atomistic models. J. Chem. Theory Comput. 2014, 10, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Wu, E.L.; Stuhlsatz, D.; Klauda, J.B.; MacKerell Jr, A.D.; Widmalm, G.; Im, W. Lipopolysaccharide membrane building and simulation. Methods Mol. Biol. 2015, 1273, 391–406. [Google Scholar] [CrossRef]
- Mortuza, S.M.; Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Zhang, Y. Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell Jr, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1998, 79, 926. [Google Scholar] [CrossRef]
- De Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2012, 9, 687–697. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Qi, Y.; Ingólfsson, H.I.; Cheng, X.; Lee, J.; Marrink, S.J.; Im, W. CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theory Comput. 2015, 11, 4486–4494. [Google Scholar] [CrossRef]
- Hsu, P.-C.; Bruininks, B.M.H.; Jefferies, D.; de Souza, P.C.T.; Lee, J.; Patel, D.S.; Marrink, S.J.; Qi, Y.; Khalid, S.; Im, W. Charmm-gui martini maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J. Comput. Chem. 2017, 38, 2354–2363. [Google Scholar] [CrossRef] [Green Version]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1998, 52, 7182. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1998, 81, 3684. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1998, 103, 8577. [Google Scholar] [CrossRef]
- Joosten, R.P.; te Beek, T.A.H.; Krieger, E.; Hekkelman, M.L.; Hooft, R.W.W.; Schneider, R.; Sander, C.; Vriend, G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011, 39, D411–D419. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Lukat, G.; Krüger, J.; Sommer, B. APL@Voro: A Voronoi-Based Membrane Analysis Tool for GROMACS Trajectories. J. Chem. Inf. Model. 2013, 53, 2908–2925. [Google Scholar] [CrossRef]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific alpha-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef]
- Reißer, S.; Strandberg, E.; Steinbrecher, T.; Ulrich, A.S. 3D hydrophobic moment vectors as a tool to characterize the surface polarity of amphiphilic peptides. Biophys. J. 2014, 106, 2385–2394. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kabelka, I.; Vácha, R. Optimal conditions for opening of membrane pore by amphiphilic peptides. J. Chem. Phys. 2015, 143, 243115. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Chen, R.; Mark, A.E. The effect of membrane curvature on the conformation of antimicrobial peptides: Implications for binding and the mechanism of action. Eur. Biophys. J. 2011, 40, 545–553. [Google Scholar] [CrossRef]
- Lee, M.-T.; Hung, W.-C.; Chen, F.-Y.; Huang, H.W. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 5087–5092. [Google Scholar] [CrossRef]
- Huang, H.W.; Charron, N.E. Understanding membrane-active antimicrobial peptides. Q. Rev. Biophys. 2017, 50, e10. [Google Scholar] [CrossRef]
- Wimley, W.C.; Hristova, K. Antimicrobial peptides: Successes, challenges and unanswered questions. J. Membr. Biol. 2011, 239, 27–34. [Google Scholar] [CrossRef]
- Bennett, W.F.D.; Hong, C.K.; Wang, Y.; Tieleman, D.P. Antimicrobial Peptide Simulations and the Influence of Force Field on the Free Energy for Pore Formation in Lipid Bilayers. J. Chem. Theory Comput. 2016, 12, 4524–4533. [Google Scholar] [CrossRef]
