
A Hydrogen-Bromate Flow Battery as a Rechargeable Chemical Power Source

 , , , , , and
, , , , , and
Abstract
:1. Introduction
BrO3− + 5 Br− + 6 H+ = 3 Br2 + 3 H2O in solution
A + Red = P + Ox in solution
2. Materials and Methods
2.1. Design of a Hydrogen-Bromate Redox Flow Battery
2.2. Methods
2.2.1. Electrochemical Methods and Operando-Spectroscopic Control of the Catholyte Composition during Electrochemical Tests of the Hydrogen-Bromate Flow Battery
2.2.2. Impedance Spectroscopy
2.2.3. Cyclic Voltammetry for Assessing the Degradation of the Positive Electrode Material
2.2.4. Investigation of a Hydrogen-Bromate Flow Battery Electrode Polarization
2.2.5. Square-Wave Voltammetry for Evaluating the Crossover of Br-Containing Compounds
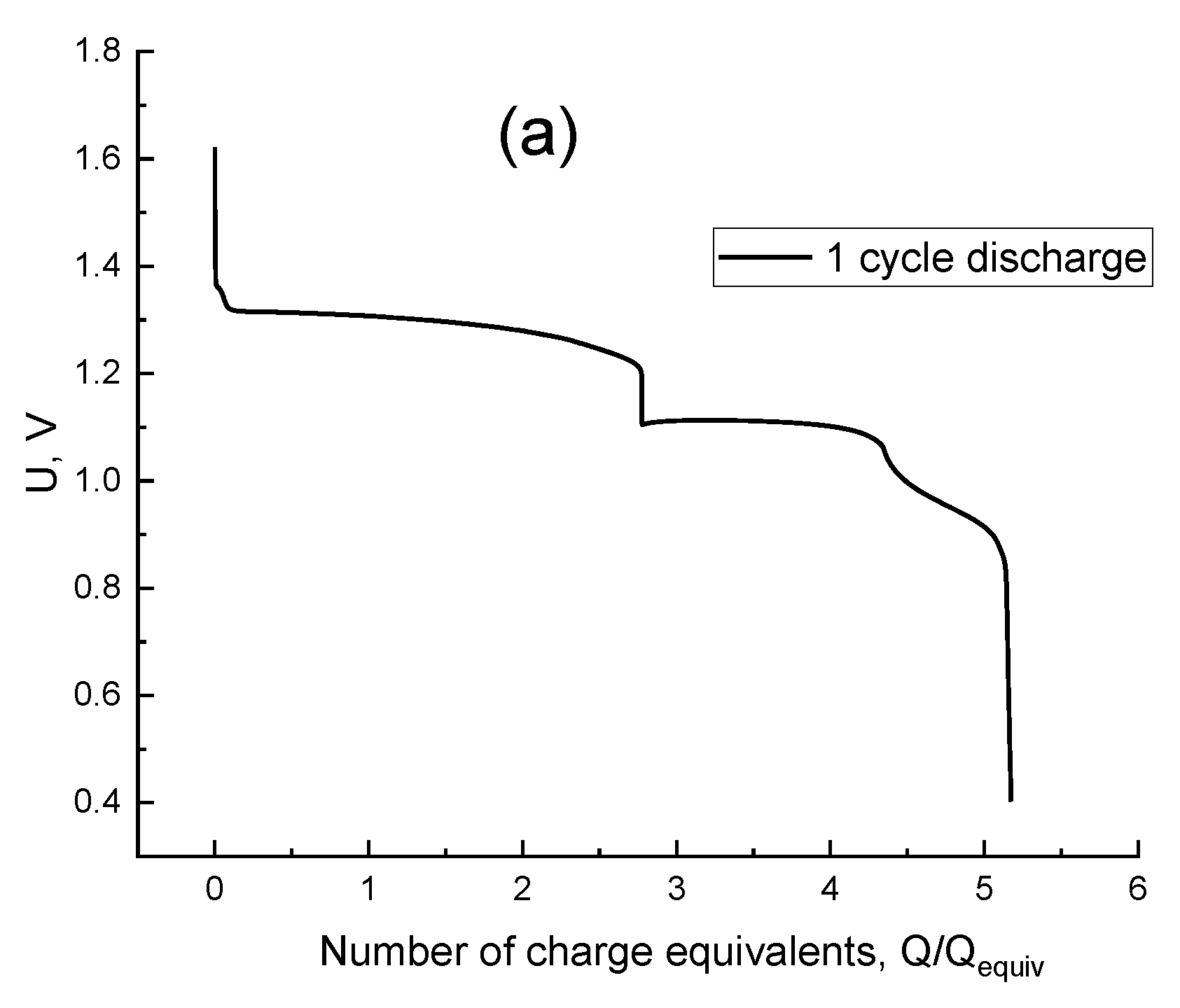
3. Results and Discussion
- Charges during the charging and discharging half-cycles, Qcharge and Qdischarge, respectively;
- Average voltage during charging and discharging half-cycles, Ucharge and Udischarge respectively;
- Coulombic efficiency (ηQ)—the ratio of the charge passed during discharge to the charge passed during charge in percent;
- Voltaic efficiency (ηU)—the ratio of the average discharge voltage to the average charge voltage in percent;
- Energy efficiency (ηE) of a hydrogen-bromate flow battery charge—discharge tests—the multiplication product of Coulombic and voltaic efficiencies;
- Capacity utilization (Qdischarge/Qtot)—the ratio of the charge received while discharging the battery to the theoretical charge in percent (6 × F × c × V, where F is Faraday’s constant, c is the concentration of electroactive compounds, V is the electrolyte volume).
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Perry, M.; Weber, A. Advanced Redox-Flow Batteries: A Perspective. J. Electrochem. Soc. 2016, 1, A5064. [Google Scholar] [CrossRef]
- Petrov, M.M.; Modestov, A.D.; Konev, D.V.; Antipov, A.E.; Loktionov, P.A. Pichugov, R.D.; Kartashova, N.V.; Glazkov, A.T.; Abunaeva, L.Z.; Andreev, V.N.; et al. Redox flow batteries: Role in modern electric power industry and comparative characteristics of the main types. Russ. Chem. Rev. 2021, 90, 677. [Google Scholar] [CrossRef]
- Leung, P.; Shah, A.A.; Sanz, L.; Flox, C.; Morante, J.R.; Xu, Q.; Mohamed, M.R.; Ponce de Leon, C.; Walsh, F.C. Recent developments in organic redox flow batteries: A critical review. J. Power Sources 2017, 360, 243. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Zhang, H. Review of the Development of First-Generation Redox Flow Batteries: Iron-Chromium System. ChemSusChem 2021, 15, e202101798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, C. Cost-effective iron-based aqueous redox flow batteries for large-scale energy storage application: A review. J. Power Sources 2021, 493, 229445. [Google Scholar] [CrossRef]
