
Nanofibrous Online Solid-Phase Extraction Coupled with Liquid Chromatography for the Determination of Neonicotinoid Pesticides in River Waters


 , , , , , and
, , , , , and
Abstract
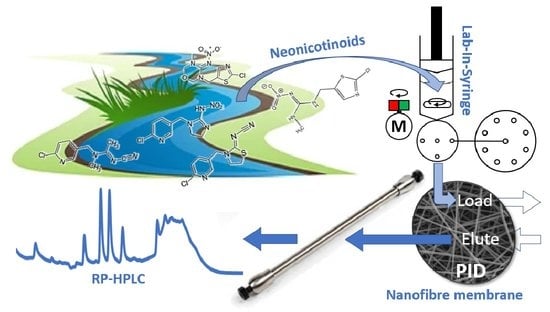
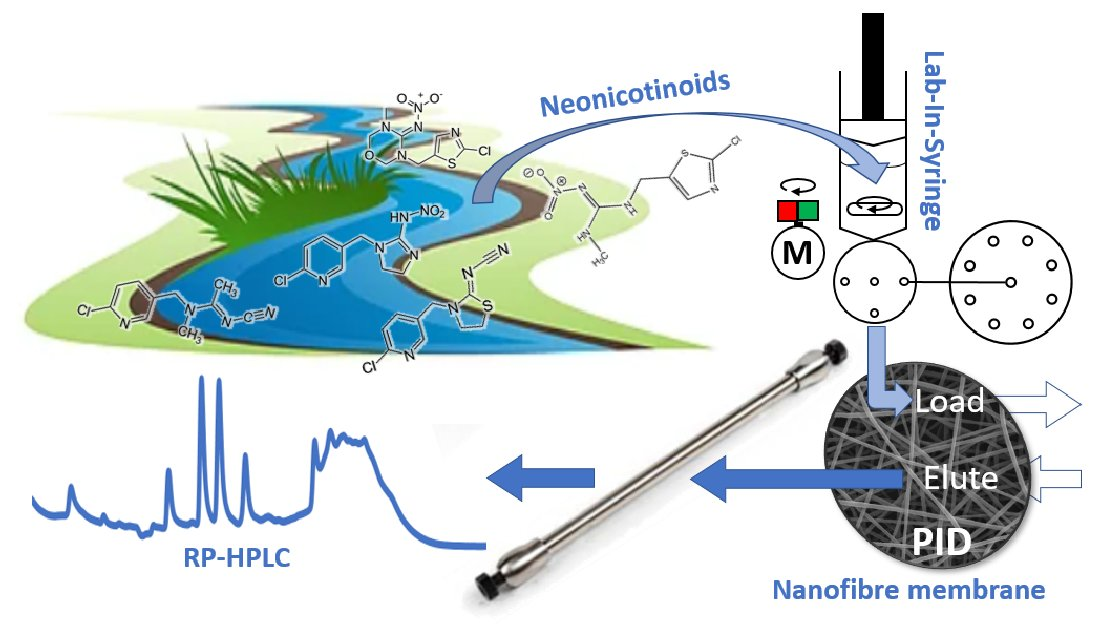
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Samples
2.2. Preparation of Nanofibers
2.3. Instrumentation
2.4. Operation
2.5. Instrumentation and Methodology Used for PID Fiber Characterization
3. Results
3.1. HPLC Conditions
3.2. Mat Holder
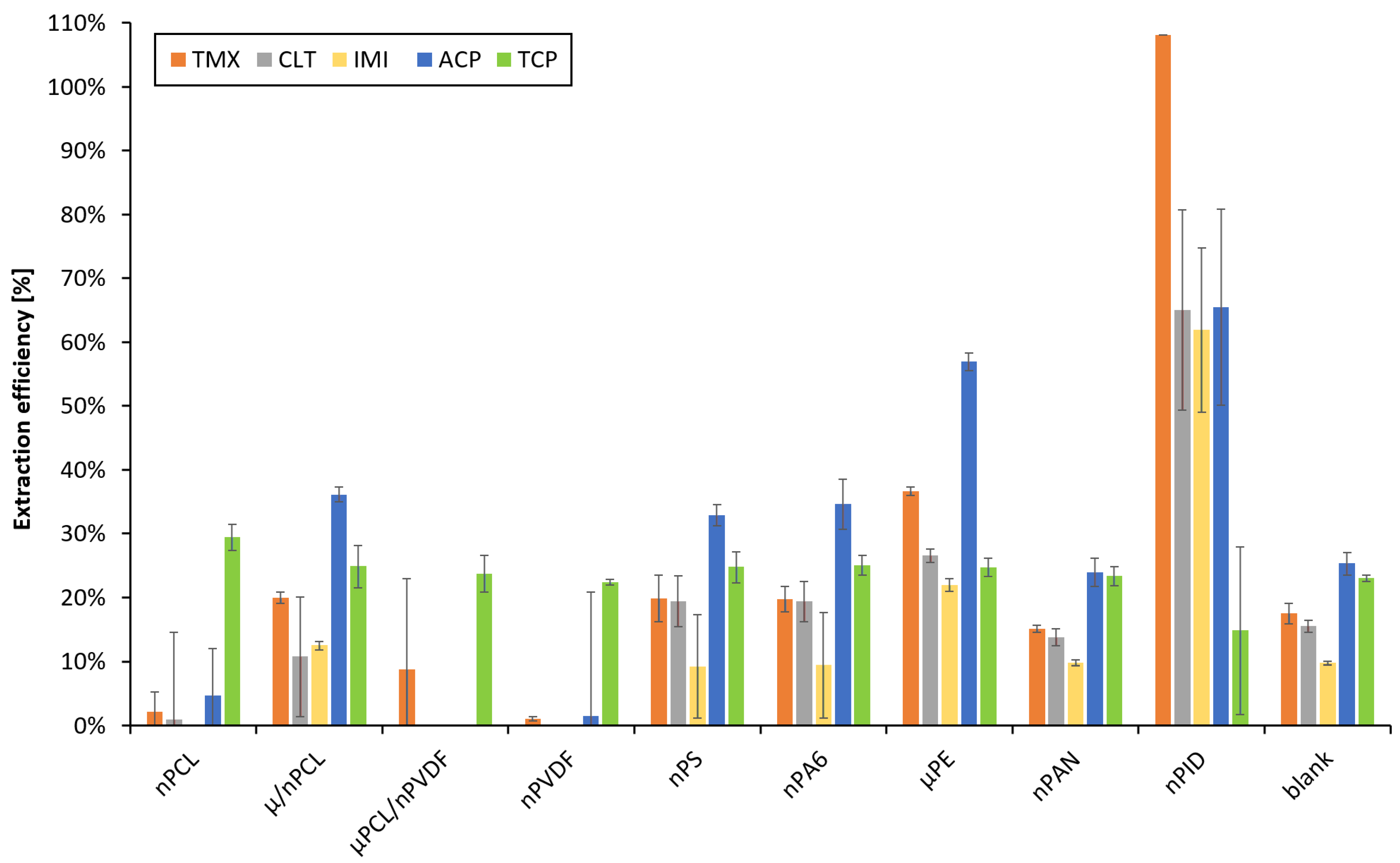
3.3. Selection of Nanofibers
3.4. Online SPE Conditions
3.4.1. Sample Volume
3.4.2. Flow Rate of Sample Loading
3.4.3. Number of PID Layers
3.4.4. Loading pH and Salt Addition
3.4.5. Washing Solution
3.5. Analytical Figures of Merit and Analysis of Surface Waters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Matsuda, K.; Buckingham, S.D.; Kleier, D.; Rauh, J.J.; Grauso, M.; Sattelle, D.B. Neonicotinoids: Insecticides acting on insect nicotinic acetylcholine receptors. Trends Pharmacol. Sci. 2001, 22, 573–580. [Google Scholar] [CrossRef]
- Kundoo, A.A.; Dar, S.A.; Mushtaq, M.; Bashir, Z.; Dar, M.S.; Gul, S.; Ali, M.T.; Gulzar, S. Role of neonicotinoids in insect pest management: A review. J. Entomol. Zool. Stud. 2018, 6, 333–339. [Google Scholar]
- Craddock, H.A.; Huang, D.; Turner, P.C.; Quirós-Alcalá, L.; Payne-Sturges, D.C. Trends in neonicotinoid pesticide residues in food and water in the United States, 1999–2015. Environ. Health 2019, 18, 7–22. [Google Scholar] [CrossRef] [Green Version]
- Elbert, A.; Haas, M.; Springer, B.; Thielert, W.; Nauen, R. Applied aspects of neonicotinoid uses in crop protection. Pest. Manag. Sci. 2008, 64, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P.; Nauen, R.; Schindler, M.; Elbert, A. Overview of the Status and Global Strategy for Neonicotinoids. J. Agric. Food Chem. 2011, 59, 2897–2908. [Google Scholar] [CrossRef] [PubMed]
- Friedli, A.; Williams, G.R.; Bruckner, S.; Neumann, P.; Straub, L. The weakest link: Haploid honey bees are more susceptible to neonicotinoid insecticides. Chemosphere 2020, 242, 125145. [Google Scholar] [CrossRef]
- Sánchez-Bayo, F. Environmental science. The trouble with neonicotinoids. Science 2014, 346, 806–807. [Google Scholar] [CrossRef]
- European Commission. Commission Implementing Regulation (EU) No 485/2013 of 24 May 2013 amending Implementing Regulation (EU) No 540/2011, as regards the conditions of approval of the active substances clothianidin, thiamethoxam and imidacloprid, and prohibiting the use and sale of seeds treated with plant protection products containing those active substances. Off. J. Eur. Union 2013, 139, 12–26. [Google Scholar]
