
Trafficking of Annexins during Membrane Repair in Human Skeletal Muscle Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of Human Skeletal Muscle Cells
2.2. Western Blot
2.3. Immunocytofluorescence
2.4. Subcellular Trafficking of ANX Fused to Fluorescent Proteins in Damaged Myotubes
2.5. Immuno-TEM
3. Results
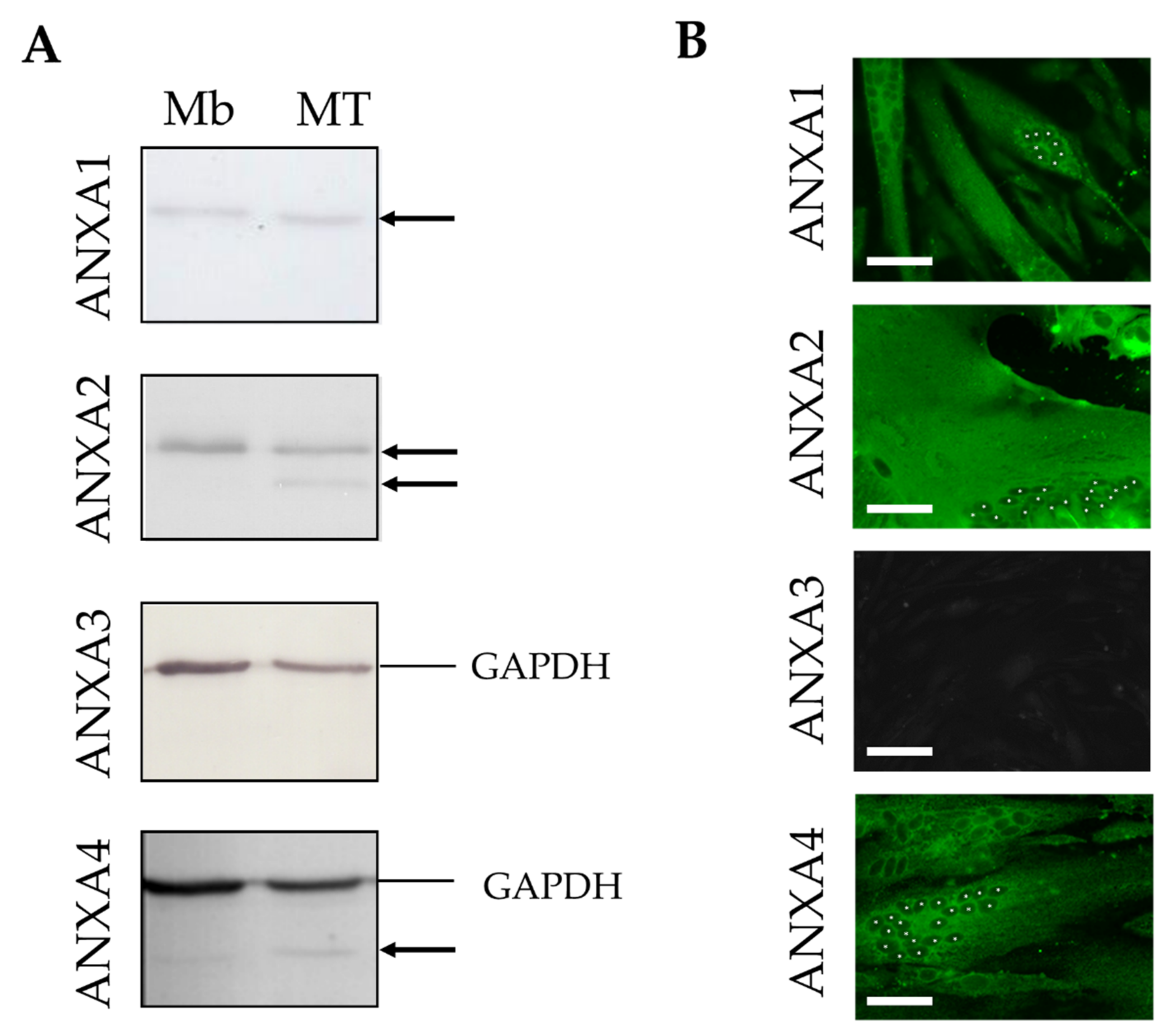
3.1. Expression of ANX in Human Skeletal Muscle Cells
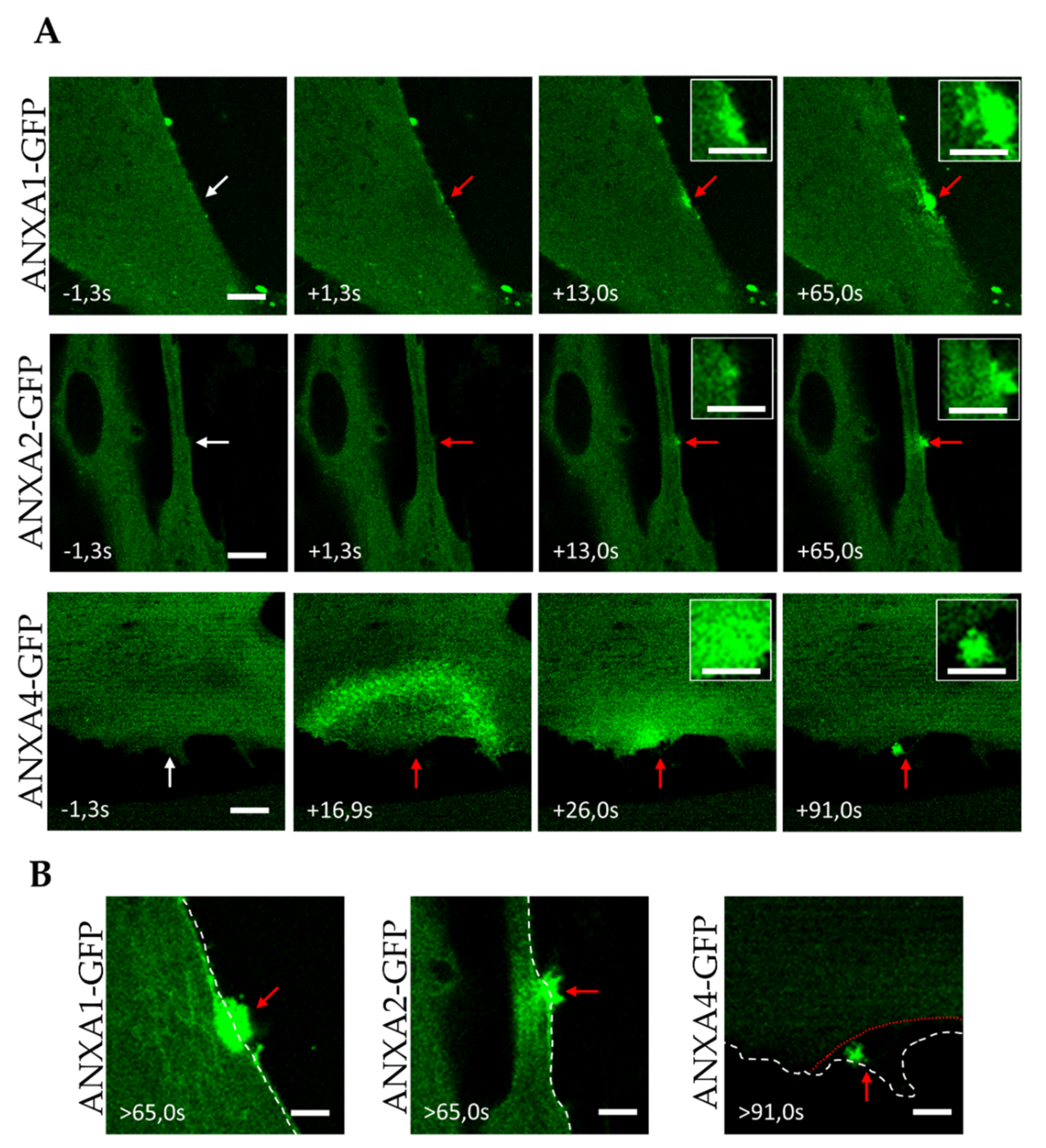
3.2. Trafficking of ANX after Sarcolemma Injury
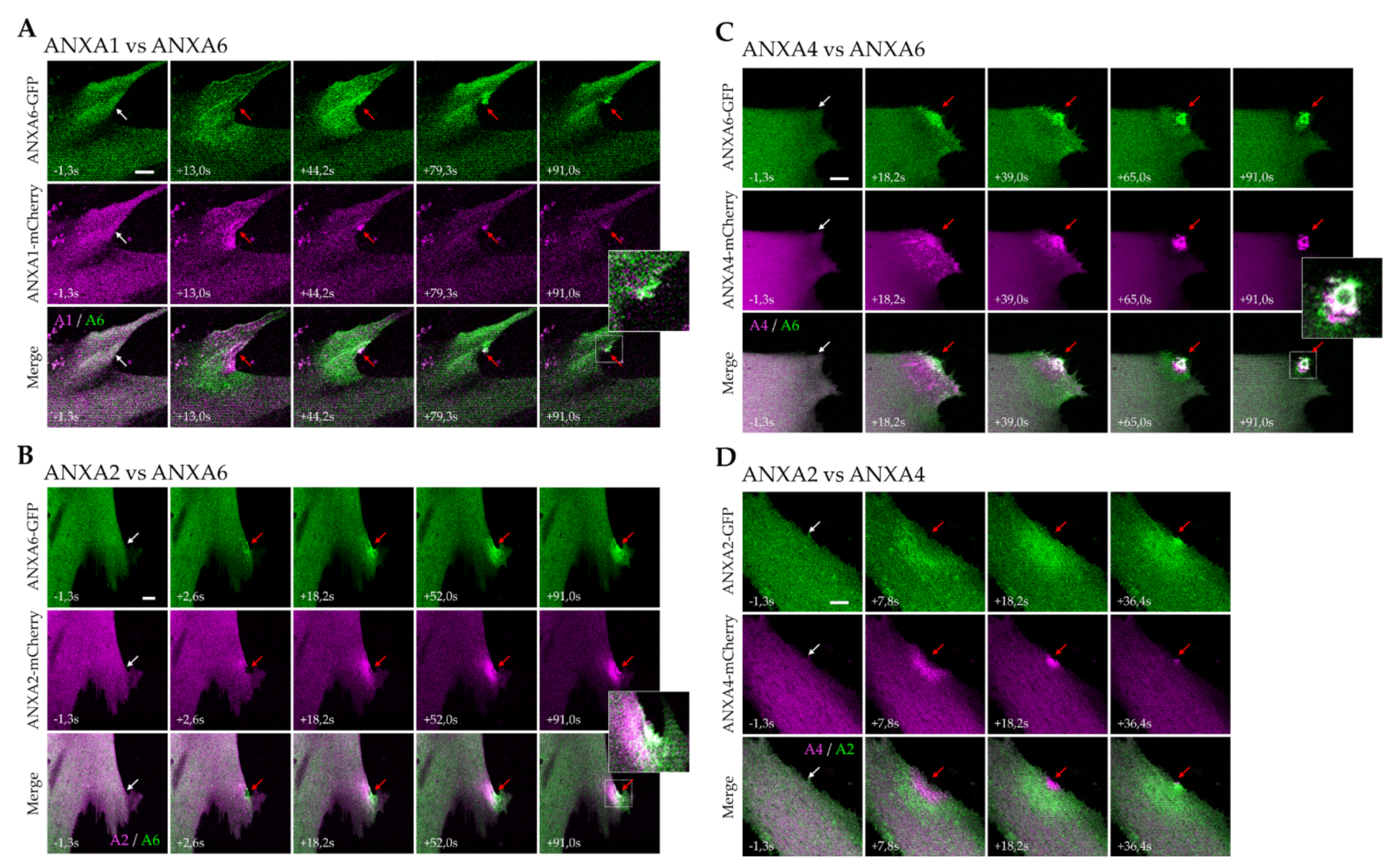
3.3. Kinetics of ANX Recruitment to the Site of Membrane Injury
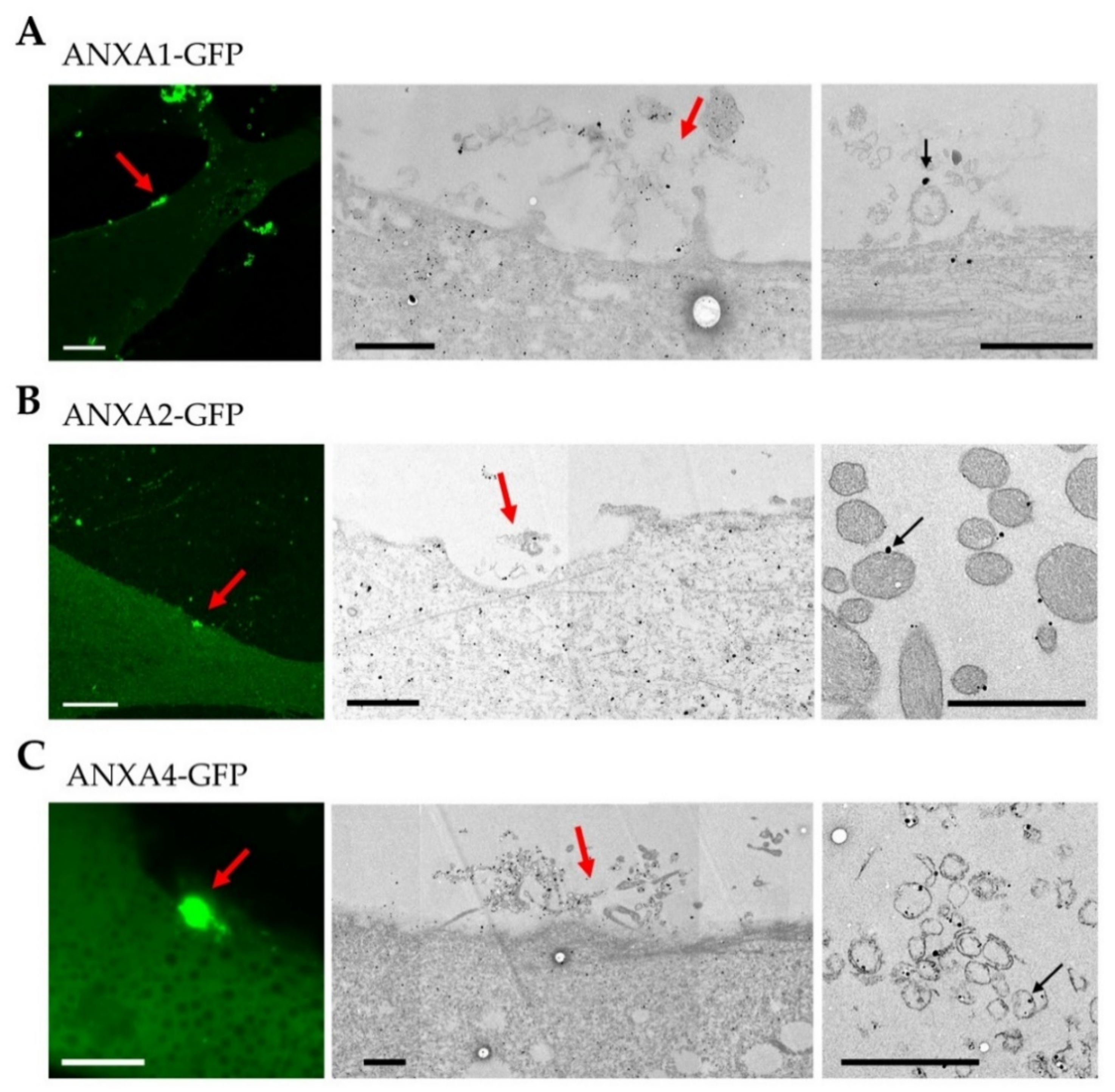
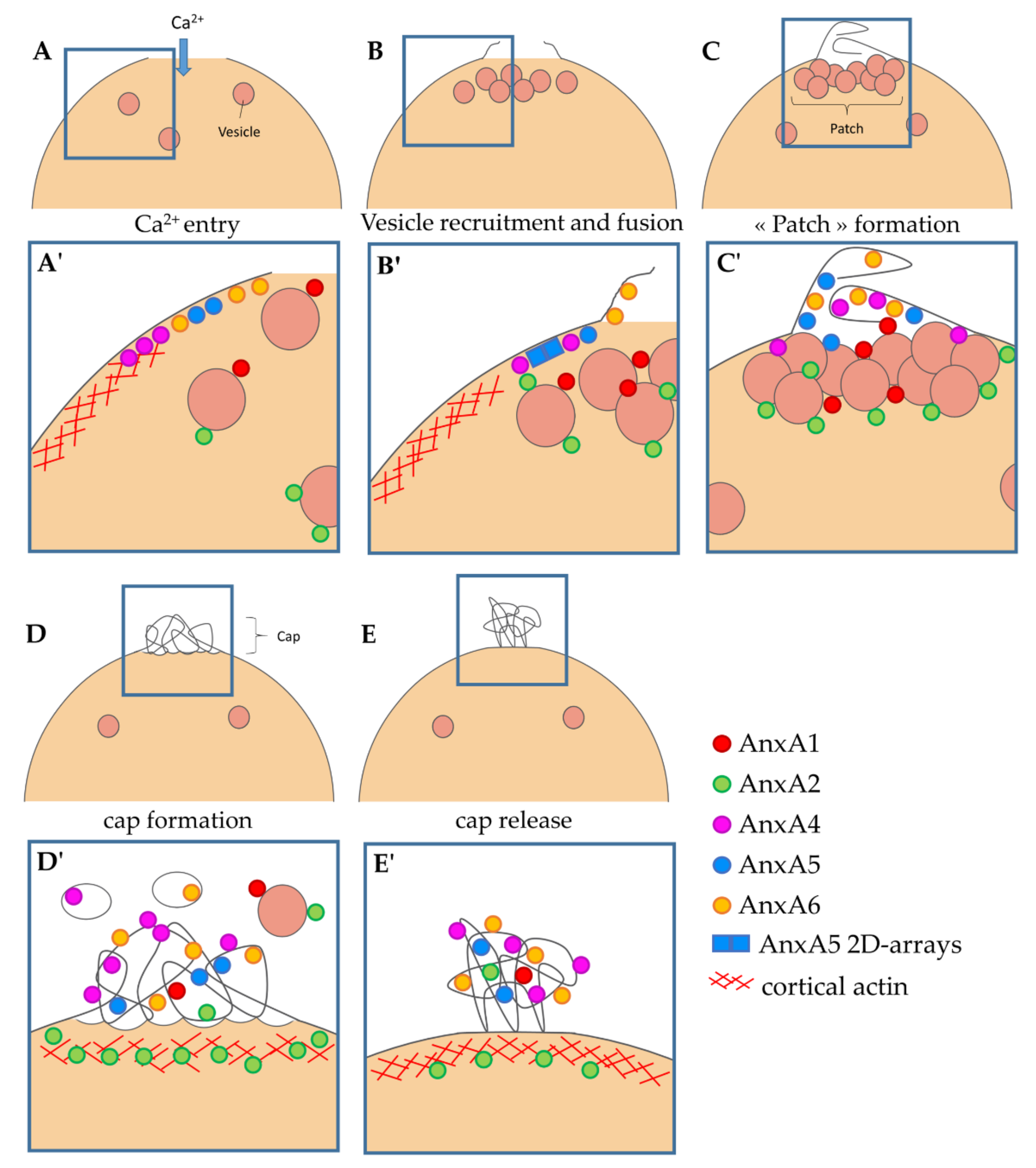
3.4. Localization of ANX at the Site of Membrane Injury Analyzed by CLEM
4. Discussion
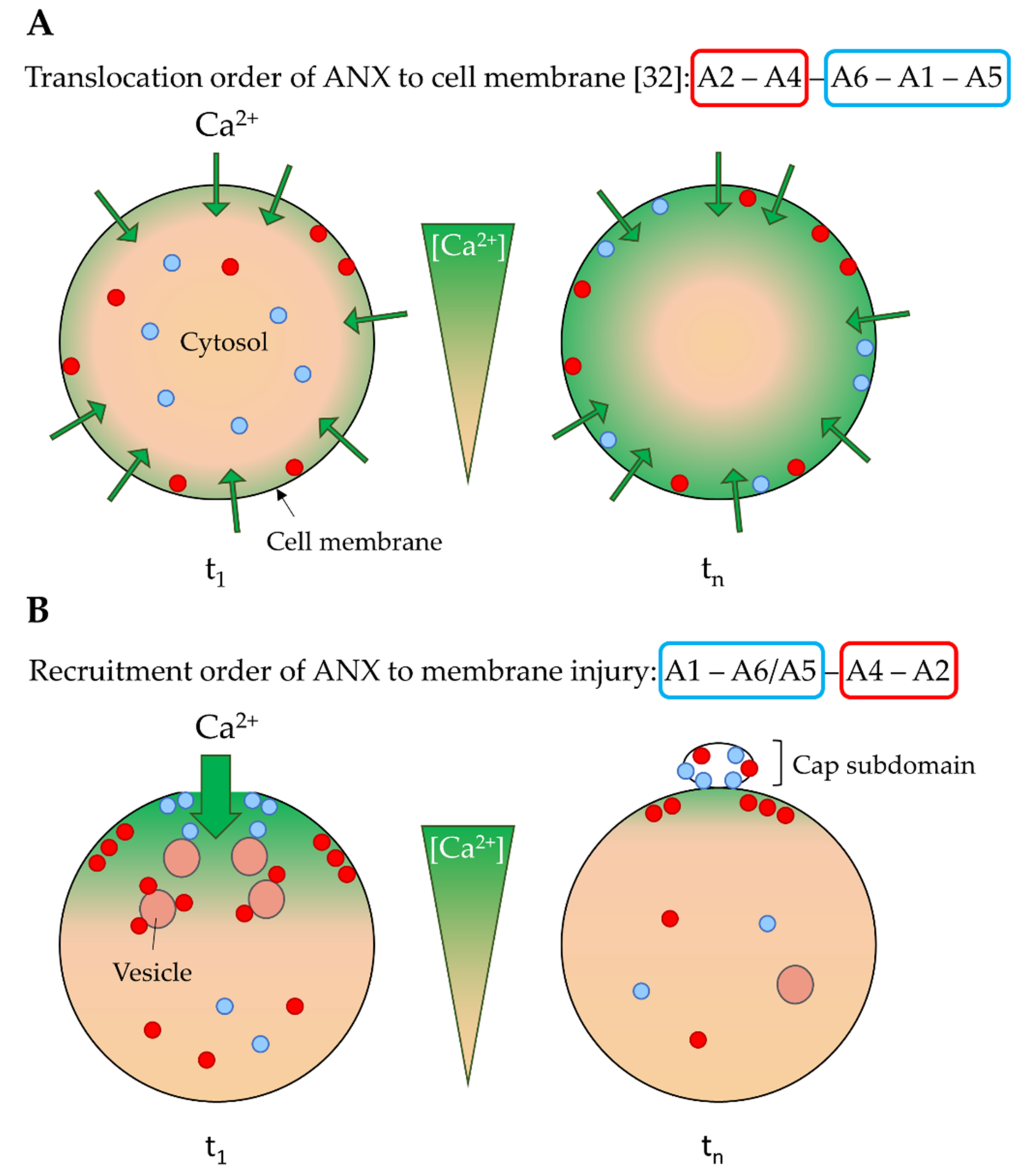
4.1. Ca2+ Sensitivity as a Driver of the ANX Machinery
4.2. Implication of ANX in Sarcolemma Repair
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McNeil, P.; Steinhardt, R.A. Plasma membrane disruption: Repair, prevention, adaptation. Annu. Rev. Cell Dev. Biol. 2003, 19, 697–731. [Google Scholar] [CrossRef]