- Glazkov, A.T.; Antipov, A.E.; Konev, D.V.; Pichugov, R.D.; Petrov, M.M.; Kartashova, N.V.; Loktionov, P.A.; Averina, J.M.; Plotko, I.I. Dataset of a vanadium redox flow battery 10 membrane-electrode assembly stack. Data Brief 2020, 31, 105840. [Google Scholar] [CrossRef]
- Petrov, M.M.; Pichugov, R.D.; Loktionov, P.A.; Antipov, A.E.; Usenko, A.A.; Konev, D.V.; Vorotyntsev, M.A.; Mintsev, V.B. Test Cell for Membrane Electrode Assembly of the Vanadium Redox Flow Battery. Dokl. Phys. Chem. 2020, 491, 39. [Google Scholar] [CrossRef]
- Pichugov, R.D.; Konev, D.V.; Petrov, M.M.; Antipov, A.E.; Loktionov, P.A.; Abunaeva, L.Z.; Usenko, A.A.; Vorotyntsev, M.A. Electrolyte Flow Field Variation: A Cell for Testing and Optimization of Membrane Electrode Assembly for Vanadium Redox Flow Batteries. ChemPlusChem 2020, 85, 1919. [Google Scholar] [CrossRef]
- Huang, Z.; Mu, A. Research and analysis of performance improvement of vanadium redox flow battery in microgrid: A technology review. Int. J. Energy Res. 2021, 45, 14170. [Google Scholar] [CrossRef]
- Loktionov, P.; Kartashova, N.; Konev, D.; Abunaeva, L.; Antipov, A.; Ruban, E.; Terent’ev, A.; Gvozdik, N.; Lyange, M.; Usenko, A. Fluoropolymer impregnated graphite foil as a bipolar plates of vanadium flow battery. Int. J. Energy Res. 2021, 46, 10123–10132. [Google Scholar] [CrossRef]
- Modestov, A.D.; Andreev, V.N.; Antipov, A.E.; Petrov, M.M. Novel Aqueous Zinc–Halogenate Flow Batteries as an Offspring of Zinc–Air Fuel Cells for Use in Oxygen-Deficient Environment. Energy Technol. 2021, 9, 2100233. [Google Scholar] [CrossRef]
- Kim, M.; Yun, D.; Jeon, J. Effect of a bromine complex agent on electrochemical performances of zinc electrodeposition and electrodissolution in Zinc–Bromide flow battery. J. Power Sources 2019, 438, 227020. [Google Scholar] [CrossRef]
- Huang, Q.; Yang, J.; Ng, C.B.; Jia, C.; Wang, Q. A redox flow lithium battery based on the redox targeting reactions between LiFePO4 and iodide. Energy Environ. Sci. 2016, 9, 917. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Mousavie, S.M.A.; Oh, T.K.; Yang, T.; Lu, Y.; Farley, C.; Bodnar, R.J.; Niu, L.; Qiao, R.; Li, Z. Sodium–Sulfur Flow Battery for Low-Cost Electrical Storage. Adv. Energy Mater. 2018, 8, 1701991. [Google Scholar] [CrossRef]
- Wei, X.; Xu, W.; Vijayakumar, M.; Cosimbescu, L.; Liu, T.; Sprenkle, V.; Wang, W. TEMPO-Based Catholyte for High-Energy Density Nonaqueous Redox Flow Batteries. Adv. Mater. 2014, 26, 7649. [Google Scholar] [CrossRef]
- Cho, K.; Ridgway, P.; Weber, A.; Haussener, S.; Battaglia, V.; Srinivasan, V. High Performance Hydrogen/Bromine Redox Flow Battery for Grid-Scale Energy Storage. J. Electrochem. Soc. 2012, 159, A1806. [Google Scholar] [CrossRef]
