
Behavior of KCNQ Channels in Neural Plasticity and Motor Disorders


Abstract
:1. Introduction
2. Modulation of Synaptic Plasticity by KCNQ Channels
3. KCNQ Channels in Dopaminergic Motor Disorders
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ach | acetylcholine |
| AIM | abnormal involuntary movements |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) |
| AP | action potential |
| CA | cornu ammonus |
| DRG | dorsal root ganglion |
| ICA 27243 | N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide |
| KCNQ | voltage-gated potassium channel |
| Kv | voltage-gated potassium channel |
| LID | L-DOPA induced dyskinesia |
| LOF | loss of function |
| LTD | long-term depression |
| LTP | long-term potentiation |
| mAChR | muscarinic acetylcholine receptors |
| MFB | medial forebrain bundle |
| NMDA | N-methyl-D-aspartate |
| 6-OHDA | 6-hydroxydopamine |
| PD | Parkinson’s disease |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| SFA | spike frequency adaptation |
| SNc | substantia nigra pars compacta |
| VGICs | voltage-gated potassium channels |
| VGKC | voltage-gated potassium channel |
| VTA | ventral tegmental area |
References
- Camerino, D.C.; Desaphy, J.F.; Tricarico, D.; Pierno, S.; Liantonio, A. Therapeutic approaches to ion channel diseases. Adv. Genet. 2008, 64, 81–145. [Google Scholar]
- Abbott, G.W. KCNQs: Ligand- and voltage-gated potassium channels. Front. Physiol. 2020, 11, 583. [Google Scholar] [CrossRef]
- Kefauver, J.M.; Ward, A.B.; Patapoutian, A. Discoveries in structure and physiology of mechanically activated ion channels. Nature 2020, 587, 567–576. [Google Scholar] [CrossRef]
- Häfner, S.; Sandoz, G. Photopharmacological approaches for dissecting potassium channel physiology. Curr. Opin. Pharmacol. 2022, 63, 102178. [Google Scholar] [CrossRef]
- Mandal, K. Review of PIP2 in cellular signaling, functions and diseases. Int. J. Mol. Sci. 2020, 21, 8342. [Google Scholar] [CrossRef]
- Harraz, O.F. PIP2: A critical regulator of vascular ion channels hiding in plain sight. Proc. Natl. Acad. Sci. USA 2020, 117, 20378–20389. [Google Scholar] [CrossRef]
- Krajnik, A.; Brazzo, J.A.; Vaidyanathan, K.; Das, T.; Redondo-Muñoz, J.; Bae, Y. Phosphoinositide signaling and mechanotransduction in cardiovascular biology and disease. Front. Cell Dev. Biol. 2020, 8, 595849. [Google Scholar] [CrossRef]
- McLean, M.A.; Stephen, A.G.; Sligar, S.G. PIP2 influences the conformational dynamics of membrane-bound KRAS4b. Biochemistry 2019, 58, 3537–3545. [Google Scholar] [CrossRef]
- Jespersen, T.; Grunnet, M.; Olesen, S.P. The KCNQ1 potassium channel: From gene to physiological function. Physiology 2005, 20, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.X.; Deng, E.Z.; Chen, W.; Lin, H. Identifying the subfamilies of voltage-gated potassium channels using feature selection technique. Int. J. Mol. Sci. 2014, 15, 12940–12951. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, R.; Logette, E.; Marani, M.; Herzog, M.; Tâche, V.; Scantamburlo, E.; Buchillier, V.; Markram, H. A Kinetic map of the homomeric voltage-gated potassium channel (Kv) family. Front. Cell. Neurosci. 2019, 13, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.A.; Adams, P.R. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 1980, 283, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Delmas, P.; Brown, D.A. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 2005, 6, 850–862. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Li, Y. KCNQ potassium channels in sensory system and neural circuits. Acta Pharmacol. Sin. 2016, 37, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eren-Koçak, E.; Dalkara, T. Ion channel dysfunction and neuroinflammation in migraine and depression. Front. Pharmacol. 2021, 12, 777607. [Google Scholar] [CrossRef] [PubMed]
- Sacco, T.; Tempia, F. A-type potassium currents active at subthreshold potentials in mouse cerebellar Purkinje cells. J. Physiol. 2002, 543, 505–520. [Google Scholar] [CrossRef]
- Brown, D.A.; Passmore, G.M. Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 2009, 156, 1185–1195. [Google Scholar] [CrossRef]
- Jentsch, T.J. Neuronal KCNQ potassium channels: Physiology and role in disease. Nat. Rev. Neurosci. 2000, 1, 21–30. [Google Scholar] [CrossRef]
- Lehmann-Horn, F.; Jurkat-Rott, K. Voltage-gated ion channels and hereditary disease. Physiol. Rev. 1999, 79, 1317–1372. [Google Scholar] [CrossRef]
- Jurkat-Rott, K.; Lehmann-Horn, F. Human muscle voltage-gated ion channels and hereditary disease. Curr. Opin. Pharmacol. 2001, 1, 280–287. [Google Scholar] [CrossRef]
- Felix, R. Channelopathies: Ion channel defects linked to heritable clinical disorders. J. Med. Genet. 2000, 37, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J. KCNQ potassium channels: Physiology, pathophysiology, and pharmacology. Pharmacol. Ther. 2001, 90, 1–19. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, L.; Liu, W.; Hu, D.; Gao, Y.; Ge, Q.; Liu, X.; Li, L.; Wang, Y.; Wang, S.; et al. Pathogenic mechanism and gene correction for LQTS-causing double mutations in KCNQ1 using a pluripotent stem cell model. Stem Cell Res. 2019, 38, 101483. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wu, J.; Liu, Z. Studying KCNQ1 mutation and drug response in type 1 long QT syndrome using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Methods Mol. Biol. 2018, 1684, 7–28. [Google Scholar]