- Blazek, A.D.; Paleo, B.J.; Weisleder, N. Plasma membrane repair: A central process for maintaining cellular homeostasis. Physiology 2015, 30, 438–448. [Google Scholar] [CrossRef]
- Croissant, C.; Carmeille, R.; Brévart, C.; Bouter, A. Annexins and membrane repair dysfunctions in muscular dystrophies. Int. J. Mol. Sci. 2021, 22, 5276. [Google Scholar] [CrossRef] [PubMed]
- Rescher, U.; Gerke, V. Annexins--unique membrane binding proteins with diverse functions. J. Cell Sci. 2004, 117, 2631–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swairjo, M.A.; Seaton, B.A. Annexin structure and membrane interactions: A molecular perspective. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 193–213. [Google Scholar] [CrossRef]
- McNeil, A.K.; Rescher, U.; Gerke, V.; McNeil, P.L. Requirement for annexin A1 in plasma membrane repair. J. Biol. Chem. 2006, 281, 35202–35207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, J.K.; Lauritzen, S.P.; Scheffer, L.; Sakaguchi, M.; Bunkenborg, J.; Simon, S.M.; Kallunki, T.; Jäättelä, M.; Nylandsted, J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014, 5, 3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623. [Google Scholar] [CrossRef] [Green Version]