- Perry, M.; Darling, R.; Zaffou, R. High Power Density Redox Flow Battery Cells. ECS Trans. 2013, 53, 7. [Google Scholar] [CrossRef]
- Oh, K.; Weber, A.Z.; Ju, H. Study of bromine species crossover in H2/Br2 redox flow batteries. Int. J. Hydrogen Energy 2017, 42, 3753–3766. [Google Scholar] [CrossRef]
- Cho, K.; Tucker, M.; Ding, M.; Ridgway, P.; Battaglia, V.; Srinivasan, V.; Weber, A. Cyclic Performance Analysis of Hydrogen/Bromine Flow Batteries for Grid-Scale Energy Storage. ChemPlusChem 2015, 80, 402. [Google Scholar] [CrossRef]
- Modestov, A.; Kartashova, N.; Pichugov, R.; Petrov, M.; Antipov, A.; Abunaeva, L. Bromine Crossover in Operando Analysis of Proton Exchange Membranes in Hydrogen−Bromate Flow Batteries. Membranes 2022, 12, 815. [Google Scholar] [CrossRef]
- Modestov, A.D.; Konev, D.V.; Tripachev, O.V.; Antipov, A.E.; Tolmachev, Y.V.; Vorotynsev, M.A. A Hydrogen–Bromate Flow Battery for Air-Deficient Environments. Energy Technol. 2018, 6, 242. [Google Scholar] [CrossRef]
- Modestov, A.D.; Konev, D.V.; Antipov, A.E.; Vorotyntsev, M.A. Hydrogen-bromate flow battery: Can one reach both high bromate utilization and specific power? J. Solid State Electrochem. 2019, 23, 3075–3088. [Google Scholar] [CrossRef]
- Tolmachev, Y.V.; Pyatkivskiy, A.; Ryzhov, V.V.; Konev, D.V.; Vorotyntsev, M.A. Energy cycle based on a high specific energy aqueous flow battery and its potential use for fully electric vehicles and for direct solar-to-chemical energy conversion. J. Solid State Electrochem. 2015, 19, 2711–2722. [Google Scholar] [CrossRef]
- Konev, D.V.; Istakova, O.I.; Vorotyntsev, M.A. Electrochemical measurement of interfacial distribution and diffusion coefficients of electroactive species for ion-exchange membranes. Membranes 2022, 12, 1041. [Google Scholar] [CrossRef]
- Konev, D.V.; Istakova, O.I.; Kartashova, N.V.; Abunaeva, L.Z.; Pyrkov, P.V.; Loktionov, P.A.; Vorotyntsev, M.A. Electrochemical Measurement of Co-Ion Diffusion Coefficient in Ion-Exchange Membranes. Russ. J. Electrochem. 2022, 58, 1103–1113. [Google Scholar] [CrossRef]
- Fleischmann, M.; Lasserre, F.; Robinson, J.; Swan, D. The application of microelectrodes to the study of homogeneous processes coupled to electrode reactions: Part I. EC0 and CE reactions. J. Electroanal. Chem. Interfacial Electrochem. 1984, 177, 97–114. [Google Scholar] [CrossRef]
- Compton, R.G.; Day, M.J.; Laing, M.E.; Northing, R.J.; Penman, J.I.; Waller, A.M. Rotating-disc electrode voltammetry. The catalytic mechanism (EC’) and its nuances. J. Chem. Soc. Faraday Trans. I 1988, 84, 2013–2025. [Google Scholar] [CrossRef]