- Ma, D.; Wei, H.; Lu, J.; Huang, D.; Liu, Z.; Loh, L.J.; Islam, O.; Liew, R.; Shim, W.; Cook, S.A. Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 2015, 6, 39. [Google Scholar] [CrossRef]
- Wuriyanghai, Y.; Makiyama, T.; Sasaki, K.; Kamakura, T.; Yamamoto, Y.; Hayano, M.; Harita, T.; Nishiuchi, S.; Chen, J.; Kohjitani, H.; et al. Complex aberrant splicing in the induced pluripotent stem cell-derived cardiomyocytes from a patient with long QT syndrome carrying KCNQ1-A344Aspl mutation. Heart Rhythm 2018, 15, 1566–1574. [Google Scholar] [CrossRef]
- García Gozalo, M.; Bermejo Arnedo, I.; De Vera McMullan, P. KCNQ1 gene mutation and epilepsy in patient with long QT syndrome. Med. Clin. 2021, 157, 456–457. [Google Scholar] [CrossRef]
- Marstrand, P.; Almatlouh, K.; Kanters, J.K.; Graff, C.; Christensen, A.H.; Bundgaard, H.; Theilade, J. Effect of moderate potassium-elevating treatment in long QT syndrome: The TriQarr potassium study. Open Heart 2021, 8, e001670. [Google Scholar] [CrossRef]
- Zhang, R.; Ding, C.; Wang, H. Treatment on arrhythmia electric storm in a Jervell and Lange-Nielsen syndrome patient by ablation of the triggering premature ventricular contraction: A case report. Ann. Palliat. Med. 2021, 10, 4938–4943. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ackerman, M.J. Prevalence and potential genetic determinants of sensorineural deafness in KCNQ1 homozygosity and compound heterozygosity. Circ. Cardiovasc. Genet. 2013, 6, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Chen, S.; Wu, X.; Zhang, W.J.; Xie, W.; Jin, Y.; Xie, L.; Xu, K.; Bai, X.; Zhang, H.M.; et al. Jervell and Lange-Nielsen syndrome due to a novel compound heterozygous KCNQ1 mutation in a Chinese family. Neural Plast. 2020, 2020, 3569359. [Google Scholar] [CrossRef] [PubMed]
- Vyas, B.; Puri, R.D.; Namboodiri, N.; Nair, M.; Sharma, D.; Movva, S.; Saxena, R.; Bohora, S.; Aggarwal, N.; Vora, A.; et al. KCNQ1 mutations associated with Jervell and Lange-Nielsen syndrome and autosomal recessive Romano-Ward syndrome in India-expanding the spectrum of long QT syndrome type 1. Am. J. Med. Genet. 2016, 170, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Tan, Q.Q.; Lan, C.J.; Lv, B.Z.; Zhou, G.M.; Zhong, W.Q.; Gu, Z.M.; Mao, Y.M.; Liao, X. The changes of KCNQ5 expression and potassium microenvironment in the retina of myopic guinea pigs. Front. Physiol. 2021, 12, 790580. [Google Scholar] [CrossRef] [PubMed]
- Mönnig, G.; Schulze-Bahr, E.; Wedekind, H.; Eckardt, L.; Kirchhof, P.; Funke, H.; Kotthoff, S.; Vogt, J.; Assmann, G.; Breithardt, G.; et al. Clinical aspects and molecular genetics of the Jervell- and Lange-Nielsen Syndrome. Z. Kardiol. 2002, 91, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Kanaumi, T.; Takashima, S.; Iwasaki, H.; Itoh, M.; Mitsudome, A.; Hirose, S. Developmental changes in KCNQ2 and KCNQ3 expression in human brain: Possible contribution to the age-dependent etiology of benign familial neonatal convulsions. Brain Dev. 2008, 30, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Devaux, J.J.; Kleopa, K.A.; Cooper, E.C.; Scherer, S.S. KCNQ2 is a nodal K+ channel. J. Neurosci. 2004, 24, 1236–1244. [Google Scholar] [CrossRef] [Green Version]
- Mary, L.; Nourisson, E.; Feger, C.; Laugel, V.; Chaigne, D.; Keren, B.; Afenjar, A.; Billette, T.; Trost, D.; Cieuta-Walti, C.; et al. Pathogenic variants in KCNQ2 cause intellectual deficiency without epilepsy: Broadening the phenotypic spectrum of a potassium channelopathy. Am. J. Med. Genet. A 2021, 185, 1803–1815. [Google Scholar] [CrossRef]
- Vanoye, C.G.; Desai, R.R.; Ji, Z.; Adusumilli, S.; Jairam, N.; Ghabra, N.; Joshi, N.; Fitch, E.; Helbig, K.L.; McKnight, D.; et al. High-throughput evaluation of epilepsy-associated KCNQ2 variants reveals functional and pharmacological heterogeneity. JCI Insight 2022, 7, e156314. [Google Scholar] [CrossRef]
- Hu, C.; Liu, D.; Luo, T.; Wang, Y.; Liu, Z. Phenotypic spectrum and long-term outcome of children with genetic early-infantile-onset developmental and epileptic encephalopathy. Epileptic Disord. 2022. [Google Scholar] [CrossRef]
- Kim, K.W.; Kim, K.; Kim, H.J.; Kim, B.I.; Baek, M.; Suh, B.C. Posttranscriptional modulation of KCNQ2 gene expression by the miR-106b microRNA family. Proc. Natl. Acad. Sci. USA 2021, 118, e2110200118. [Google Scholar] [CrossRef]
- Monni, L.; Kraus, L.; Dipper-Wawra, M.; Soares-Da-Silva, P.; Maier, N.; Schmitz, D.; Holtkamp, M.; Fidzinski, P. In vitro and in vivo anti-epileptic efficacy of eslicarbazepine acetate in a mouse model of KCNQ2-related self-limited epilepsy. Br. J. Pharmacol. 2022, 179, 84–102. [Google Scholar] [CrossRef] [PubMed]
- Nissenkorn, A.; Kornilov, P.; Peretz, A.; Blumkin, L.; Heimer, G.; Ben-Zeev, B.; Attali, B. Personalized treatment with retigabine for pharmacoresistant epilepsy arising from a pathogenic variant in the KCNQ2 selectivity filter. Epileptic Disord. 2021, 23, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Chang, T.M.; Liang, J.S.; Li, S.Y. KCNQ2 mutations in childhood nonlesional epilepsy: Variable phenotypes and a novel mutation in a case series. Mol. Genet. Genom. Med. 2019, 7, e00816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milh, M.; Lacoste, C.; Cacciagli, P.; Abidi, A.; Sutera-Sardo, J.; Tzelepis, I.; Colin, E.; Badens, C.; Afenjar, A.; Coeslier, A.D.; et al. Variable clinical expression in patients with mosaicism for KCNQ2 mutations. Am. J. Med. Genet. A 2015, 167, 2314–2318. [Google Scholar] [CrossRef] [PubMed]
