
Interaction of Oxicam Derivatives with the Artificial Models of Biological Membranes—Calorimetric and Fluorescence Spectroscopic Study

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Experimental
2.2.1. Differential Scanning Calorimetry (DSC)
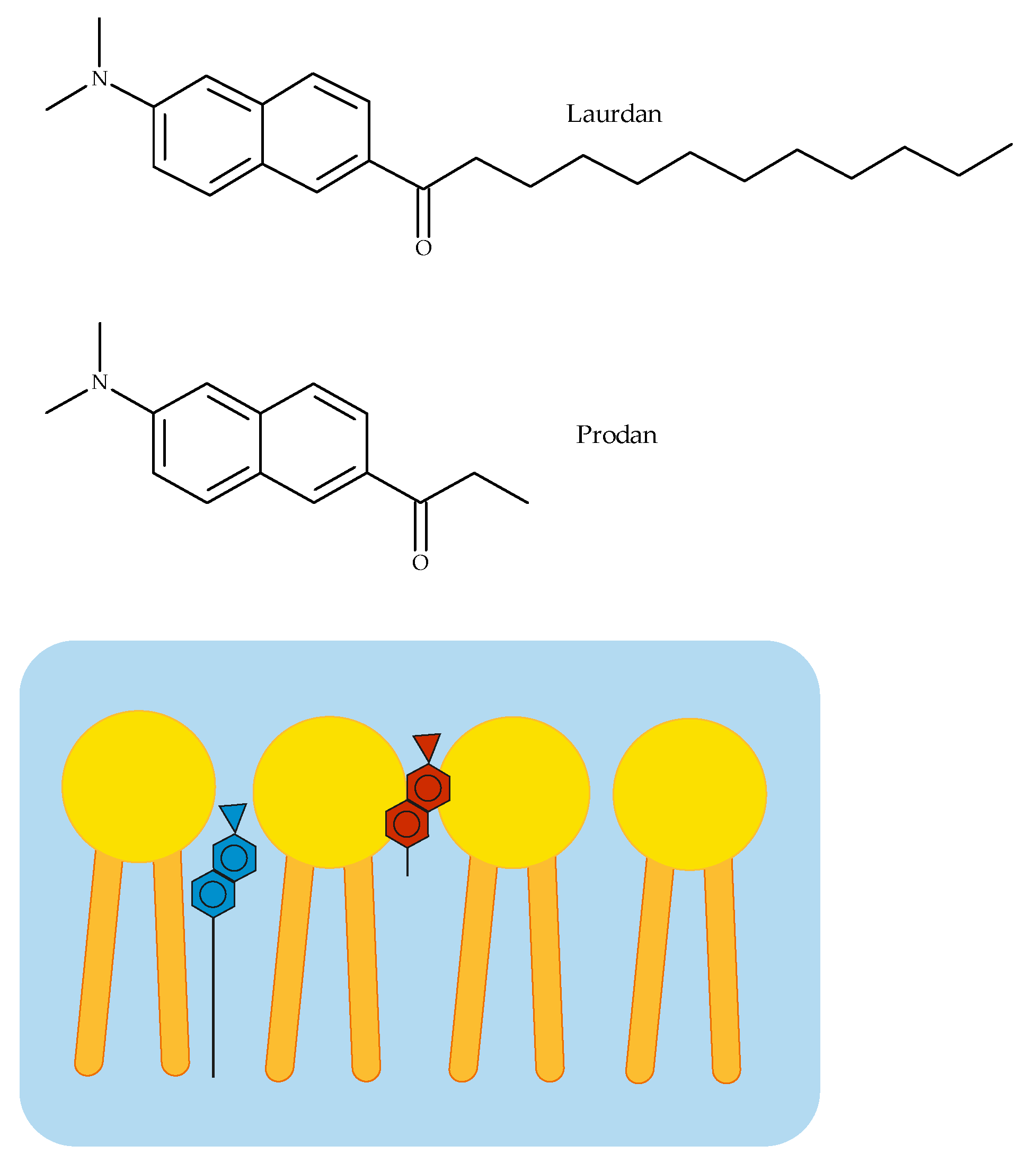
2.2.2. Fluorescence Spectroscopy
2.2.3. Prediction of ADMET Properties
3. Results
3.1. Differential Scanning Calorimetry
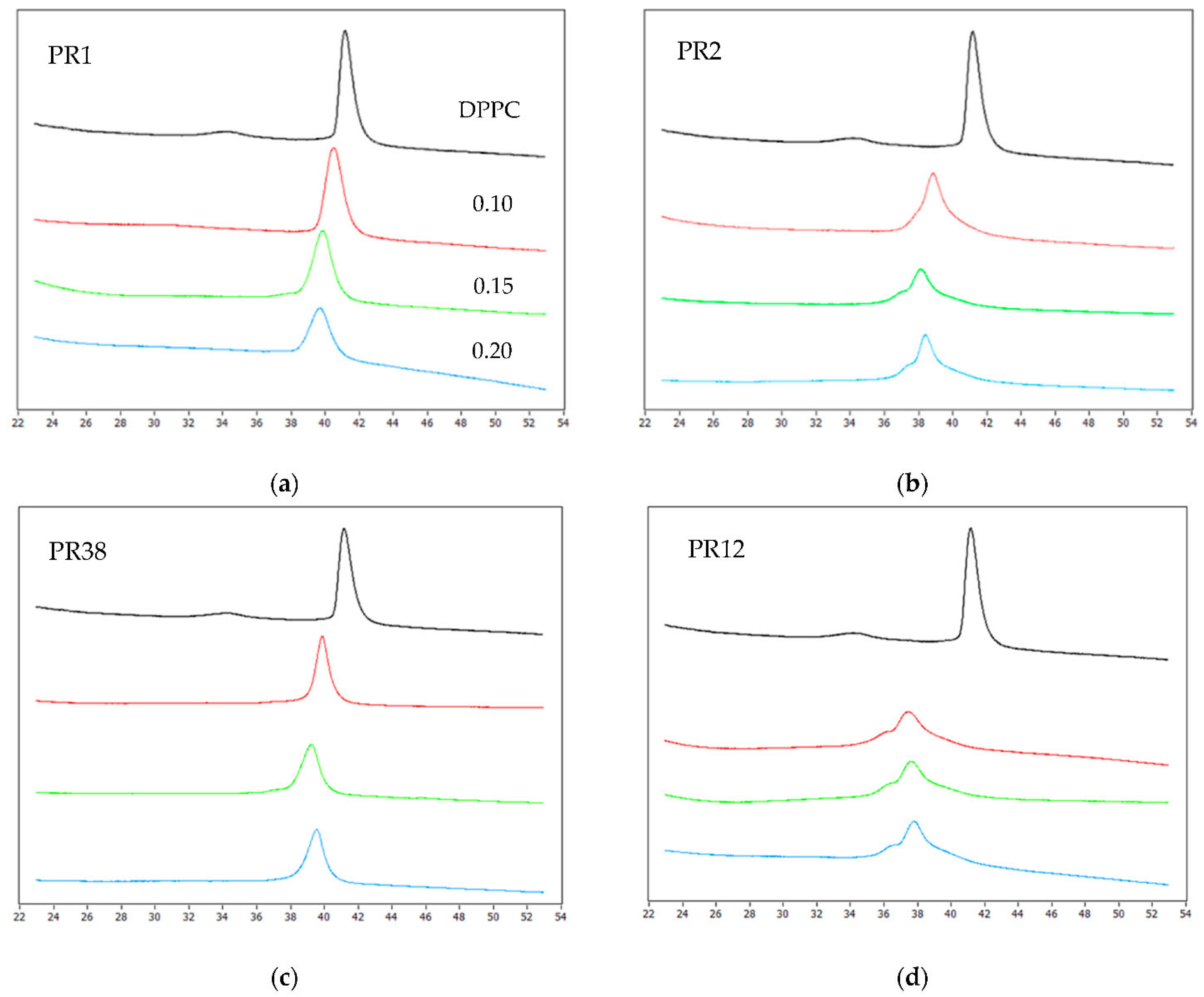
3.1.1. DSC Measurements of DPPC with Studied Compounds
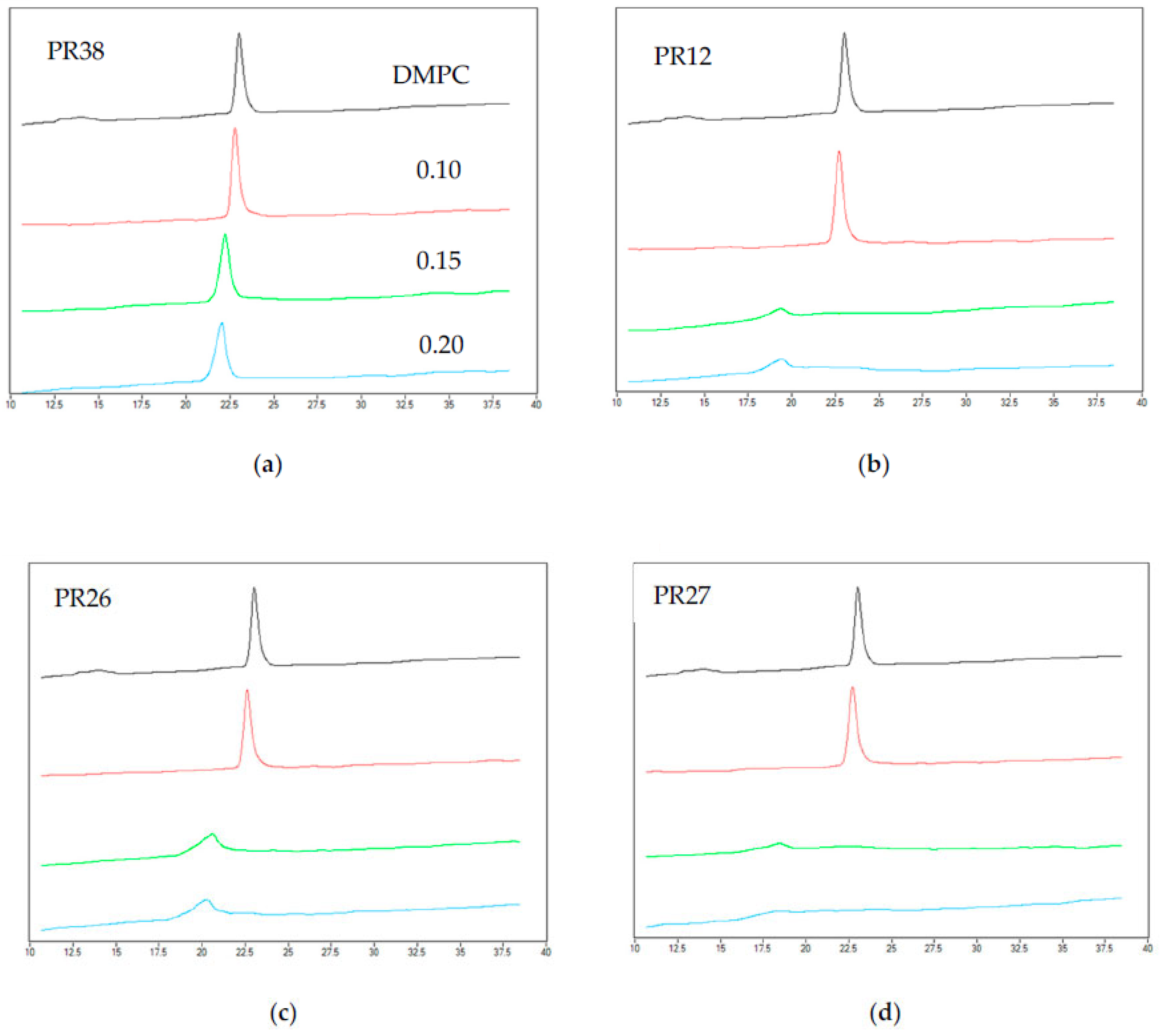
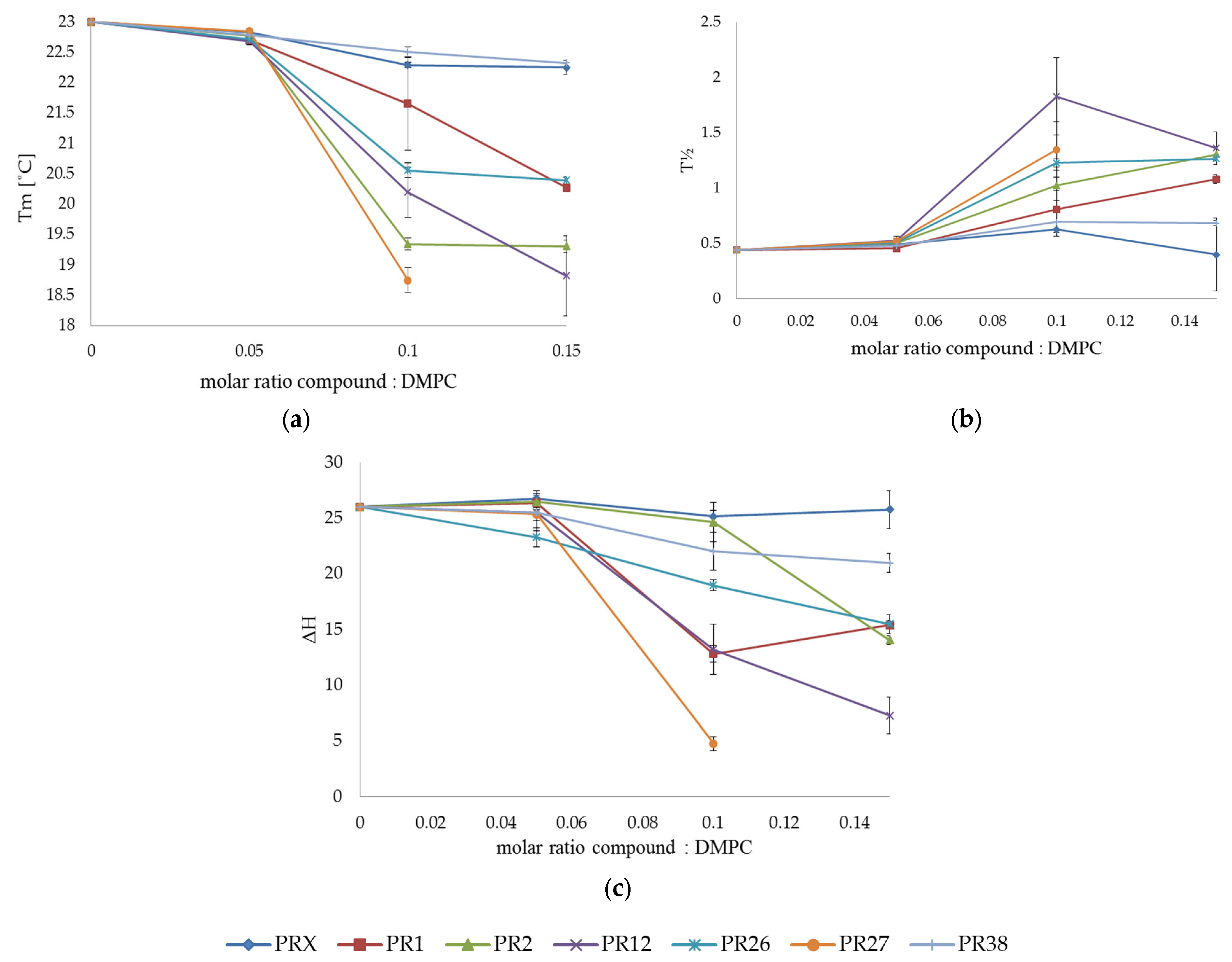
3.1.2. DSC Measurements of DMPC with Studied Compounds
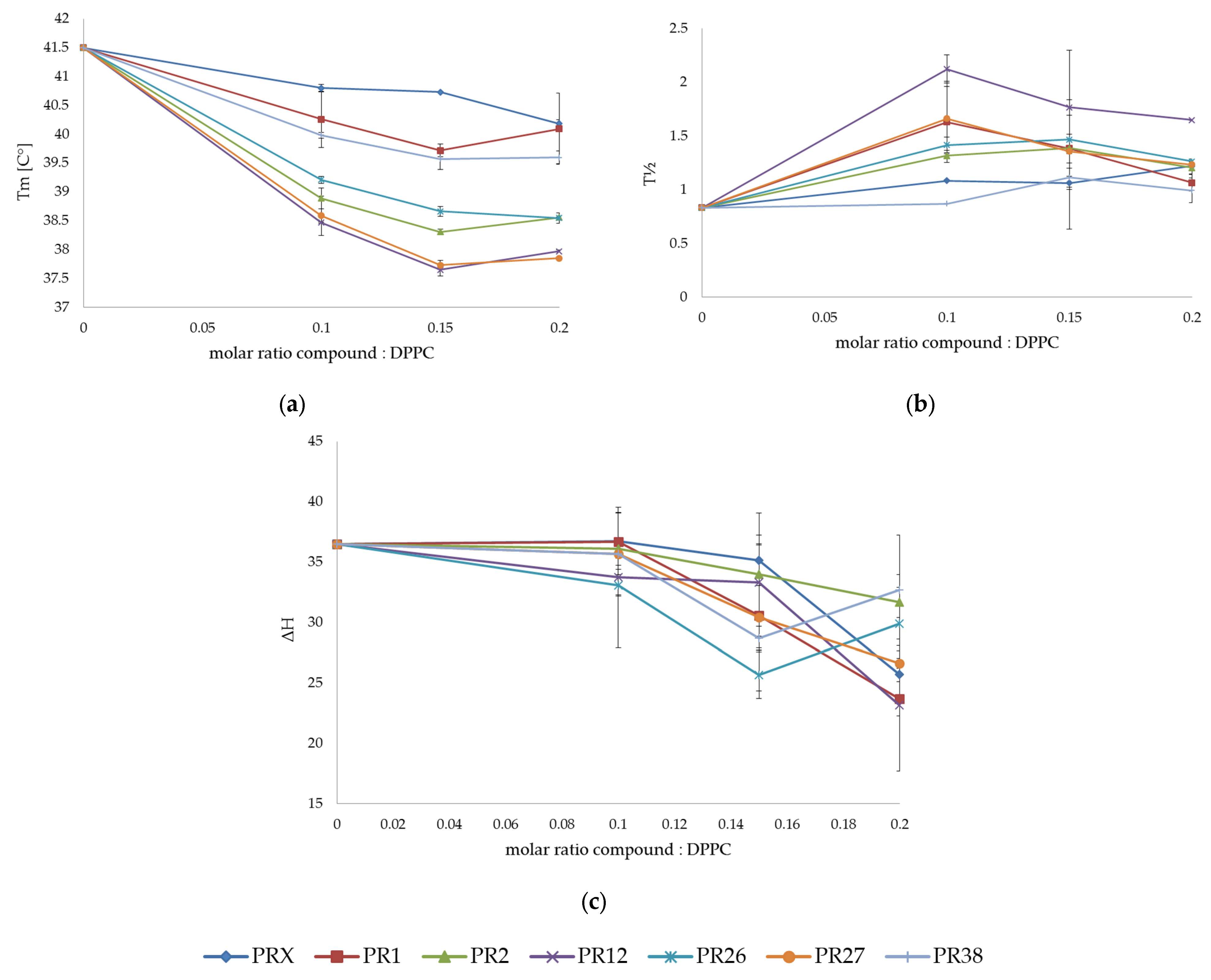
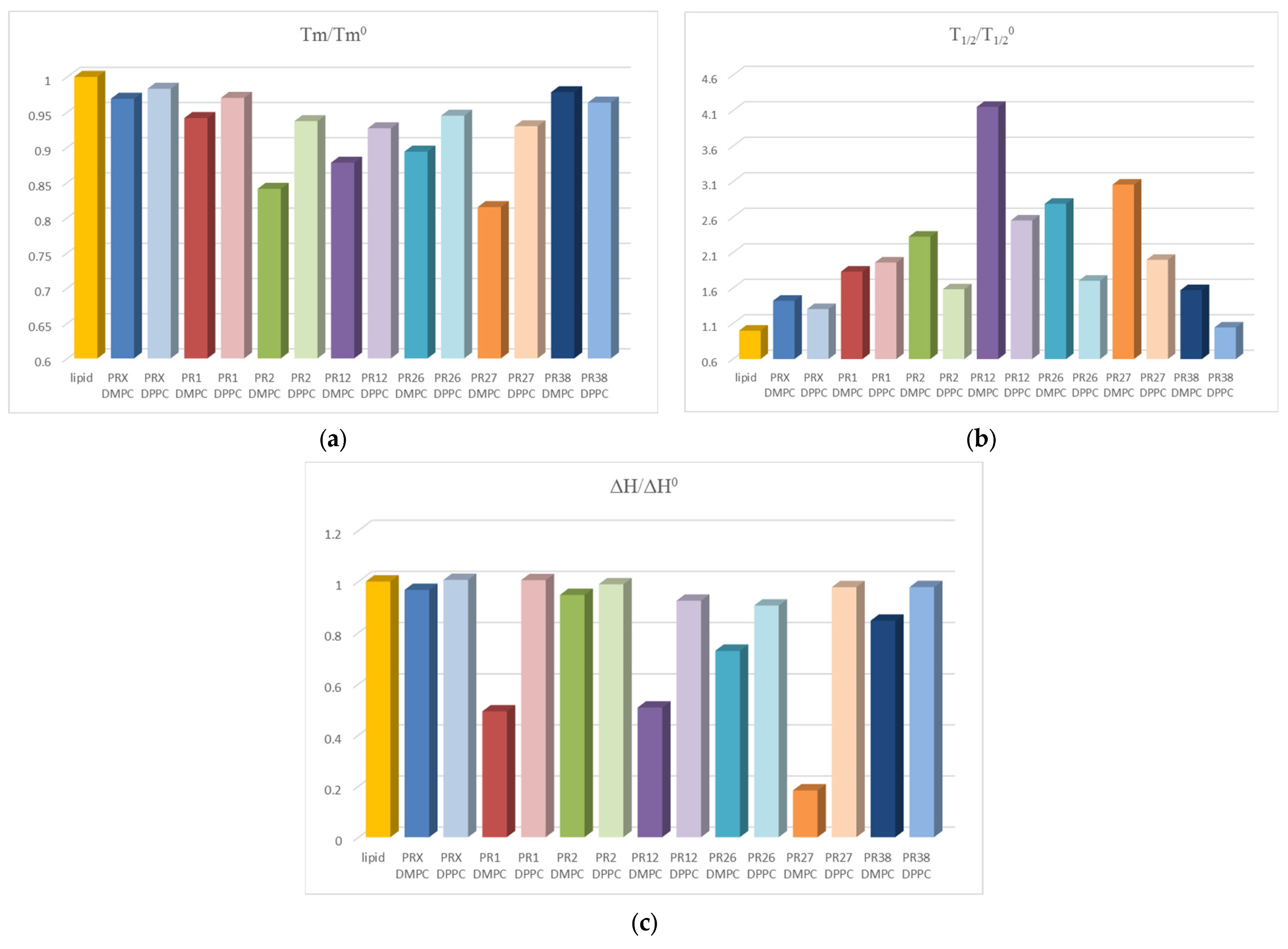
3.1.3. Comparison of the Results Obtained in DSC Studies for DPPC and DMPC
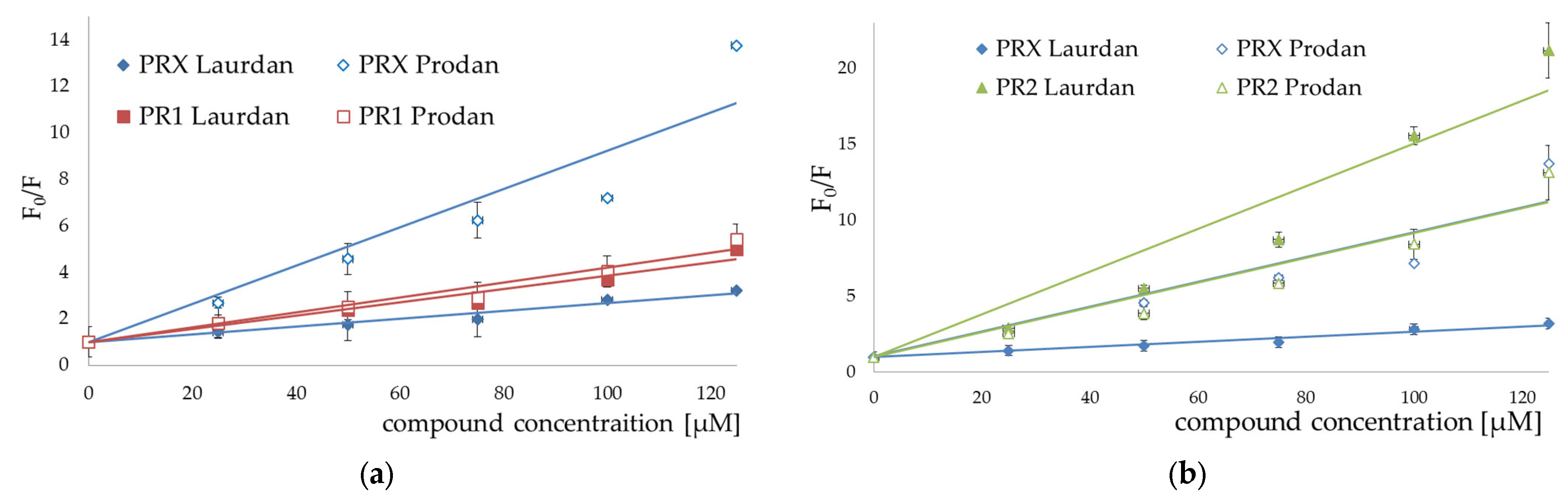
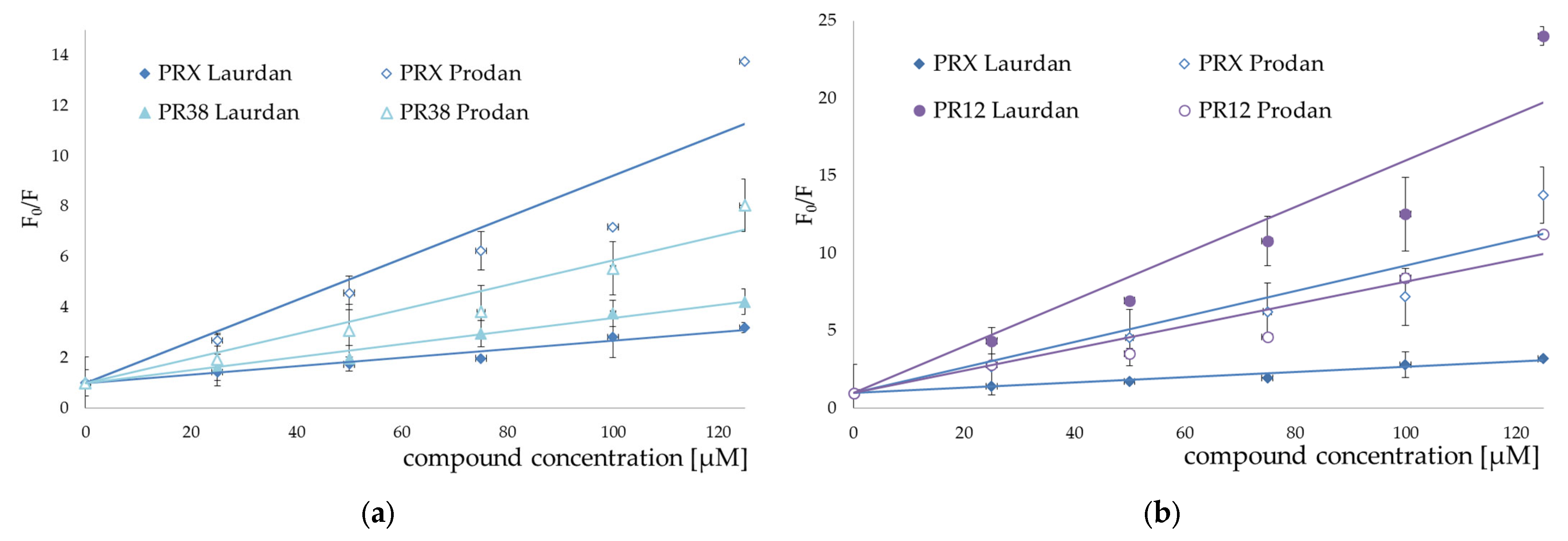
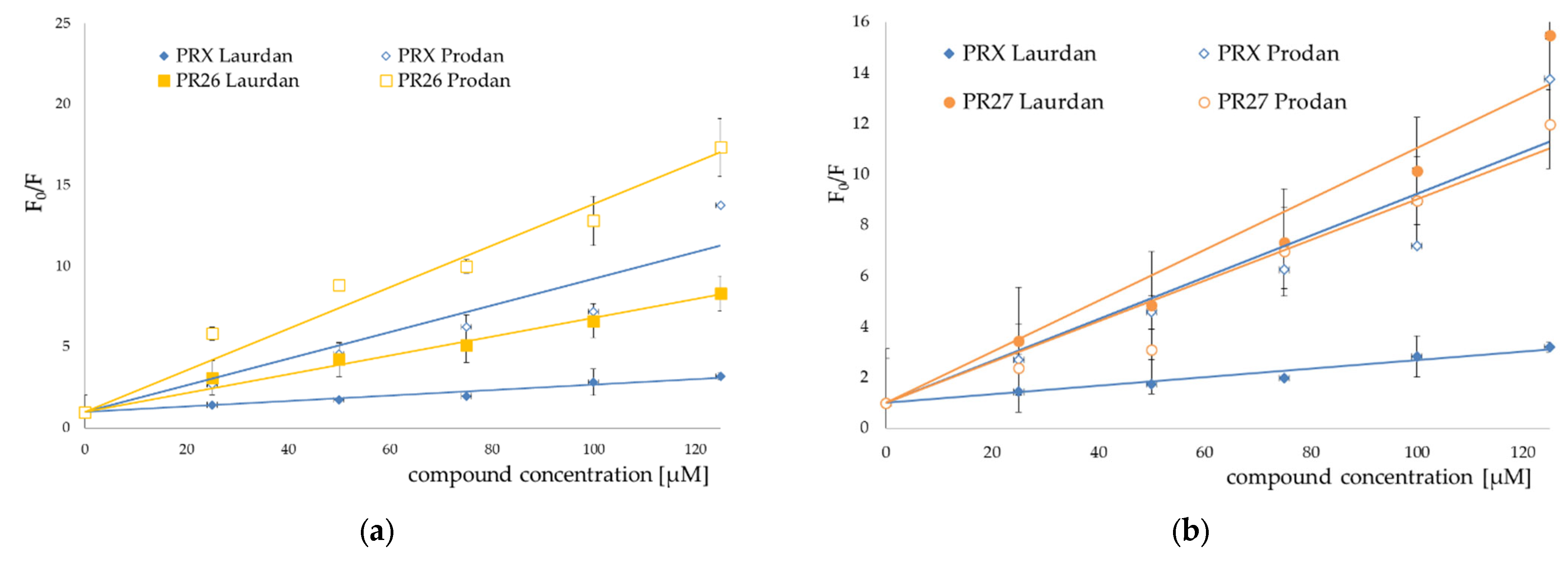
3.2. Fluorescence Spectroscopy
3.3. Prediction of ADMET Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mescola, A.; Ragazzini, G.; Alessandrini, A. Daptomycin Strongly Affects the Phase Behavior of Model Lipid Bilayers. J. Phys. Chem. B 2020, 124, 8562–8571. [Google Scholar] [CrossRef] [PubMed]
- Balleza, D.; Mescola, A.; Alessandrini, A. Model lipid systems and their use to evaluate the phase state of biomembranes, their mechanical properties and the effect of non - conventional antibiotics: The case of daptomycin. Eur. Biophys. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lucio, M.; Lima, J.L.F.C.; Reis, S. Drug-Membrane Interactions: Significance for Medicinal Chemistry. Curr. Med. Chem. 2010, 17, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Venerito, M.; Wex, T.; Malfertheiner, P. Nonsteroidal Anti-Inflammatory Drug-Induced Gastroduodenal Bleeding: Risk Factors and Prevention Strategies. Pharmaceuticals 2010, 3, 2225–2237. [Google Scholar] [CrossRef] [PubMed]
- Kaduševičius, E. Novel applications of nsaids: Insight and future perspectives in cardiovascular, neurodegenerative, diabetes and cancer disease therapy. Int. J. Mol. Sci. 2021, 22, 6637. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Keeton, A.B.; Tinsley, H.N.; Whitt, J.D.; Gary, B.D.; Mathew, B.; Singh, R.; Grizzle, W.E.; Reynolds, R.C. NSAIDs: Old Drugs Reveal New Anticancer Targets. Pharmaceuticals 2010, 3, 1652–1667. [Google Scholar] [CrossRef] [PubMed]
- Maniewska, J.; Jeżewska, D. Non-Steroidal Anti-Inflammatory Drugs in Colorectal Cancer Chemoprevention. Cancers 2021, 13, 594. [Google Scholar] [CrossRef]
- Baek, S.J.; Eling, T.; Kolawole, O.R.; Kashfi, K. NSAIDs and Cancer Resolution: New Paradigms beyond Cyclooxygenase. Int. J. Mol. Sci 2022, 2022, 1432. [Google Scholar]
- Kazberuk, A.; Chalecka, M.; Palka, J.; Surazynski, A. Nonsteroidal Anti-Inflammatory Drugs as PPARγ Agonists Can Induce PRODH/POX-Dependent Apoptosis in Breast Cancer Cells: New Alternative Pathway in NSAID-Induced Apoptosis. Int. J. Mol. Sci. 2022, 23, 1510. [Google Scholar] [CrossRef]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-inflammatory drugs as anticancer agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef]
- Moore, A.H.; Bigbee, M.J.; Boynton, G.E.; Wakeham, C.M.; Rosenheim, H.M.; Staral, C.J.; Morrissey, J.L.; Hund, A.K. Non-Steroidal Anti-Inflammatory Drugs in Alzheimer’s Disease and Parkinson’s Disease: Reconsidering the Role of Neuroinflammation. Pharmaceuticals 2010, 3, 1812–1841. [Google Scholar] [CrossRef]