- Denuault, G.; Fleischmann, M.; Pletcher, D.; Tutty, O.R. Development of the theory for the interpretation of steady state limiting currents at a microelectrode: EC’ processes: First and second order reactions. J. Electroanal. Chem. Interfacial Electrochem. 1990, 280, 243–254. [Google Scholar] [CrossRef]
- Denuault, G.; Pletcher, D. Improvement to the equation for the steady state limiting currents at a microelectrode: EC’ processes (1st and 2nd order reactions). J. Electroanal. Chem. Interfacial Electrochem. 1991, 305, 131–134. [Google Scholar] [CrossRef]
- Lavagnini, I.; Pastore, P.; Magno, F. Digital simulation of steady state and non-steady state voltammetric responses for electrochemical reactions occurring at an inlaid microdisk electrode: Application to ECirr, EC’ and CE first-order reactions. J. Electroanal. Chem. 1993, 358, 193–201. [Google Scholar] [CrossRef]
- Tutty, O.R. Second-order kinetics for steady state EC0 reactions at a disc microelectrode. J. Electroanal. Chem. 1994, 377, 39–51. [Google Scholar] [CrossRef]
- Molina, A. Analytical solution corresponding to the i/t response to a multipotential step for a catalytic mechanism. J. Electroanal. Chem. 1998, 443, 163–167. [Google Scholar] [CrossRef]
- Molina, A.; Serna, C.; Martinez-Ortiz, F. Square wave voltammetry for a pseudo-first-order catalytic process at spherical electrodes. J. Electroanal. Chem. 2000, 486, 9–15. [Google Scholar] [CrossRef]
- Mirceski, V.; Gulaboski, R. Surface catalytic mechanism in square-wave voltammetry. Electroanalysis 2001, 13, 1326–1334. [Google Scholar] [CrossRef]
- Mirceski, V.; Gulaboski, R. The surface catalytic mechanism: A comparative study with square-wave and staircase cyclic voltammetry. J. Solid State Electrochem. 2003, 7, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Compton, R.; Banks, C.E. Understanding Voltammetry, 2nd ed.Imperial College Press: London, UK, 2011. [Google Scholar]
- Molina, A.; Gonzalez, J.; Laborda, E.; Wang, Y.; Compton, R.G. Analytical theory of the catalytic mechanism in square wave voltammetry at disc electrodes. Phys. Chem. Chem. Phys. 2011, 13, 16748–16755. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.R.; Lawrence, N.S.; Hartshorne, R.S.; Compton, R.G. Cyclic voltammetry of the EC’ mechanism at hemispherical particles and their arrays: The split wave. J. Phys. Chem. C 2011, 115, 11204–11215. [Google Scholar] [CrossRef]
- Calhoun, R.L.; Bard, A.J. Study of the EC’ mechanism by scanning electrochemical microscopy (SECM). ECS Trans. 2011, 35, 39–51. [Google Scholar] [CrossRef]