- Lazo, P.A.; García, J.L.; Gómez-Puertas, P.; Marcos-Alcalde, Í.; Arjona, C.; Villarroel, A.; González-Sarmiento, R.; Fons, C. Novel dominant KCNQ2 exon 7 partial in-frame duplication in a complex epileptic and neurodevelopmental delay syndrome. Int. J. Mol. Sci. 2020, 21, 4447. [Google Scholar] [CrossRef]
- Kaminsky, Z.; Jones, I.; Verma, R.; Saleh, L.; Trivedi, H.; Guintivano, J.; Akman, R.; Zandi, P.; Lee, R.S.; Potash, J.B. DNA methylation and expression of KCNQ3 in bipolar disorder. Bipolar Disord. 2015, 17, 150–159. [Google Scholar] [CrossRef]
- Mittal, K.; Rafiq, M.A.; Rafiullah, R.; Harripaul, R.; Ali, H.; Ayaz, M.; Aslam, M.; Naeem, F.; Amin-Ud-Din, M.; Waqas, A.; et al. Mutations in the genes for thyroglobulin and thyroid peroxidase cause thyroid dyshormonogenesis and autosomal-recessive intellectual disability. J. Hum. Genet. 2016, 61, 867–872. [Google Scholar] [CrossRef]
- Rim, J.H.; Choi, J.Y.; Jung, J.; Gee, H.Y. Activation of KCNQ4 as a therapeutic strategy to treat hearing loss. Int. J. Mol. Sci. 2021, 22, 2510. [Google Scholar] [CrossRef]
- Lee, S.Y.; Choi, H.B.; Park, M.; Choi, I.S.; An, J.; Kim, A.; Kim, E.; Kim, N.; Han, J.H.; Kim, M.Y.; et al. Novel KCNQ4 variants in different functional domains confer genotype- and mechanism-based therapeutics in patients with nonsyndromic hearing loss. Exp. Mol. Med. 2021, 53, 1192–1204. [Google Scholar] [CrossRef]
- Thorpe, R.K.; Walls, W.D.; Corrigan, R.; Schaefer, A.; Wang, K.; Huygen, P.; Casavant, T.L.; Smith, R.J.H. AudioGene: Refining the natural history of KCNQ4, GSDME, WFS1, and COCH-associated hearing loss. Hum. Genet. 2022, 141, 877–887. [Google Scholar] [CrossRef]
- Yen, T.T.; Chen, I.C.; Hua, M.W.; Wei, C.Y.; Shih, K.H.; Li, J.L.; Lin, C.H.; Hsiao, T.H.; Chen, Y.M.; Jiang, R.S. A KCNQ4 c.546C>G Genetic variant associated with late onset non-syndromic hearing loss in a Taiwanese population. Genes 2021, 12, 1711. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Wasano, K.; Takahashi, S.; Homma, K. Cell death-inducing cytotoxicity in truncated KCNQ4 variants associated with DFNA2 hearing loss. Dis. Model Mech. 2021, 14, dmm049015. [Google Scholar] [CrossRef] [PubMed]
- Peixoto-Pinheiro, B.; Vona, B.; Löwenheim, H.; Rüttiger, L.; Knipper, M.; Adel, Y. Age-related hearing loss pertaining to potassium ion channels in the cochlea and auditory pathway. Dis. Model Mech. 2021, 473, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Borgini, M.; Mondal, P.; Liu, R.; Wipf, P. Chemical modulation of Kv7 potassium channels. RSC Med. Chem. 2021, 12, 483–537. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Sun, H.; Li, M. Zinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutants. Nat. Chem. Biol. 2007, 3, 287–296. [Google Scholar] [CrossRef]
- Tompson, D.J.; Buraglio, M.; Andrews, S.M.; Wheless, J.W. Adolescent clinical development of ezogabine/retigabine as adjunctive therapy for partial-onset seizures: Pharmacokinetics and tolerability. J. Pediatr. Pharmacol. Ther. 2016, 21, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Gunthorpe, M.J.; Large, C.H.; Sankar, R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 2012, 53, 412–424. [Google Scholar] [CrossRef]
- Bayasgalan, T.; Stupniki, S.; Kovács, A.; Csemer, A.; Szentesi, P.; Pocsai, K.; Dionisio, L.; Spitzmaul, G.; Pál, B. Alteration of mesopontine cholinergic function by the lack of KCNQ4 subunit. Front. Cell. Neurosci. 2021, 15, 707789. [Google Scholar] [CrossRef]
- Niu, X.; Yu, K.; He, B. Transcranial focused ultrasound induces sustained synaptic plasticity in rat hippocampus. Brain Stimul. 2022, 15, 352–359. [Google Scholar] [CrossRef]
- Caragea, V.M.; Manahan-Vaughan, D. Bidirectional regulation of hippocampal synaptic plasticity and modulation of cumulative spatial memory by dopamine D2-like receptors. Front. Behav. Neurosci. 2022, 15, 803574. [Google Scholar] [CrossRef]
- Sahu, G.; Turner, R.W. The molecular basis for the calcium-dependent slow afterhyperpolarization in CA1 hippocampal pyramidal neurons. Front. Physiol. 2021, 12, 759707. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Schmitt, N.; Calloe, K.; Dalby, B.W.; Grunnet, M.; Olesen, S.P. The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology 2006, 51, 1068–1077. [Google Scholar] [CrossRef]
- Blom, S.M.; Schmitt, N.; Jensen, H.S. The acrylamide (S)-2 as a positive and negative modulator of Kv7 channels expressed in Xenopus laevis oocytes. PLoS ONE 2009, 4, e8251. [Google Scholar] [CrossRef]
- Zhang, X.; An, H.; Li, J.; Zhang, Y.; Liu, Y.; Jia, Z.; Zhang, W.; Chu, L.; Zhang, H. Selective activation of vascular Kv 7.4/Kv 7.5 K+ channels by fasudil contributes to its vasorelaxant effect. Br. J. Pharmacol. 2016, 173, 3480–3491. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, D.; Hughes, B.A. KCNQ5/K(v)7.5 potassium channel expression and subcellular localization in primate retinal pigment epithelium and neural retina. Am. J. Physiol. Cell Physiol. 2011, 301, C1017–C1026. [Google Scholar] [CrossRef] [Green Version]
- Fogwe, L.A.; Reddy, V.; Mesfin, F.B. Neuroanatomy, Hippocampus; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482171 (accessed on 15 January 2020).