- Ramos-Martín, F.; Herrera-León, C.; Antonietti, V.; Sonnet, P.; Sarazin, C.; D’amelio, N. Antimicrobial Peptide K11 Selectively Recognizes Bacterial Biomimetic Membranes and Acts by Twisting Their Bilayers. Pharmaceuticals 2021, 14, 1. [Google Scholar] [CrossRef]
- Zhang, L.; Dhillon, P.; Yan, H.; Farmer, S.; Hancock, R.E.W. Interactions of Bacterial Cationic Peptide Antibiotics with Outer and Cytoplasmic Membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3317–3321. [Google Scholar] [CrossRef]
- Gong, H.; Hu, X.; Liao, M.; Fa, K.; Ciumac, D.; Clifton, L.A.; Sani, M.-A.; King, S.M.; Maestro, A.; Separovic, F.; et al. Structural Disruptions of the Outer Membranes of Gram-Negative Bacteria by Rationally Designed Amphiphilic Antimicrobial Peptides. ACS Appl. Mater. Interfaces 2021, 13, 16062–16074. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Chapple, D.S. Peptide Antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar] [CrossRef]
- Weerakoon, D.; Petrov, K.; Pedebos, C.; Khalid, S. Polymyxin B1 within the E. coli cell envelope: Insights from molecular dynamics simulations. Biophys. Rev. 2021, 13, 1061–1070. [Google Scholar] [CrossRef]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial Peptides in Action. J. Amer. Chem. Soc. 2006, 9, 12156–12161. [Google Scholar] [CrossRef]
- Rončević, T.; Gerdol, M.; Mardirossian, M.; Maleš, M.; Cvjetan, S.; Benincasa, M.; Maravić, A.; Gajski, G.; Krce, L.; Aviani, I.; et al. Anisaxins, helical antimicrobial peptides from marine parasites, kill resistant bacteria by lipid extraction and membrane disruption. Acta Biomater. 2022, 146, 131–144. [Google Scholar] [CrossRef]
- Juretić, D.; Sonavane, Y.; Ilić, N.; Gajski, G.; Goić-Barišić, I.; Tonkić, M.; Kozic, M.; Maravić, A.; Pellay, F.-X.; Zoranic, L. Designed peptide with a flexible central motif from ranatuerins adapts its conformation to bacterial membranes. Biochim. Biophys. Acta BBA Biomembr. 2018, 1860, 2655–2668. [Google Scholar] [CrossRef]
- Boags, A.; Hsu, P.-C.; Samsudin, F.; Bond, P.J.; Khalid, S. Progress in Molecular Dynamics Simulations of Gram-Negative Bacterial Cell Envelopes. J. Phys. Chem. Lett. 2017, 8, 2513–2518. [Google Scholar] [CrossRef]
- Ulmschneider, J.P.; Smith, J.C.; Ulmschneider, M.B.; Ulrich, A.S.; Strandberg, E. Reorientation and dimerization of the membrane-bound antimicrobial peptide pgla from microsecond all-atom MD simulations. Biophys. J. 2012, 103, 472–482. [Google Scholar] [CrossRef]
- Petkov, P.; Lilkova, E.; Ilieva, N.; Litov, L. Self-association of antimicrobial peptides: A molecular dynamics simulation study on bombinin. Int. J. Mol. Sci. 2019, 20, 5450. [Google Scholar] [CrossRef]
- Zou, R.; Zhu, X.; Tu, Y.; Wu, J.; Landry, M.P. Activity of Antimicrobial Peptide Aggregates Decreases with Increased Cell Membrane Embedding Free Energy Cost. Biochemistry 2018, 57, 2606–2610. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, Alpha-Helical Antimicrobial Peptides. Biopolymers 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Bechinger, B.; Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Juretić, D.; Golemac, A.; Strand, D.E.; Chung, K.; Ilić, N.; Goić-Barišić, I.; Pellay, F.-X. The spectrum of design solutions for improving the activity-selectivity product of peptide antibiotics against multidrug-resistant bacteria and prostate cancer PC-3 cells. Molecules 2020, 25, 3526. [Google Scholar] [CrossRef] [PubMed]