- Pereira-Leite, C.; Nunes, C.; Reis, S. Interaction of nonsteroidal anti-inflammatory drugs with membranes: In vitro assessment and relevance for their biological actions. Prog. Lipid Res. 2013, 52, 571–584. [Google Scholar] [CrossRef]
- Luckey, M. Membrane Structural Biology; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Lichtenberger, L.M.; Wang, Z.M.; Romero, J.J.; Ulloa, C.; Perez, J.C.; Giraud, M.N.; Barreto, J.C. Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nature 1995, 2, 983–989. [Google Scholar] [CrossRef]
- Banerjee, R.; Chakraborty, H.; Sarkar, M. Photophysical studies of oxicam group of NSAIDs: Piroxicam, meloxicam and tenoxicam. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2003, 59, 1213–1222. [Google Scholar] [CrossRef]
- Chakraborty, H.; Sarkar, M. Interaction of piroxicam and meloxicam with DMPG/DMPC mixed vesicles: Anomalous partitioning behavior. Biophys. Chem. 2007, 125, 306–313. [Google Scholar] [CrossRef]
- Chakraborty, H.; Chakraborty, P.K.; Raha, S.; Mandal, P.C.; Sarkar, M. Interaction of piroxicam with mitochondrial membrane and cytochrome c. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1138–1146. [Google Scholar] [CrossRef]
- Kyrikou, I.; Hadjikakou, S.K.; Kovala-Demertzi, D.; Viras, K.; Mavromoustakos, T. Effects of non-steroid anti-inflammatory drugs in membrane bilayers. Chem. Phys. Lipids 2004, 132, 157–169. [Google Scholar] [CrossRef]
- Lúcio, M.; Bringezu, F.; Reis, S.; Lima, J.L.F.C.; Brezesinski, G. Binding of Nonsteroidal Anti-inflammatory Drugs to DPPC: Structure and Thermodynamic Aspects. Langmuir 2008, 24, 4132–4139. [Google Scholar] [CrossRef]