- Hu, H.; Vervaeke, K.; Storm, J.F. M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J. Neurosci. 2007, 27, 1853–1867. [Google Scholar] [CrossRef] [Green Version]
- Cooper, E.C.; Harrington, E.; Jan, Y.N.; Jan, L.Y. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci. 2001, 21, 9529–9540. [Google Scholar] [CrossRef] [Green Version]
- Tzingounis, A.V.; Heidenreich, M.; Kharkovets, T.; Spitzmaul, G.; Jensen, H.S.; Nicoll, R.A.; Jentsch, T.J. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc. Natl. Acad. Sci. USA 2010, 107, 10232–10237. [Google Scholar] [CrossRef] [Green Version]
- Boscia, F.; Elkjaer, M.L.; Illes, Z.; Kukley, M. Altered expression of ion channels in white matter lesions of progressive multiple sclerosis: What do we know about their function? Front. Cell. Neurosci. 2021, 15, 685703. [Google Scholar] [CrossRef]
- Schultz, C.; Engelhardt, M. Anatomy of the hippocampal formation. Front. Neurol. Neurosci. 2014, 34, 6–17. [Google Scholar]
- Ito, H.T.; Schuman, E.M. Functional division of hippocampal area CA1 via modulatory gating of entorhinal cortical inputs. Hippocampus 2012, 22, 372–387. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.C.; Li, L.; Zhang, M.; Li, W.B.; Li, Q.J.; Zhao, L. Division of CA1, CA3 and DG regions of the hippocampus of Wistar rat. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2012, 28, 189–192. [Google Scholar]
- Watson, C.; Binks, D. Elongation of the CA1 field of the septal hippocampus in ungulates. J. Comp. Neurol. 2019, 527, 818–832. [Google Scholar] [CrossRef]
- Van Groen, T.; Wyss, J.M. Extrinsic projections from area CA1 of the rat hippocampus: Olfactory, cortical, subcortical, and bilateral hippocampal formation projections. J. Comp. Neurol. 1990, 302, 515–528. [Google Scholar] [CrossRef]
- De La Rosa-Prieto, C.; Ubeda-Banon, I.; Mohedano-Moriano, A.; Pro-Sistiaga, P.; Saiz-Sanchez, D.; Insausti, R.; Martinez-Marcos, A. Subicular and CA1 hippocampal projections to the accessory olfactory bulb. Hippocampus 2009, 19, 124–129. [Google Scholar] [CrossRef]
- Bliss, T.V.; Cooke, S.F. Long-term potentiation and long-term depression: A clinical perspective. Clinics 2011, 66 (Suppl. 1), 3–17. [Google Scholar] [CrossRef] [Green Version]
- Bliss, T.V.; Lomo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef]
- Kemp, A.; Manahan-Vaughan, D. Hippocampal long-term depression and long-term potentiation encode different aspects of novelty acquisition. Proc. Natl. Acad. Sci. USA 2004, 101, 8192–8197. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T. Long-term depression and other synaptic plasticity in the cerebellum. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Wiegert, J.S.; Pulin, M.; Gee, C.E.; Oertner, T.G. The fate of hippocampal synapses depends on the sequence of plasticity-inducing events. Elife 2018, 7, e39151. [Google Scholar] [CrossRef]
- Sakurai, M. Synaptic modification of parallel fibre-Purkinje cell transmission in in vitro guinea-pig cerebellar slices. J. Physiol. 1987, 394, 463–480. [Google Scholar] [CrossRef]
- Wiegert, J.S.; Oertner, T.G. Long-term depression triggers the selective elimination of weakly integrated synapses. Proc. Natl. Acad. Sci. USA 2013, 110, E4510–E4519. [Google Scholar] [CrossRef] [Green Version]
- Lezmy, J.; Gelman, H.; Katsenelson, M.; Styr, B.; Tikochinsky, E.; Lipinsky, M.; Peretz, A.; Slutsky, I.; Attali, B. M-current inhibition in hippocampal excitatory neurons triggers intrinsic and synaptic homeostatic responses at different temporal scales. J. Neurosci. 2020, 40, 3694–3706. [Google Scholar] [CrossRef]
- Petrovic, M.M.; Nowacki, J.; Olivo, V.; Tsaneva-Atanasova, K.; Randall, A.D.; Mellor, J.R. Inhibition of post-synaptic Kv7/KCNQ/M channels facilitates long-term potentiation in the hippocampus. PLoS ONE 2012, 7, e30402. [Google Scholar] [CrossRef]
- Huang, P.; Li, C.; Fu, T.; Zhao, D.; Yi, Z.; Lu, Q.; Guo, L.; Xu, X. Flupirtine attenuates chronic restraint stress-induced cognitive deficits and hippocampal apoptosis in male mice. Behav. Brain Res. 2015, 288, 1–10. [Google Scholar] [CrossRef]