- Sønder, S.L.; Boye, T.L.; Tölle, R.; Dengjel, J.; Maeda, K.; Jäättelä, M.; Simonsen, A.C.; Jaiswal, J.K.; Nylandsted, J. Annexin A7 is required for ESCRT III-mediated plasma membrane repair. Sci. Rep. 2019, 9, 6726. [Google Scholar] [CrossRef]
- Koerdt, S.N.; Gerke, V. Annexin A2 is involved in Ca2+-dependent plasma membrane repair in primary human endothelial cells. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 1046–1053. [Google Scholar] [CrossRef]
- Carmeille, R.; Degrelle, S.A.; Plawinski, L.; Bouvet, F.; Gounou, C.; Evain-Brion, D.; Brisson, A.R.; Bouter, A. Annexin-A5 promotes membrane resealing in human trophoblasts. Biochim. Biophys. Acta 2015, 1853, 2033–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouter, A.; Gounou, C.; Bérat, R.; Tan, S.; Gallois, B.; Granier, T.; D’Estaintot, B.L.B.L.; Pöschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roostalu, U.; Strähle, U. In vivo imaging of molecular interactions at damaged sarcolemma. Dev. Cell 2012, 22, 515–529. [Google Scholar] [CrossRef] [Green Version]
- Carmeille, R.; Bouvet, F.; Tan, S.; Croissant, C.; Gounou, C.; Mamchaoui, K.; Mouly, V.; Brisson, A.R.; Bouter, A. Membrane repair of human skeletal muscle cells requires Annexin-A5. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2267–2279. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef]
- Croissant, C.; Gounou, C.; Bouvet, F.; Tan, S.; Bouter, A. Annexin-A6 in membrane repair of human skeletal muscle cell: A role in the cap subdomain. Cells 2020, 9, 1742. [Google Scholar] [CrossRef]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 mediates dysferlin accumulation and muscle cell membrane repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.-C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Griffin, D.A.; Johnson, R.W.; Whitlock, J.M.; Pozsgai, E.R.; Heller, K.N.; Grose, W.E.; Arnold, W.D.; Sahenk, Z.; Hartzell, H.C.; Rodino-Klapac, L.R. Defective membrane fusion and repair in Anoctamin5 -deficient muscular dystrophy. Hum. Mol. Genet. 2016, 25, 1900–1911. [Google Scholar] [CrossRef] [Green Version]
- Cagliani, R.; Magri, F.; Toscano, A.; Merlini, L.; Fortunato, F.; Lamperti, C.; Rodolico, C.; Prelle, A.; Sironi, M.; Aguennouz, M.; et al. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum. Mutat. 2005, 26, 283. [Google Scholar] [CrossRef]