- Food Safety-European Commission. 2019. Available online: https://ec.europa.eu/food/plant/pesticides/approval_active_substances/approval_renewal/neonicotinoids_en (accessed on 13 December 2021).
- Van der Sluijs, J.P.; Amaral-Rogers, V.; Belzunces, L.P.; Bijleveld van Lexmond, M.F.I.J.; Bonmatin, J.M.; Chagnon, M.; Downs, C.A.; Furlan, L.; Gibbons, D.W.; Giorio, C.; et al. Conclusions of the worldwide integrated assessment on the risks of neonicotinoids and fipronil to biodiversity and ecosystem functioning. Environ. Sci. Pollut. Res. Int. 2015, 22, 148–154. [Google Scholar] [CrossRef]
- Abdel-Ghany, M.F.; Hussein, L.A.; El Azab, N.F. Multiresidue analysis of five neonicotinoid insecticides and their primary metabolite in cucumbers and soil using high-performance liquid chromatography with diode-array detection. J. AOAC Int. 2017, 100, 176–188. [Google Scholar] [CrossRef]
- Campillo, N.; Viñas, P.; Férez-Melgarejo, G.; Hernández-Córdoba, M. Liquid chromatography with diode array detection and tandem mass spectrometry for the determination of neonicotinoid insecticides in honey samples using dispersive liquid-liquid microextraction. J. Agric. Food Chem. 2013, 61, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Rozas, L.; Lara, F.J.; del Olmo Iruela, M.; García-Campaña, A.M. A novel approach based on capillary liquid chromatography for the simultaneous determination of neonicotinoid residues in cereal samples. Microchem. J. 2021, 161, 105756. [Google Scholar] [CrossRef]
- Martel, A.-C.; Lair, C. Validation of a highly sensitive method for the determination of neonicotinoid insecticides residues in honeybees by liquid chromatography with electrospray tandem mass spectrometry. Intern. J. Environm. Anal. Chem. 2011, 91, 978–988. [Google Scholar] [CrossRef]
- Suganthi, A.; Bhuvaneswari, K.; Ramya, M. Determination of neonicotinoid insecticide residues in sugarcane juice using LCMSMS. Food Chem. 2018, 241, 275–280. [Google Scholar] [CrossRef]
- Valverde, S.; Ares, A.M.; Arribas, M.; Bernal, J.L.; Nozal, M.J.; Bernal, J. Development and validation of UHPLC–MS/MS methods for determination of neonicotinoid insecticides in royal jelly-based products. J. Food Compos. Anal. 2018, 70, 105–113. [Google Scholar] [CrossRef]