- Stanton, P.K. LTD, LTP, and the sliding threshold for long-term synaptic plasticity. Hippocampus 1996, 6, 35–42. [Google Scholar] [CrossRef]
- McCutchen, E.; Scheiderer, C.L.; Dobrunz, L.E.; McMahon, L.L. Coexistence of muscarinic long-term depression with electrically induced long-term potentiation and depression at CA3-CA1 synapses. J. Neurophysiol. 2006, 96, 3114–3121. [Google Scholar] [CrossRef] [Green Version]
- Milner, A.J.; Cummings, D.M.; Spencer, J.P.; Murphy, K.P. Bi-directional plasticity and age-dependent long-term depression at mouse CA3-CA1 hippocampal synapses. Neurosci. Lett. 2004, 367, 1–5. [Google Scholar] [CrossRef]
- Fontán-Lozano, A.; Suárez-Pereira, I.; Delgado-García, J.M.; Carrión, A.M. The M-current inhibitor XE991 decreases the stimulation threshold for long-term synaptic plasticity in healthy mice and in models of cognitive disease. Hippocampus 2011, 21, 22–32. [Google Scholar] [CrossRef]
- Milh, M.; Roubertoux, P.; Biba, N.; Chavany, J.; Spiga Ghata, A.; Fulachier, C.; Collins, S.C.; Wagner, C.; Roux, J.C.; Yalcin, B.; et al. A knock-in mouse model for KCNQ2-related epileptic encephalopathy displays spontaneous generalized seizures and cognitive impairment. Epilepsia 2020, 61, 868–878. [Google Scholar] [CrossRef] [Green Version]
- Baculis, B.C.; Zhang, J.; Chung, H.J. The role of Kv7 channels in neural plasticity and behavior. Front. Physiol. 2020, 11, 568667. [Google Scholar] [CrossRef]
- Thomann, P.A.; Seidl, U.; Brinkmann, J.; Hirjak, D.; Traeger, T.; Wolf, R.C.; Essig, M.; Schroder, J. Hippocampal morphology and autobiographic memory in mild cognitive impairment and Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 507–515. [Google Scholar] [CrossRef]
- Hirjak, D.; Wolf, R.C.; Remmele, B.; Seidl, U.; Thomann, A.K.; Kubera, K.M.; Schröder, J.; Maier-Hein, K.H.; Thomann, P.A. Hippocampal formation alterations differently contribute to autobiographic memory deficits in mild cognitive impairment and Alzheimer’s disease. Hippocampus 2017, 27, 702–715. [Google Scholar] [CrossRef]
- Li, X.T. Alzheimer’s disease therapy based on acetylcholinesterase inhibitor/blocker effects on voltage-gated potassium channels. Metab. Brain Dis. 2022, 37, 581–587. [Google Scholar] [CrossRef]
- Spoleti, E.; Krashia, P.; La Barbera, L.; Nobili, A.; Lupascu, C.A.; Giacalone, E.; Keller, F.; Migliore, M.; Renzi, M.; D’Amelio, M. Early derailment of firing properties in CA1 pyramidal cells of the ventral hippocampus in an Alzheimer’s disease mouse model. Exp. Neurol. 2021, 350, 113969. [Google Scholar] [CrossRef]
- Moriguchi, S.; Inagaki, R.; Fukunaga, K. Memantine improves cognitive deficits via KATP channel inhibition in olfactory bulbectomized mice. Mol. Cell. Neurosci. 2021, 117, 103680. [Google Scholar] [CrossRef]
- Djebari, S.; Iborra-Lázaro, G.; Temprano-Carazo, S.; Sánchez-Rodríguez, I.; Nava-Mesa, M.O.; Múnera, A.; Gruart, A.; Delgado-García, J.M.; Jiménez-Díaz, L.; Navarro-López, J.D. G-Protein-gated inwardly rectifying potassium (Kir3/GIRK) channels govern synaptic plasticity that supports hippocampal-dependent cognitive functions in male mice. J. Neurosci. 2021, 41, 7086–7102. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M. Mitochondrial ion channels in aging and related diseases. Curr. Aging Sci. 2022. [Google Scholar] [CrossRef]
- Islas, Á.A.; Scior, T.; Torres-Ramirez, O.; Salinas-Stefanon, E.M.; Lopez-Lopez, G.; Flores-Hernandez, J. Computational molecular characterization of the interaction of acetylcholine and the NMDA receptor to explain the direct glycine-competitive potentiation of NMDA-mediated neuronal currents. ACS Chem. Neurosci. 2022, 13, 229–244. [Google Scholar] [CrossRef]
- Cocozza, G.; Garofalo, S.; Capitani, R.; D’Alessandro, G.; Limatola, C.; Taylor, K.I. Microglial potassium channels: From homeostasis to neurodegeneration. Biomolecules 2021, 11, 1774. [Google Scholar] [CrossRef]
- Sharma, K.; Pradhan, S.; Duffy, L.K.; Yeasmin, S.; Bhattarai, N.; Schulte, M.K. Role of receptors in relation to plaques and tangles in Alzheimer’s disease pathology. Int. J. Mol. Sci. 2021, 22, 12987. [Google Scholar] [CrossRef]