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Manera, J.D.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 2019, 10, 2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, M.L.; Weinheimer-Haus, E.M.; Koh, T.J. Macrophage activation and skeletal muscle healing following traumatic injury. J. Pathol. 2014, 232, 344–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.-H.; Mouly, V.; Cooper, R.N.; Mamchaoui, K.; Bigot, A.; Shay, J.W.; Di Santo, J.P.; Butler-Browne, G.S.; Wright, W.E. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: Consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell 2007, 6, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Geissbuehler, M.; Lasser, T. How to display data by color schemes compatible with red-green color perception deficiencies. Opt. Express 2013, 21, 9862. [Google Scholar] [CrossRef] [Green Version]
- Bouvet, F.; Ros, M.; Bonedeau, E.; Croissant, C.; Frelin, L.; Saltel, F.; Moreau, V.; Bouter, A. Defective membrane repair machinery impairs survival of invasive cancer cells. Sci Rep 2020, 10, 21821. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Monastyrskaya, K.; Burkhard, F.C.; Wray, S.; Draeger, A. Modulating signaling events in smooth muscle: Cleavage of annexin 2 abolishes its binding to lipid rafts. FASEB J. 2002, 16, 1177–1184. [Google Scholar] [CrossRef]
- Wang, C.Y.; Lin, C.F. Annexin A2: Its molecular regulation and cellular expression in cancer development. Dis. Markers 2014, 2014, 308976. [Google Scholar] [CrossRef] [Green Version]
- Grindheim, A.K.; Saraste, J.; Vedeler, A. Protein phosphorylation and its role in the regulation of Annexin A2 function. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 2515–2529. [Google Scholar] [CrossRef]
- Gabel, M.; Delavoie, F.; Royer, C.; Tahouly, T.; Gasman, S.; Bader, M.F.; Vitale, N.; Chasserot-Golaz, S. Phosphorylation cycling of Annexin A2 Tyr23 is critical for calcium-regulated exocytosis in neuroendocrine cells. Biochim. Biophys. Acta-Mol. Cell Res. 2019, 1866, 1207–1217. [Google Scholar] [CrossRef]
- Bizzarro, V.; Fontanella, B.; Franceschelli, S.; Pirozzi, M.; Christian, H.; Parente, L.; Petrella, A. Role of Annexin A1 in mouse myoblast cell differentiation. J. Cell. Physiol. 