- Da Silva Sousa, J.; Oliveira do Nascimento, H.; Hiago de Oliveira Gomes, H.; Ferreira do Nascimento, R. Pesticide residues in groundwater and surface water: Recent advances in solid-phase extraction and solid-phase microextraction sample preparation methods for multiclass analysis by gas chromatography-mass spectrometry. Microchem. J. 2021, 168, 106359. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, X.; Yin, X.; Wang, C.; Wang, Z. Dispersive liquid–liquid microextraction combined with sweeping micellar electrokinetic chromatography for the determination of some neonicotinoid insecticides in cucumber samples. Food Chem. 2012, 133, 544–550. [Google Scholar] [CrossRef]
- Jovanov, P.; Guzsvany, V.; Franko, M.; Lazic, S.; Sakac, M.; Saric, B.; Banjac, V. Multi-residue method for determination of selected neonicotinoid insecticides in honey using optimized dispersive liquid-liquid microextraction combined with liquid chromatography-tandem mass spectrometry. Talanta 2013, 111, 125–133. [Google Scholar] [CrossRef]
- Lachat, L.; Glauser, G. Development and Validation of an ultra-sensitive UHPLC-MS/MS method for neonicotinoid analysis in milk. J. Agric. Food Chem. 2018, 66, 8639–8646. [Google Scholar] [CrossRef]
- David, A.; Botias, C.; Abdul-Sada, A.; Goulson, D.; Hill, E.M. Sensitive determination of mixtures of neonicotinoid and fungicide residues in pollen and single bumblebees using a scaled down QuEChERS method for exposure assessment. Anal. Bioanal. Chem. 2015, 407, 8151–8162. [Google Scholar] [CrossRef]
- Song, S.; Zhang, C.; Chen, Z.; He, F.; Wei, J.; Tan, H.; Li, X. Simultaneous determination of neonicotinoid insecticides and insect growth regulators residues in honey using LC-MS/MS with anion exchanger-disposable pipette extraction. J. Chromatogr. A 2018, 1557, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, Y.; Xu, Z.; Ji, Y.; Si, X.; Lin, T.; Liu, H.; Liu, Z. Generic imprinted fiber array strategy for high-throughput and ultrasensitive simultaneous determination of multiple neonicotinoids. Food Chem. 2022, 382, 132407. [Google Scholar] [CrossRef] [PubMed]
- Di Ottavio, F.; Della Pelle, F.; Montesano, C.; Scarpone, R.; Escarpa, A.; Compagnone, D.; Sergi, M. Determination of Pesticides in Wheat Flour Using Microextraction on Packed Sorbent Coupled to Ultra-High Performance Liquid Chromatography and Tandem Mass Spectrometry. Food Anal. Methods 2017, 10, 1699–1708. [Google Scholar] [CrossRef] [Green Version]
- Ettiene, G.; Bauza, R.; Plata, M.R.; Contento, A.M.; Ríos, Á. Determination of neonicotinoid insecticides in environmental samples by micellar electrokinetic chromatography using solid-phase treatments. Electrophoresis 2012, 33, 2969–2977. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Rozas, L.; Lara, F.J.; del Olmo Iruela, M.; García-Campaña, A.M. Micellar electrokinetic chromatography as efficient alternative for the multiresidue determination of seven neonicotinoids and 6-chloronicotinic acid in environmental samples. Anal. Bioanal. Chem. 2020, 412, 6231–6240. [Google Scholar] [CrossRef]