- Moreno, M.M.; Garidel, P.; Suwalsky, M.; Howe, J.; Brandenburg, K. The membrane-activity of Ibuprofen, Diclofenac, and Naproxen: A physico-chemical study with lecithin phospholipids. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1296–1303. [Google Scholar] [CrossRef]
- Nunes, C.; Brezesinski, G.; Lima, J.L.F.C.; Reis, S.; Lúcio, M. Synchrotron SAXS and WAXS study of the interactions of NSAIDS with lipid membranes. J. Phys. Chem. B 2011, 115, 8024–8032. [Google Scholar] [CrossRef]
- Wilkosz, N.; Rissanen, S.; Cyza, M.; Szybka, R.; Nowakowska, M.; Bunker, A.; Róg, T.; Kepczynski, M. Effect of piroxicam on lipid membranes: Drug encapsulation and gastric toxicity aspects. Eur. J. Pharm. Sci. 2017, 100, 116–125. [Google Scholar] [CrossRef]
- Pereira-Leite, C.; Figueiredo, M.; Burdach, K.; Nunes, C.; Reis, S. Unraveling the role of drug-lipid interactions in nsaids-induced cardiotoxicity. Membranes 2021, 11, 24. [Google Scholar] [CrossRef]
- Sun, X.; Xue, Z.; Yasin, A.; He, Y.; Chai, Y.; Li, J.; Zhang, K. Colorectal cancer and adjacent normal mucosa differ in apoptotic and inflammatory protein expression. Eng. Regen. 2022, 2, 279–287. [Google Scholar] [CrossRef]
- Knobloch, J.; Suhendro, D.K.; Zieleniecki, J.L.; Shapter, J.G.; Köper, I. Membrane-drug interactions studied using model membrane systems. Saudi J. Biol. Sci. 2015, 22, 714–718. [Google Scholar] [CrossRef]
- Krzyzak, E.; Szkatuła, D.; Szczȩśniak-Siȩga, B.; Malinka, W. Synthesis and DSC study a new pyridinedicarboximide diones derivatives, obtained under various conditions. J. Therm. Anal. Calorim. 2015, 120, 847–853. [Google Scholar] [CrossRef]