- Hirni, D.I.; Kivisaari, S.L.; Monsch, A.U.; Taylor, K.I. Distinct neuroanatomical bases of episodic and semantic memory performance in Alzheimer’s disease. Neuropsychologia 2013, 51, 930–937. [Google Scholar] [CrossRef] [Green Version]
- Bomilcar, I.; Bertrand, E.; Morris, R.G.; Mograbi, D.C. The seven selves of dementia. Front. Psychiatry 2021, 12, 646050. [Google Scholar] [CrossRef]
- Liu, H.; Jia, L.; Chen, X.; Shi, L.; Xie, J. The Kv7/KCNQ channel blocker XE991 protects nigral dopaminergic neurons in the 6-hydroxydopamine rat model of Parkinson’s disease. Brain Res. Bull. 2018, 137, 132–139. [Google Scholar] [CrossRef]
- Sibille, K.T.; Bartsch, F.; Reddy, D.; Fillingim, R.B.; Keil, A. Increasing neuroplasticity to bolster chronic pain treatment: A role for intermittent fasting and glucose administration? J. Pain. 2016, 17, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Paventi, G.; Virginia Soldovieri, M.; Servettini, I.; Barrese, V.; Miceli, F.; Josè Sisalli, M.; Ambrosino, P.; Mosca, I.; Vinciguerra, I.; Testai, L.; et al. Kv7.4 channels regulate potassium permeability in neuronal mitochondria. Biochem. Pharmacol. 2022, 197, 114931. [Google Scholar] [CrossRef]
- Bloms-Funke, P.; Bankstahl, M.; Bankstahl, J.; Kneip, C.; Schröder, W.; Löscher, W. The novel dual-mechanism Kv7 potassium channel/TSPO receptor activator GRT-X is more effective than the Kv7 channel opener retigabine in the 6-Hz refractory seizure mouse model. Neuropharmacology 2022, 203, 108884. [Google Scholar] [CrossRef]
- Clark, S.; Antell, A.; Kaufman, K. New antiepileptic medication linked to blue discoloration of the skin and eyes. Ther. Adv. Drug Saf. 2015, 6, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, Y.; Hu, F.; Yang, J.; Guo, X.; Hou, X.; Ju, C.; Wang, K. Activation of neuronal voltage-gated potassium Kv7/KCNQ/M-current by a novel channel opener SCR2682 for alleviation of chronic pain. J. Pharmacol. Exp. Ther. 2021, 377, 20–28. [Google Scholar] [CrossRef]
- Rivera-Arconada, I.; Vicente-Baz, J.; Lopez-Garcia, J.A. Targeting Kv7 channels in pain pathways. Oncotarget 2017, 8, 12554–12555. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Baz, J.; Lopez-Garcia, J.A.; Rivera-Arconada, I. Implication of Kv7 channels in the spinal antinociceptive actions of celecoxib. J. Pharmacol. Exp. Ther. 2019, 370, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Baz, J.; Rivera-Arconada, I. Spinal actions of the NSAID diclofenac on nociceptive transmission in comparison to the Kv7 channel opener flupirtine. Neuroscience 2020, 440, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.; Cumming, J.; Martin, M.; Ballard, C.; Ertan-Lyons, D.; Sultzer, D.; Devanand, D.; Weintraub, D.; McEvoy, B.; Youakim, J.; et al. Trial of pimvanserin in dementia-related psychosis. N. Engl. J. Med. 2021, 385, 3099–3319. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Baz, J.; Lopez-Garcia, J.A.; Rivera-Arconada, I. Effects of novel subtype selective M-current activators on spinal reflexes in vitro: Comparison with retigabine. Neuropharmacology 2016, 109, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Baz, J.; Lopez-Garcia, J.A.; Rivera-Arconada, I. Central sensitization of dorsal root potentials and dorsal root reflexes: An in vitro study in the mouse spinal cord. Eur. J. Pain. 2021, 26, 356–369. [Google Scholar] [CrossRef]
- Teng, B.C.; Song, Y.; Zhang, F.; Ma, T.Y.; Qi, J.L.; Zhang, H.L.; Li, G.; Wang, K. Activation of neuronal Kv7/KCNQ/M-channels by the opener QO58-lysine and its anti-nociceptive effects on inflammatory pain in rodents. Acta Pharmacol. Sin. 2016, 37, 1054–1062. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.R.; Hu, S.W.; Zhang, S.; Song, Y.; Wang, X.Y.; Wang, L.; Li, Y.Y.; Yu, Y.M.; Liu, H.; Liu, D.; et al. KCNQ channels in the mesolimbic reward circuit regulate nociception in chronic pain in mice. Neurosci. Bull. 2021, 37, 597–610. [Google Scholar] [CrossRef]
- Chinta, S.J.; Andersen, J.K. Dopaminergic neurons. Int. J. Biochem. Cell Biol. 2005, 37, 942–946. [Google Scholar] [CrossRef]