- Gulaboski, R.; Mihajlov, L. Catalytic mechanism in successive two-step protein- film voltammetry—Theoretical study in square-wave voltammetry. Biophys. Chem. 2011, 155, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gulaboski, R.; Mirceski, V.; Bogeski, I.; Hoth, M. Protein film voltammetry: Electrochemical enzymatic spectroscopy: A review on recent progress. J. Solid State Electrochem. 2012, 16, 2315–2328. [Google Scholar] [CrossRef]
- Yue, D.; Jia, Y.; Yao, Y.; Sun, J.; Jing, Y. Structure and electrochemical behavior of ionic liquid analogue based on choline chloride and urea. Electrochim. Acta 2012, 65, 30–36. [Google Scholar] [CrossRef]
- Gulaboski, R.; Mirceski, V. New aspects of the electrochemical-catalytic (EC’) mechanism in square-wave voltammetry. Electrochim. Acta 2015, 167, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Vorotyntsev, M.A.; Konev, D.V.; Tolmachev, Y.V. Electroreduction of halogen oxoanions via autocatalytic redox mediation by halide anions: Novel EC” mechanism. Theory for stationary 1D regime. Electrochim. Acta 2015, 173, 779. [Google Scholar] [CrossRef]
- Vorotyntsev, M.A.; Antipov, A.E. Reduction of bromate anion via autocatalytic redox-mediation by Br2/Br− redox couple. Theory for stationary 1D regime. Effect of different Nernst layer thicknesses for reactants. J. Electroanal. Chem. 2016, 779, 146. [Google Scholar] [CrossRef]
- Vorotyntsev, M.A.; Antipov, A.E.; Konev, D.V. Bromate anion reduction: Novel autocatalytic (EC″) mechanism of electrochemical processes. Its implication for redox flow batteries of high energy and power densities. Pure Appl. Chem. 2017, 89, 1429. [Google Scholar] [CrossRef]
- Vorotyntsev, M.A.; Antipov, A.E. Bromate electroreduction from acidic solution at spherical microelectrode under steady-state conditions: Theory for the redox-mediator autocatalytic (EC″) mechanism. Electrochim. Acta 2017, 258, 544. [Google Scholar] [CrossRef]
- Vorotyntsev, M.A.; Antipov, A.E. Bromate electroreduction from acidic solution at rotating disc electrode. Theoretical study of the steady-state convective-diffusion transport for excess of bromate ions compared to proton. Electrochim. Acta 2018, 261, 113. [Google Scholar] [CrossRef]
- Vorotyntsev, M.A.; Volgin, V.M.; Davydov, A.D. Halate electroreduction from acidic solution at rotating disc electrode. Theoretical study of the steady-state convective-migration-diffusion transport for comparable concentrations of halate ions and protons. Electrochim. Acta 2022, 409, 139961. [Google Scholar] [CrossRef]
- Modestov, A.D.; Konev, D.V.; Antipov, A.E.; Petrov, M.M.; Pichugov, R.D.; Vorotynsev, M.A. Bromate electroreduction from sulfuric acid solution at rotating disk electrode: Experimental study. Electrochim. Acta 2018, 259, 655. [Google Scholar] [CrossRef]