- Szczęśniak-Sięga, B.; Gębczak, K.; Gębarowski, T.; Maniewska, J. Synthesis, COX-1/2 inhibition and antioxidant activities of new oxicam analogues designed as potential chemopreventive agents. Acta Biochim. Pol. 2018, 65, 199–207. [Google Scholar] [CrossRef]
- Szczęśniak-Sięga, B.M.; Mogilski, S.; Wiglusz, R.J.; Janczak, J.; Maniewska, J.; Malinka, W.; Filipek, B. Synthesis and pharmacological evaluation of novel arylpiperazine oxicams derivatives as potent analgesics without ulcerogenicity. Bioorganic Med. Chem. 2019, 27, 1619–1628. [Google Scholar] [CrossRef]
- Maniewska, J.; Szcześniak-Si Ga, B.; Poła, A.; Sroda-Pomianek, K.; Malinka, W.; Michalak, K. The interaction of new piroxicam analogues with lipid bilayers-A calorimetric and fluorescence spectroscopic study. Acta Pol. Pharm. Drug Res. 2014, 71, 1004–1012. [Google Scholar]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Jain, M.K.; Wu, N.M. Effect of small molecules on the dipalmitoyl lecithin liposomal bilayer: III. Phase transition in lipid bilayer. J. Membr. Biol. 1977, 34, 157–201. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Quenching of Fluorescence. In Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 1999; pp. 237–265. [Google Scholar]
- Parasassi, T.; Krasnowska, E.K.; Bagatolli, L.; Gratton, E. Laurdan and Prodan as Polarity-Sensitive Fluorescent Membrane Probes. J. Fluoresc. 1998, 8, 365–373. [Google Scholar] [CrossRef]
- Raguz, M.; Brnjas-Kraljevic, J. Resolved Fluorescence Emission Spectra of PRODAN in Ethanol/Buffer Solvents. J. Chem. Inf. Model. 2005, 45, 1636–1640. [Google Scholar] [CrossRef] [PubMed]