2010, 224, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Skrahina, T.; Piljić, A.; Schultz, C. Heterogeneity and timing of translocation and membrane-mediated assembly of different annexins. Exp. Cell Res. 2008, 314, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Piljić, A.; Schultz, C. Annexin A4 self-association modulates general membrane protein mobility in living cells. Mol. Biol. Cell 2006, 17, 3318–3328. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.-K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Kaetzel, M.A.; Mo, Y.D.; Mealy, T.R.; Campos, B.; Bergsma-Schutter, W.; Brisson, A.; Dedman, J.R.; Seaton, B.A. Phosphorylation mutants elucidate the mechanism of annexin IV-mediated membrane aggregation. Biochemistry 2001, 40, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- Croissant, C.; Bouvet, F.; Tan, S.; Bouter, A. Imaging Membrane Repair in Single Cells Using Correlative Light and Electron Microscopy. Curr. Protoc. Cell Biol. 2018, 81, e55. [Google Scholar] [CrossRef]
- Togo, T.; Krasieva, T.B.; Steinhardt, R.A. A Decrease in Membrane Tension Precedes Successful Cell-Membrane Repair. Mol. Biol. Cell 2000, 11, 4339–4346. [Google Scholar] [CrossRef] [Green Version]
- Boye, T.L.; Jeppesen, J.C.; Maeda, K.; Pezeshkian, W.; Solovyeva, V.; Nylandsted, J.; Simonsen, A.C. Annexins induce curvature on free-edge membranes displaying distinct morphologies. Sci. Rep. 2018, 8, 10309. [Google Scholar] [CrossRef]
- Middel, V.; Zhou, L.; Takamiya, M.; Beil, T.; Shahid, M.; Roostalu, U.; Grabher, C.; Rastegar, S.; Reischl, M.; Nienhaus, G.U.; et al. Dysferlin-mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nat. Commun. 2016, 7, 12875. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Croissant, C.; Gounou, C.; Bouvet, F.; Tan, S.; Bouter, A. Trafficking of Annexins during Membrane Repair in Human Skeletal Muscle Cells. Membranes 2022, 12, 153. https://doi.org/10.3390/membranes12020153
Croissant C, Gounou C, Bouvet F, Tan S, Bouter A. Trafficking of Annexins during Membrane Repair in Human Skeletal Muscle Cells. Membranes. 2022; 12(2):153. https://doi.org/10.3390/membranes12020153
Chicago/Turabian StyleCroissant, Coralie, Céline Gounou, Flora Bouvet, Sisareuth Tan, and Anthony Bouter. 2022. "Trafficking of Annexins during Membrane Repair in Human Skeletal Muscle Cells" Membranes 12, no. 2: 153. https://doi.org/10.3390/membranes12020153