- Di Muccio, A.; Fidente, P.; Barbini, D.A.; Dommarco, R.; Seccia, S.; Morrica, P. Application of solid-phase extraction and liquid chromatography-mass spectrometry to the determination of neonicotinoid pesticide residues in fruit and vegetables. J. Chromatogr. A 2006, 1108, 1–6. [Google Scholar] [CrossRef]
- Yáñez, K.P.; Bernal, J.L.; Nozal, M.J.; Martín, M.T.; Bernal, J. Determination of seven neonicotinoid insecticides in beeswax by liquid chromatography coupled to electrospray-mass spectrometry using a fused-core column. J. Chromatogr. A 2013, 1285, 110–117. [Google Scholar] [CrossRef]
- Montiel-León, J.M.; Duy, S.V.; Munoz, G.; Amyot, M.; Sauvé, S. Evaluation of on-line concentration coupled to liquid chromatography tandem mass spectrometry for the quantification of neonicotinoids and fipronil in surface water and tap water. Anal. Bioanal. Chem. 2018, 410, 2765–2779. [Google Scholar] [CrossRef]
- Háková, M.; Havlíková, L.C.; Chvojka, J.; Erben, J.; Solich, P.; Švec, F. Šatínský, D. Polycaprolactone nanofibers functionalized with a dopamine coating for on-line solid phase extraction of bisphenols, betablockers, nonsteroidal drugs, and phenolic acids. Microchim. Acta 2019, 186, 710. [Google Scholar] [CrossRef]
- Háková, M.; Chocholouš, P.; Valachovič, A.; Erben, J.; Chvojka, J.; Solich, P.; Švec, F.; Šatínský, D. On-line polydopamine coating as a new way to functionalize polypropylene fiber sorbent for solid phase extraction. Talanta 2020, 219, 121189. [Google Scholar] [CrossRef]
- Háková, M.; Havlíková, L.C.; Švec, F.; Solich, P.; Šatínský, D. Nanofibers as advanced sorbents for on-line solid phase extraction in liquid chromatography: A tutorial. Anal. Chim. Acta 2020, 1121, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Šrámková, I.H.; Carbonell-Rozas, L.; Horstkotte, B.; Háková, M.; Erben, J.; Chvojka, J.; Švec, F.; Solich, P.; García-Campaña, A.M.; Šatínský, D. Screening of extraction properties of nanofibers in a sequential injection analysis system using a 3D printed device. Talanta 2019, 197, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Maya, F.; Horstkotte, B.; Estela, J.M.; Cerda, V. Lab in a syringe: Fully automated dispersive liquid-liquid microextraction with integrated spectrophotometric detection. Anal. Bioanal. Chem. 2012, 404, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Horstkotte, B.; Solich, P. The automation technique Lab-In-Syringe: A practical guide. Molecules 2020, 25, 1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šrámková, I.H.; Horstkotte, B.; Erben, J.; Chvojka, J.; Švec, F.; Solich, P.; Šatínský, D. 3D-printed magnetic stirring cages for semidispersive extraction of bisphenols from water using polymer micro- and nanofibers. Anal. Chem. 2020, 92, 3964–3971. [Google Scholar] [CrossRef]
- Háková, M.; Havlíková, L.C.; Chvojka, J.; Švec, F.; Solich, P.; Šatínský, D. Nanofiber polymers as novel sorbents for on-line solid phase extraction in chromatographic system: A comparison with monolithic reversed phase C18 sorbent. Anal. Chim. Acta 2018, 1018, 26–34. [Google Scholar] [CrossRef]
- Háková, M.; Havlíková, L.C.; Chvojka, J.; Erben, J.; Solich, P.; Švec, F.; Šatínský, D. A comparison study of nanofiber, microfiber, and new composite nano/microfiber polymers used as sorbents for on-line solid phase extraction in chromatography system. Anal. Chim. Acta 2018, 1023, 44–52. [Google Scholar] [CrossRef]
- Háková, M.; Havlíková, L.C.; Švec, F.; Solich, P.; Erben, J.; Chvojka, J.; Šatínský, D. Novel nanofibrous sorbents for the extraction and determination of resveratrol in wine. Talanta 2020, 206, 120181. [Google Scholar] [CrossRef]
- Fikarová, K.; Horstkotte, B.; Machián, D.; Sklenářová, H.; Solich, P. Lab-In-Syringe for automated double-stage sample preparation by coupling salting out liquid-liquid extraction with online solid-phase extraction and liquid chromatographic separation for sulfonamide antibiotics from urine. Talanta 2021, 221, 121427. [Google Scholar] [CrossRef]
- Cocovi-Solberg, D.J.; Miro, M. CocoSoft: Educational software for automation in the analytical chemistry laboratory. Anal. Bioanal. Chem. 2015, 407, 6227–6233. [Google Scholar] [CrossRef] [Green Version]
- Vichapong, J.; Burakham, R.; Srijaranai, S. Vortex-assisted surfactant-enhanced-emulsification liquid-liquid microextraction with solidification of floating organic droplet combined with HPLC for the determination of neonicotinoid pesticides. Talanta 2013, 117, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Pihlström, T.; Fernández-Alba, A.R.; Gamón, M.; Amate, C.F.; Poulsen, M.E.; Lippold, R.; Anastassiades, M. Analytical quality control and method validation procedures for pesticide residues analysis in food and feed. Sante 2017, 11813, 21–22. Available online: https://www.eurl-pesticides.eu/userfiles/file/EurlALL/SANTE_11312_2021.pdf (accessed on 2 April 2022).