- Bagatolli, L.A.; Parasassi, T.; Fidelio, G.D.; Gratton, E. A Model for the Interaction of 6-Lauroyl-2-(N,N-dimethylamino)naphthalene with Lipid Environments: Implications for Spectral Properties. Photochem. Photobiol. 1999, 70, 557. [Google Scholar] [CrossRef]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef]
- Clark, D.E. In silico prediction of blood-brain barrier permeation. Drug Discov. Today 2003, 8, 927–933. [Google Scholar] [CrossRef]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Escribá, P.V.; González-Ros, J.M.; Goñi, F.M.; Kinnunen, P.K.J.; Vigh, L.; Sánchez-Magraner, L.; Fernández, A.M.; Busquets, X.; Horváth, I.; Barceló-Coblijn, G. Membranes: A meeting point for lipids, proteins and therapies. J. Cell. Mol. Med. 2008, 12, 829–875. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Zhou, Y.; Jayaraman, V.; Doyen, J.R.; O’Neil, R.G.; Dial, E.J.; Volk, D.E.; Gorenstein, D.G.; Boggara, M.B.; Krishnamoorti, R. Insight into NSAID-induced membrane alterations, pathogenesis and therapeutics: Characterization of interaction of NSAIDs with phosphatidylcholine. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 994–1002. [Google Scholar] [CrossRef]
- Szczęśniak-Sięga, B.; Maniewska, J.; Poła, A.; Środa-Pomianek, K.; Malinka, W.; Michalak, K. Synthesis of new piroxicam derivatives and their influence on lipid bilayers. Acta Pol. Pharm. Drug Res. 2014, 71, 1045–1050. [Google Scholar]
- Maniewska, J.; Gąsiorowska, J.; Szczęśniak-Sięga, B.; Michalak, K. The interaction of new oxicam derivatives with lipid bilayers as measured by calorimetry and fluorescence spectroscopy. Acta Biochim. Pol. 2018, 65, 185–191. [Google Scholar] [CrossRef]