- William, M.; Singh, S.; Chu, X.-P. Commentary: Large acid-evoked currents, mediated by ASIC1a, accompany differentiation in human dopaminergic neurons. Front. Cell. Neurosci. 2021, 15, 789354. [Google Scholar] [CrossRef]
- Hayes, M.T. Parkinson’s disease and parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson disease: A review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xue, B.; Wang, J.; Liu, H.; Shi, L.; Xie, J. Potassium channels: A potential therapeutic target for Parkinson’s disease. Neurosci. Bull. 2018, 34, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Weikop, P.; Mikkelsen, M.D.; Rode, F.; Mikkelsen, J.D. The pan-Kv7 (KCNQ) channel opener retigabine inhibits striatal excitability by direct action on striatal neurons in vivo. Basic Clin. Pharmacol. Toxicol. 2017, 120, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Srivanitchapoom, P. Levodopa-induced dyskinesia: Clinical features, pathophysiology, and medical management. Ann. Indian Acad. Neurol. 2017, 20, 190–198. [Google Scholar]
- Tronci, E.; Francardo, V. Animal models of L-DOPA-induced dyskinesia: The 6-OHDA-lesioned rat and mouse. J. Neural. Transm. 2018, 125, 1137–1144. [Google Scholar] [CrossRef]
- Sander, S.E.; Lemm, C.; Lange, N.; Hamann, M.; Richter, A. Retigabine, a K(V)7 (KCNQ) potassium channel opener, attenuates L-DOPA-induced dyskinesias in 6-OHDA-lesioned rats. Neuropharmacology 2012, 62, 1052–1061. [Google Scholar] [CrossRef]
- Sander, S.E.; Lambrecht, C.; Richter, A. The K(V)7.2/3 preferring channel opener ICA 27243 attenuates L-DOPA-induced dyskinesia in hemiparkinsonian rats. Neurosci. Lett. 2013, 545, 59–63. [Google Scholar] [CrossRef]
- Hansen, H.H.; Waroux, O.; Seutin, V.; Jentsch, T.J.; Aznar, S.; Mikkelsen, J.D. Kv7 channels: Interaction with dopaminergic and serotonergic neurotransmission in the CNS. J. Physiol. 2008, 586, 1823–1832. [Google Scholar] [CrossRef]
- McCutcheon, R.A.; Abi-Dargham, A.; Howes, O.D. Schizophrenia, dopamine and the striatum: From biology to symptoms. Trends Neurosci. 2019, 42, 205–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattmann, M.E.; Yu, H.; Lin, Z.; Xu, K.; Huang, X.; Long, S.; Wu, M.; McManus, O.B.; Engers, D.W.; Le, U.M.; et al. Identification of (R)-N-(4-(4-methoxyphenyl) thiazol-2-yl)-1-tosylpiperidine-2-carboxamide, ML277, as a novel, potent and selective K(v)7.1 (KCNQ1) potassium channel activator. Bioorg. Med. Chem. Lett. 2012, 22, 5936–5941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Lin, Z.; Mattmann, M.E.; Zou, B.; Terrenoire, C.; Zhang, H.; Wu, M.; McManus, O.B.; Kass, R.S.; Lindsley, C.W.; et al. Dynamic subunit stoichiometry confers a progressive continuum of pharmacological sensitivity by KCNQ potassium channels. Proc. Natl. Acad. Sci. USA 2013, 110, 8732–8737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, M.G.; Halaszovich, C.R.; Oliver, D. Aminoglycosides inhibit KCNQ4 channels in cochlear outer hair cells via depletion of phosphatidylinositol (4,5) bisphosphate. Mol. Pharmacol. 2011, 79, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Abbott, G.W.; Redford, K.E.; Yoshimura, R.F.; Manville, R.W.; Moreira, L.; Tran, K.; Arena, G.; Kookootsedes, A.; Lasky, E.; Gunnison, E. KCNQ and KCNE isoform-dependent pharmacology rationalizes native American dual use of specific plants as both analgesics and gastrointestinal therapeutics. Front. Physiol. 2021, 12, 777057. [Google Scholar] [CrossRef]
- Volberg, W.A.; Koci, B.J.; Su, W.; Lin, J.; Zhou, J. Blockade of human cardiac potassium channel human ether-a-go-go-related gene (HERG) by macrolide antibiotics. J. Pharmacol. Exp. Ther. 2002, 302, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; McBride, B.F.; Leake, B.F.; Kim, R.B.; Roden, D.M. Modulation of drug block of the cardiac potassium channel KCNA5 by the drug transporters OCTN1 and MDR1. Br. J. Pharmacol. 2010, 161, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Grünert, S.C.; Bodi, I.; Odening, K.E. Possible mechanisms for sensorineural hearing loss and deafness in patients with propionic acidemia. Orphanet J. Rare Dis. 2017, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Naffaa, M.M.; Al-Ewaidat, O.A. Ligand modulation of KCNQ-encoded (KV7) potassium channels in the heart and nervous system. Eur. J. Pharmacol. 2021, 906, 174278. [Google Scholar] [CrossRef]