- Kachangoon, R.; Vichapong, J.; Burakham, R.; Santaladchaiyakit, Y.; Srijaranai, S. Ultrasonically modified amended-cloud point extraction for simultaneous pre-concentration of neonicotinoid insecticide residues. Molecules 2018, 23, 1165. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yang, X.; Wang, J.; Cui, J.; Dong, A.J.; Zhao, H.T.; Zhang, L.W.; Wang, Z.Y.; Xu, R.B.; Li, W.J.; et al. Multi-residue method for determination of seven neonicotinoid insecticides in grains using dispersive solid-phase extraction and dispersive liquid-liquid micro-extraction by high performance liquid chromatography. Food Chem. 2012, 134, 1691–1698. [Google Scholar] [CrossRef]
- Moyakao, K.; Santaladchaiyakit, Y.; Srijaranai, S.; Vichapong, J. Preconcentration of trace neonicotinoid insecticide residues using vortex-assisted dispersive micro solid-phase extraction with montmorillonite as an efficient sorbent. Molecules 2018, 23, 883. [Google Scholar] [CrossRef] [Green Version]
- Badawy, M.E.I.; Ismail, A.M.E.; Ibrahim, A.I.H. Quantitative analysis of acetamiprid and imidacloprid residues in tomato fruits under greenhouse conditions. J. Environm. Sci. Health Part B 2019, 54, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Jovanov, P.; Guzsvány, V.; Lazić, S.; Franko, M.; Sakač, M.; Šarić, L.; Kos, J. Development of HPLC-DAD method for determination of neonicotinoids in honey. J. Food Compos. Anal. 2015, 40, 106–113. [Google Scholar] [CrossRef]
- Mahdavi, V.; Garshasbi, Z.; Farimani, M.M.; Farhadpour, M.; Aboul-Enein, H.Y. Health risk assessment of neonicotinoid insecticide residues in pistachio using a QuEChERS-based method in combination with HPLC-UV. Biomed. Chromatogr. 2020, 34, e4747. [Google Scholar] [CrossRef]
- Farajzadeh, M.A.; Bamorowat, M.; Mogaddam, M.R.A. Ringer tablet-based ionic liquid phase microextraction: Application in extraction and preconcentration of neonicotinoid insecticides from fruit juice and vegetable samples. Talanta 2016, 160, 211–216. [Google Scholar] [CrossRef]
- Vichapong, J.; Burakham, R.; Santaladchaiyakit, Y.; Srijaranai, S. A preconcentration method for analysis of neonicotinoids in honey samples by ionic liquid-based cold-induced aggregation microextraction. Talanta 2016, 155, 216–221. [Google Scholar] [CrossRef]
- Chen, W.; Wu, S.; Zhang, J.; Yu, F.; Hou, J.; Miao, X.; Tu, X. Matrix-induced sugaring-out: A simple and rapid sample preparation method for the determination of neonicotinoid pesticides in honey. Molecules 2019, 24, 2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogaddam, M.R.A.; Farajzadeh, M.A.; Khodadadeian, F.; Nemati, M.; Mohebbi, A. Development of simultaneously salt and ultrasonic-assisted liquid phase microextraction for the extraction of neonicotinoid insecticides from fresh fruit juices and fruit juices. Int. J. Environ. Anal. Chem. 2020, 102, 1697–1708. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, Y.; Li, H.; Zeng, E.Y.; You, J. Application of Box–Behnken design to optimize multi-sorbent solid phase extraction for trace neonicotinoids in water containing high level of matrix substances. Talanta 2017, 170, 392–398. [Google Scholar] [CrossRef]