- Szczęśniak-Sięga, B.M.; Wiatrak, B.; Czyżnikowska, Ż.; Janczak, J.; Wiglusz, R.J.; Maniewska, J. Synthesis and biological evaluation as well as in silico studies of arylpiperazine-1,2-benzothiazine derivatives as novel anti-inflammatory agents. Bioorg. Chem. 2021, 106, 104476. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Symbol | Chemical Structure |
|---|---|
| PR1 |  |
| PR2 |  |
| PR12 |  |
| PR26 |  |
| PR27 |  |
| PR38 |  |
| PRX (piroxicam) |  |
| Parameter/ Optimal Value | Compound | |||||
|---|---|---|---|---|---|---|
| PR1 | PR2 | PR12 | PR26 | PR27 | PR38 | |
| MW (molecular weight) optimal 100–600 | 503.19 | 503.15 | 521.14 | 521.18 | 521.14 | 521.18 |
| nHA (number of hydrogen bond acceptors) optimal 0–12 | 7 | 8 | 8 | 7 | 8 | 7 |
| nHD (number of hydrogen bond donors) optimal 0–7 | 0 | 0 | 0 | 0 | 0 | 0 |
| TPSA (topological polar surface area) optimal 0–140 | 78 | 95 | 95 | 78 | 95 | 78 |
| nRot (number of rotatable bonds) optimal 0–11 | 7 | 6 | 6 | 7 | 6 | 7 |
| nRing (number of rings) optimal 0–6 | 5 | 5 | 5 | 5 | 5 | 5 |
| nHet (number of heteroatoms) optimal 1–15 | 8 | 9 | 10 | 9 | 10 | 9 |
| logP (log of the octanol/water partition coefficient) optimal 0–3 | 3.9 | 3.1 | 3.3 | 4.0 | 3.3 | 4.0 |
| logD (logP at physiological pH) optimal 1–3 | 3.3 | 2.4 | 2.5 | 3.3 | 2.4 | 3.2 |
| Parameter/ Optimal Value | Compound | |||||
| PR1 | PR2 | PR12 | PR26 | PR27 | PR38 | |
| QED (measure of drug-likeness based on the concept of desirability; attractive > 0.67, unattractive 0.49–0.67, too complex < 0.34) | 0.4 | 0.4 | 0.4 | 0.3 | 0.4 | 0.3 |