- Barrese, V.; Stott, J.B.; Baldwin, S.N.; Mondejar-Parreño, G.; Greenwood, I.A. SMIT (Sodium-Myo-Inositol Transporter) 1 regulates arterial contractility through the modulation of vascular Kv7 channels. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2468–2480. [Google Scholar]
- Larsson, J.E.; Frampton, D.J.A.; Liin, S.I. Polyunsaturated fatty acids as modulators of KV7 channels. Front. Physiol. 2020, 11, 641. [Google Scholar] [CrossRef] [PubMed]
- Peixoto-Neves, D.; Kanthakumar, P.; Afolabi, J.M.; Soni, H.; Buddington, R.K.; Adebiyi, A. KV7.1 channel blockade inhibits neonatal renal autoregulation triggered by a step decrease in arterial pressure. Am. J. Physiol. Renal Physiol. 2022, 322, F197–F207. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.W.; Wu, H.M.; Bai, Y.; Zeng, Q.; Cao, Z.M.; Wu, X.S.; Tang, M. Potassium channel Shaker play a protective role against cardiac aging in Drosophila. Yi Chuan 2021, 43, 94–99. [Google Scholar] [PubMed]
- Stott, J.B.; Barrese, V.; Suresh, M.; Masoodi, S.; Greenwood, I.A. Investigating the role of G protein βγ in Kv7-dependent relaxations of the rat vasculature. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2091–2102. [Google Scholar] [CrossRef] [Green Version]
- Blandin, C.E.; Gravez, B.J.; Hatem, S.N.; Balse, E. Remodeling of ion channel trafficking and cardiac arrhythmias. Orphanet J. Rare Dis. 2021, 10, 2417. [Google Scholar] [CrossRef]
- Gallego, M.; Zayas-Arrabal, J.; Alquiza, A.; Apellaniz, B.; Casis, O. Electrical features of the diabetic myocardium. Arrhythmic and cardiovascular safety considerations in diabetes. Front. Pharmacol. 2021, 12, 687256. [Google Scholar] [CrossRef]
- Herrera-Pérez, S.; Campos-Ríos, A.; Rueda-Ruzafa, L.; Lamas, J.A. Contribution of K2P potassium channels to cardiac physiology and pathophysiology. Int. J. Mol. Sci. 2021, 22, 6635. [Google Scholar] [CrossRef]
- Yang, K.; Xue, Y.; Yu, M.; Jiao, H.; Li, Y.; Wei, X.; Liu, W.; Sun, Y.; Chen, N.; Song, L.; et al. Protective effect of trimetazidine on potassium ion homeostasis in myocardial tissue in mice with heart failure. Biomed. Res. Int. 2022, 2022, 2387860. [Google Scholar] [CrossRef]



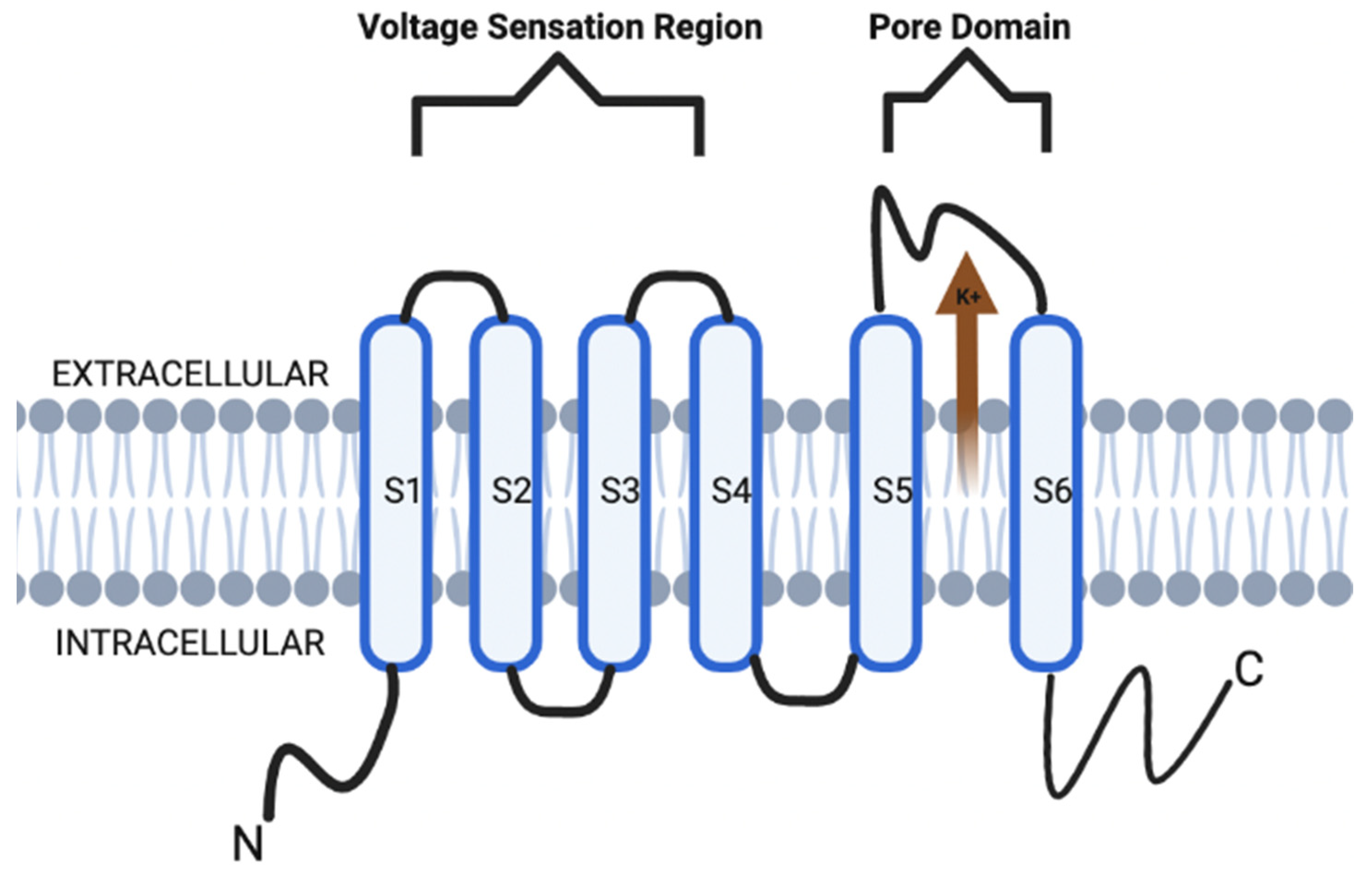{kind=link}
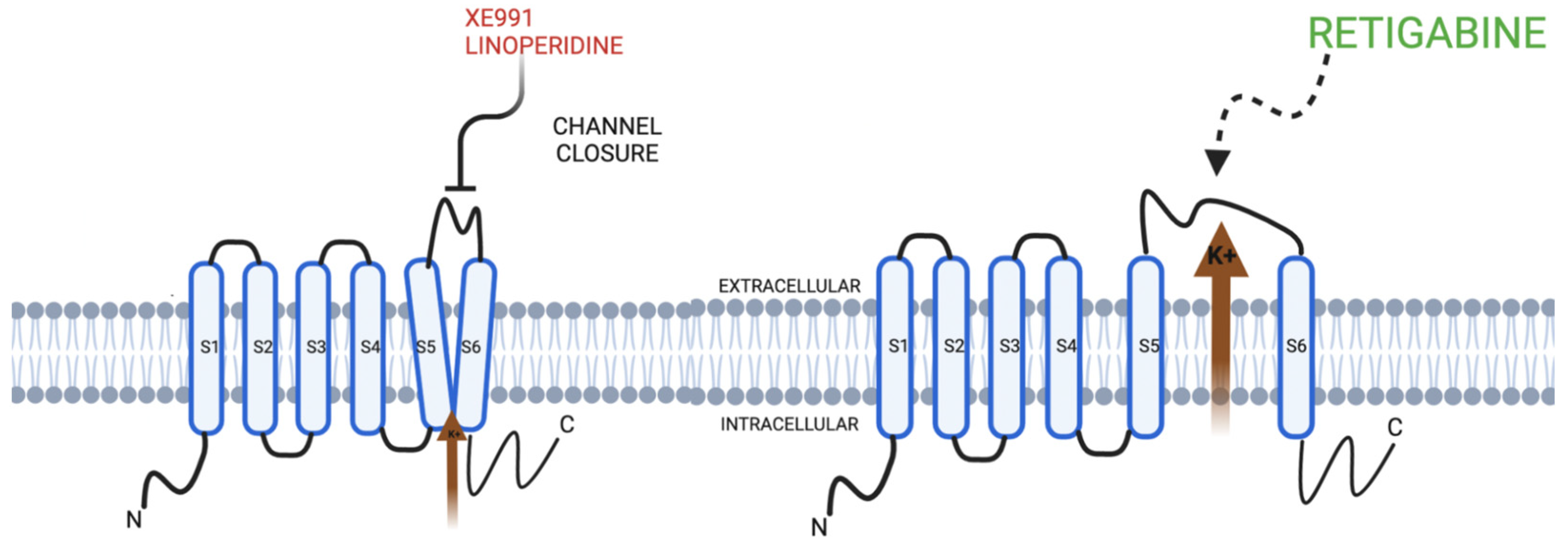{kind=link}
| Gene | Expression Distribution | Associated Pathologies |
|---|---|---|
| KCNQ1 | Cochlea | Type 1 long QT syndrome |
| Heart | ||
| KCNQ2 | Cerebellum | Benign familial neonatal seizures |
| Hippocampus Medulla | Early onset epileptic encephalopathy | |
| KCNQ3 | Cerebellum | Benign familial neonatal seizures |
| Hippocampus Medulla | Early onset epileptic encephalopathy Bipolar Disorder | |
| KCNQ4 | Cochlea Trigeminal ganglia | Deafness |
| KCNQ5 | Retinal pigment epithelium | * |
| Action on Channel | Mechanisms | Agents |
|---|---|---|
| Opening | Phosphatidyl inositol 4, 5 biphosphate (PIP 2) Gamma-amino butyric acid (GABA) Inositol 1, 4, 5 triphosphate (IP3) a. PIP2 synthesis b. Suppression of muscarinic current (M) receptor at acetyl choline receptor | Retigabine Flupirtine ICA 27243 |
| Closing | Muscarinic current (M) associated with acetyl choline receptor Pathological Mechanisms a. Movement Disorders | Linoperidine XE991 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.P.; William, M.; Malavia, M.; Chu, X.-P. Behavior of KCNQ Channels in Neural Plasticity and Motor Disorders. Membranes 2022, 12, 499. https://doi.org/10.3390/membranes12050499
Singh SP, William M, Malavia M, Chu X-P. Behavior of KCNQ Channels in Neural Plasticity and Motor Disorders. Membranes. 2022; 12(5):499. https://doi.org/10.3390/membranes12050499
Chicago/Turabian StyleSingh, Som P., Matthew William, Mira Malavia, and Xiang-Ping Chu. 2022. "Behavior of KCNQ Channels in Neural Plasticity and Motor Disorders" Membranes 12, no. 5: 499. https://doi.org/10.3390/membranes12050499