- Iancu, V.-I.; Radum, G.-L. Occurrence of neonicotinoids in waste water from the Bucharest treatment plant. Anal. Methods 2018, 10, 2691–2700. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Sensitivity/Calibration Slope (n = 3) [mV·L·µg·−1] | Linear Range [µg·L−1] | LOD [µg·L−1] | LOQ [µg·L−1] | Repeatability (% RSD, n = 3, 10 µg·L−1 Level) | Preconcentration Factor |
|---|---|---|---|---|---|---|
| TMX | 2.16 × 103 ± 24 | 2.0–100.0 | 0.55 | 1.82 | 6.8 | 70 |
| CLT | 6.50 × 103 ± 240 | 2.0–100.0 | 0.56 | 1.88 | 0.4 | 76 |
| IMI | 7.00 × 103 ± 56 | 2.3–100.0 | 0.67 | 2.25 | 2.8 | 82 |
| ACP | 2.90 × 103 ± 225 | 5.5–100.0 | 1.65 | 5.49 | 4.4 | 81 |
| TCP | 2.35 × 103 ± 119 | 1.0–100.0 | 0.36 | 1.21 | 4.2 | 76 |
| Recovery [%] | Sample 1 | Sample 2 | Sample 3 | Sample 4 | ||||
|---|---|---|---|---|---|---|---|---|
| Analyte | 5 µg·L−1 | 20 µg·L−1 | 5 µg·L−1 | 20 µg·L−1 | 5 µg·L−1 | 20 µg·L−1 | 5 µg·L−1 | 20 µg·L−1 |
| TMX | 101.7 ± 12.8 | 97.3 ± 4.5 | 113.3 ± 0.9 | 95.4 ± 4.6 | n.e. | 83.3 ± 4.9 | 90.2 ± 10.0 | 103.6 ± 22.6 |
| CLT | 74.1 ± 6.0 | 91.7 ± 6.8 | 59.0 ± 5.7 | 82.6 ± 2.0 | 37.6 ± 12.8 | 65.1 ± 0.7 | 54.1 ± 4.8 | 84.8 ± 8.3 |
| IMI | 85.0 ± 1.4 | 91.7 ± 1.0 | 81.2 ± 7.2 | 81.5 ± 0.9 | 83.1 ± 5.1 | 80.0 ± 7.3 | 83.3 ± 7.3 | 92.9 ± 5.7 |
| TCP | 60.7 ± 14.5 | 76.1 ± 8.0 | 60.5 ± 3.0 | 62.8 ± 7.1 | 53.8 ± 15.8 | 66.7 ± 3.5 | 53.9 ± 5.8 | 68.5 ± 2.4 |
| Analyte | Sample Type, Quantity | HPLC Mode and Column | Injection Volume [µL] | Extraction Method | Time [min] | EF | LOD LOQ | Recovery [%] | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| ACP, CLT, IMI, TCP, TMX | Honey, 2 g | Gradient Spherisorb 0DS2 (150 mm × 4 mm, 5 µm) | 20 | SPE with C1, then DLLME with CHCl3 in the ACN extract | 10 | 13 * | 0.2–1.0 µg kg−1 07–3.3 µg kg−1 | 90–104 | [12] |
| ACP, CLT, DNT, IMI, NTP, TCP, TMX | Grain (brown rice, maize, millet, oat), 10 g | Isocratic Agilent TC-C18 (250 × 4.6 mm, 5 µm) | 20 | QuEChERS with clean-up with PSA, C18, and graphitized carbon black followed by DLLME with CHCl3 + CH2Cl2 | 28 | 5 * | 2–5 µg kg−1 7–18 µg kg−1 | 76–123 | [45] |
| DNT, NTP, ACP, CLT, IMI, TMX | Tea, honey, 0.1 g | Isocratic Agilent Zorbax Eclipse Plus C18 (2.1 × 100 mm, 1.8 μm) | 20 | MIP-SPME | 25 | 10–56 | 0.03–0.58 µg L−1 0.09–1.93 µg L−1 | 85.4–116.8 | [23] |
| ACP, CLT, IMI, TCP, TMX | Fruit juice, surface waters, 13 mL | Isocratic LiChrospher®100 RP-18 ec (4.6 mm × 150 mm, 5.0 µm) | 20 | DµSPE using montmorillonite | 13 | 8–176 | 0.005–0.065 µg L−1 0.008–0.263 µg L−1 | 70–138 | [46] |
| ACP, IMI, FNC, NTP, TCP, 6-CNA | Cucumber, soil, 10 g | Isocratic Synergi Hydro RP C18 (250 × 4.6 mm, 4 µm) | 50 | Modified QuEChERS, clean-up of ACN extract with C18 | 20 | 1 * | 6–122 μg kg−1 18–366 μg kg−1 | 77–120 | [11] |
| ACP, IMI | Tomato, 2 g | Isocratic ZORBAX Eclipse Plus C18 (250 × 4.6 mm, 5 µm) | 5 | QuEChERS | 4 | 1 * | 3.31–8.53 µg kg−1 11–28 µg kg−1 | 83–97 | [47] |