| SA score (synthetic accessibility score is designed to estimate ease of synthesis of drug-like molecules; ≥6—difficult, <6—easy to synthesize) | 3.0 | 3.0 | 3.0 | 3.1 | 3.0 | 3.1 |
| Fsp3 (number of sp3 hybridized carbons/total carbon count, correlating with melting point and solubility; ≥0.42 is considered a suitable value) | 0.3 | 0.2 | 0.2 | 0.3 | 0.2 | 0.2 |
| Lipinski Rule (MW ≤ 500; logP ≤ 5; Hacc ≤ 10; Hdon ≤ 5; if two properties are out of range, a poor absorption or permeability is possible, one is acceptable) | all accepted | |||||
| Pfizer Rule (compounds with a high log p (>3) and low TPSA (<75) are likely to be toxic) | all accepted | |||||
| GSK Rule (MW ≤ 400; logP ≤ 4; compounds satisfying the GSK rule may have a more favorable ADMET profile) | all rejected | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maniewska, J.; Czyżnikowska, Ż.; Szczęśniak-Sięga, B.M.; Michalak, K. Interaction of Oxicam Derivatives with the Artificial Models of Biological Membranes—Calorimetric and Fluorescence Spectroscopic Study. Membranes 2022, 12, 791. https://doi.org/10.3390/membranes12080791
Maniewska J, Czyżnikowska Ż, Szczęśniak-Sięga BM, Michalak K. Interaction of Oxicam Derivatives with the Artificial Models of Biological Membranes—Calorimetric and Fluorescence Spectroscopic Study. Membranes. 2022; 12(8):791. https://doi.org/10.3390/membranes12080791
Chicago/Turabian StyleManiewska, Jadwiga, Żaneta Czyżnikowska, Berenika M. Szczęśniak-Sięga, and Krystyna Michalak. 2022. "Interaction of Oxicam Derivatives with the Artificial Models of Biological Membranes—Calorimetric and Fluorescence Spectroscopic Study" Membranes 12, no. 8: 791. https://doi.org/10.3390/membranes12080791