- Kamech, N.; Vukičević, D.; Ladram, A.; Piesse, C.; Vasseur, J.; Bojović, V.; Simunić, J.; Juretić, D. Improving the selectivity of antimicrobial peptides from anuran skin. J. Chem. Inf. Model. 2012, 52, 3341–3351. [Google Scholar] [CrossRef] [PubMed]
- Robles-loaiza, A.A.; Pinos-Tamayo, E.A.; Mendes, B.; Ortega-Pila, J.A.; Proaño-Bolaños, C.; Plisson, F.; Teixeira, C.; Gomes, P.; Almeida, J.R. Traditional and Computational Screening of Non-Toxic Peptides and Approaches to Improving Selectivity. Pharmaceuticals 2022, 15, 323. [Google Scholar] [CrossRef]
- Kucerka, N.; Van Oosten, B.; Pan, J.; Heberle, F.; Harroun, T.; Katsaras, J. Molecular Structures of Fluid Phosphatidylethanolamine Bilayers Obtained from Simulation-to-Experiment Comparisons and Experimental Scattering Density Profiles. J. Phys. Chem. B 2014, 119, 1947–1956. [Google Scholar] [CrossRef]
- Pan, J.; Heberle, F.A.; Tristram-Nagle, S.; Szymanski, M.; Koepfinger, M.; Katsaras, J.; Kučerka, N. Molecular structures of fluid phase phosphatidylglycerol bilayers as determined by small angle neutron and X-ray scattering. Biochim. Biophys. Acta BBA Biomembr. 2012, 1818, 2135–2148. [Google Scholar] [CrossRef]
- Kučerka, N.; Nieh, M.-P.; Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim. Biophys. Acta BBA Biomembr. 2011, 1808, 2761–2771. [Google Scholar] [CrossRef]
- Piggot, T.J.; Holdbrook, D.A.; Khalid, S. Electroporation of the E. coli and S. Aureus Membranes: Molecular Dynamics Simulations of Complex Bacterial Membranes. J. Phys. Chem. B 2011, 115, 13381–13388. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | No. of Peptides | Gram − | POPC | Gram + | Gram − Outer Membrane | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time | No. of Lipids | Time | No. of Lipids | Time | No. of. Lipids | 2 O-Antigens | 0 O-Antigens | No. of Lipids and LPSs | ||
| Time | Time | |||||||||
| AA-0 | 0 | 0.5 | 192 POPE 64 POPG | - | 256 POPC | 0.5 | 146 POPG 96 Lys-PG 14 PVCL2 | 0.5 | 0.5 | Up: 50 LPS Down: 105 PVPE 30 PVPG 8 PVCL2 |
| AA-1 a * | 1 | 1 | 0.5 | 1 | 0.5 | 0.1 | ||||
| AA-1 b * | 1 | 1.6 | - | 1.6 | - | - | ||||
| AA-2 | 2 | 1.5 | - | 1.5 | - | - | ||||
| AA-12 a * | 12 | 1 | 384 POPE 128 POPG | 0.5 | 512 POPC | 1 | 292 POPG 192 Lys-PG 28 PVCL2 | - | 0.1 | |
| AA-12 b * | 12 | 1 | 0.5 | 1 | 0.4 | 0.2 | ||||
| CG-12 | 12 | 25 | 384 POPE 128 POPG | |||||||
| CG-24 | 24 | 42.5 | ||||||||
| CG2AA-12 ** | 12 | 0.5 | ||||||||
| CG2AA-24 ** | 24 | 0.5 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maleš, M.; Zoranić, L. Simulation Study of the Effect of Antimicrobial Peptide Associations on the Mechanism of Action with Bacterial and Eukaryotic Membranes. Membranes 2022, 12, 891. https://doi.org/10.3390/membranes12090891
Maleš M, Zoranić L. Simulation Study of the Effect of Antimicrobial Peptide Associations on the Mechanism of Action with Bacterial and Eukaryotic Membranes. Membranes. 2022; 12(9):891. https://doi.org/10.3390/membranes12090891
Chicago/Turabian StyleMaleš, Matko, and Larisa Zoranić. 2022. "Simulation Study of the Effect of Antimicrobial Peptide Associations on the Mechanism of Action with Bacterial and Eukaryotic Membranes" Membranes 12, no. 9: 891. https://doi.org/10.3390/membranes12090891