| ACP, CLT, DNT, IMI, NTP, TCP, TMX | Honey, 5 mL | Gradient ZORBAX Eclipse XDB-C18 (50 × 4.6 mm, 1.8 µm) | Not given | DLLME with ACN and dichlormethane; QuEChERS | 7 | 10 * | 1.5–2.5 µg kg−1 2.0–2.5 µg kg−1 | 73.1–118.3 | [48] |
| ACP, IMI | Pistachio, 5 g | Isocratic Alltima C18 (250 × 4.6 mm, 5 μm) | 20 | Modified QuEChERS | 10 | 5 * | 10–20 μg L−1 33–60 μg L−1 | 70–114 | [49] |
| ACP, IMI, TMX | Fruit juice and vegetables, 10 mL | Isocratic STR–ODS (II) (150 × 4.6 mm, 5 µm) | 5 | Effervescence-assisted DLLE using an ionic liquid | 8 | 6.65–8.4 | 0.12–0.33 µg L−1 0.41–1.1 µg L−1 | 66–84 | [50] |
| ACP, CLT, IMI, TCP, TMX | Surface water, 10 mL | Isocratic Chromolith® HR RP-18 ec (4.6 × 100 mm) | 20 | Ultrasonically modified CPE with Triton X-114 | 9 | 20–333 | 0.3–2 µg L−1 3–6 µg L−1 | 64–120 | [44] |
| ACP, CLT, IMI, NTP, TMX | Water and fruit juice, 10 mL | Isocratic Atlantis dC18column (150 × 4.6 mm,5 μm) | 20 | VSLLME-SFO ** with octanol and SDS | 8 | 20–100 | 0.1–0.5 µg L−1 2–3 µg L−1 | 85–105 | [42] |
| ACP, CLT, IMI, TCP | Honey, 8 mL | Isocratic LiChrosphers 100RP-18 ec (150 × 4.6 mm, 5 µm) | 20 | Effervescence-assisted DLLE using ionic liquid | 12 | 50 * | 0.01 µg L−1 0.03 µg L−1 | 86–100 | [51] |
| ACP, IMI, TCP | Honey, 2 g | Isocratic WondaSil C18 (250 × 4.6 mm, 5 µm) | 20 | Matrix-induced sugaring-out method SULLE with ACN | 15 | - | 21–27 μg kg−1 70–90 μg kg−1 | 91–98 | [52] |
| ACP, CLT, IMI, NTP, TCP | Commercial fruit juices, 50 mL | Isocratic Zorbax SB-Aq (150 × 4.6 mm i.d., 5 μm) | 10 | Ultrasound-assisted DLLME with toluene | 9 | 34–40 | 0.08–0.31 µg L−1 0.27–0.92 µg L−1 | 68–80 | [53] |
| ACP, CLT, DNF, IMI, TCP, TMX | Natural waters, 2 mL | Gradient, Kinetex RP-C18 (4.6 × 150, 2.1 µm, 100 Å). | n.a. | Automated online SPE using nanofibers as sorbent membrane | 16 | 70–82 | 0.36–1.65 µg L−1 1.21–5.49 µg L−1 | 63–120 ** | Present method |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šrámková, I.H.; Horstkotte, B.; Carbonell-Rozas, L.; Erben, J.; Chvojka, J.; Lara, F.J.; García-Campaña, A.M.; Šatínský, D. Nanofibrous Online Solid-Phase Extraction Coupled with Liquid Chromatography for the Determination of Neonicotinoid Pesticides in River Waters. Membranes 2022, 12, 648. https://doi.org/10.3390/membranes12070648
Šrámková IH, Horstkotte B, Carbonell-Rozas L, Erben J, Chvojka J, Lara FJ, García-Campaña AM, Šatínský D. Nanofibrous Online Solid-Phase Extraction Coupled with Liquid Chromatography for the Determination of Neonicotinoid Pesticides in River Waters. Membranes. 2022; 12(7):648. https://doi.org/10.3390/membranes12070648
Chicago/Turabian StyleŠrámková, Ivana H., Burkhard Horstkotte, Laura Carbonell-Rozas, Jakub Erben, Jiří Chvojka, Francisco J. Lara, Ana M. García-Campaña, and Dalibor Šatínský. 2022. "Nanofibrous Online Solid-Phase Extraction Coupled with Liquid Chromatography for the Determination of Neonicotinoid Pesticides in River Waters" Membranes 12, no. 7: 648. https://doi.org/10.3390/membranes12070648