- Konev, D.V.; Antipov, A.E.; Petrov, M.M.; Shamraeva, M.A.; Vorotynsev, M.A. Surprising dependence of the current density of bromate electroreduction on the microelectrode radius as manifestation of the autocatalytic redox-cycle (EC″) reaction mechanism. Electrochem. Commun. 2018, 86, 76. [Google Scholar] [CrossRef]
- Petrov, M.M.; Konev, D.V.; Kuznetsov, V.V.; Antipov, A.E.; Glazkov, A.T.; Vorotyntsev, M.A. Electrochemically driven evolution of Br-containing aqueous solution composition. J. Electroanal. Chem. 2019, 836, 125–133. [Google Scholar] [CrossRef]
- Petrov, M.M.; Konev, D.V.; Antipov, A.E.; Kartashova, N.V.; Kuznetsov, V.V.; Vorotyntsev, M.A. Theoretical Analysis of Changes in the Solution Composition during Anodic Electrolysis of Bromide. Russ. J. Electrochem. 2019, 55, 1058. [Google Scholar] [CrossRef]
- Petrov, M.M.; Konev, D.V.; Antipov, A.E.; Kartashova, N.V.; Kuznetsov, V.V.; Vorotyntsev, M.A. Theoretical Analysis of Changes in the System’s Composition in the Course of Oxidative Electrolysis of Bromide Solution: pH Dependence. Russ. J. Electrochem 2020, 56, 883. [Google Scholar] [CrossRef]
- Pichugov, R.D.; Konev, D.V.; Speshilov, I.O.; Abunaeva, L.Z.; Petrov, M.M.; Vorotyntsev, M.A. Analysis of the composition of bromide anion oxidation products in aqueous solutions with different pH via rotating ring-disk electrode method. Membranes 2022, 12, 820. [Google Scholar] [CrossRef] [PubMed]
- Grgur, B.N. Electrochemical Oxidation of Bromides on DSA/RuO2 Anode in the Semi-Industrial Batch Reactor for On-Site Water Disinfection. J. Electrochem. Soc. 2019, 166, E50. [Google Scholar] [CrossRef]
- Cettou, P.; Robertson, P.; Ibl, N. On the electrolysis of aqueous bromide solutions to bromate. Electrochim. Acta 1984, 29, 875. [Google Scholar] [CrossRef]
- Osuga, T.; Sugino, K. Electrolytic Production of Bromates. J. Electrochem. Soc. 1957, 104, 448. [Google Scholar] [CrossRef]
- Pavlovic, O.Z. Formation of bromates at a RuO2/TiO2 titanium anode. Surf. Coat. Technol. 1988, 37, 177. [Google Scholar] [CrossRef]
- Henry Bergmann, M.E.; Iourtchouk, T.; Rollin, J. The occurrence of bromate and perbromate on BDD anodes during electrolysis of aqueous systems containing bromide: First systematic experimental studies. J. Appl. Electrochem. 2011, 41, 1109. [Google Scholar] [CrossRef]
- Vacca, A.; Mascia, M.; Palmas, S.; Mais, L.; Rizzardini, S. On the formation of bromate and chlorate ions during electrolysis with boron doped diamond anode for seawater treatment. J. Chem. Technol. Biotechnol. 2013, 88, 2244. [Google Scholar] [CrossRef]
- Konev, D.V.; Antipov, A.E.; Vorotyntsev, M.A.; Loktionov, P.A.; Glazkov, A.T.; Pichugov, R.D.; Petrov, M.M. Cell for Spectrophotometry of Electrolytes in the Process of Electrochemical Research. Russia Patent 190893, 22 January 2019. [Google Scholar]
- Konev, D.V.; Vorotyntsev, M.A.; Loktionov, P.A.; Kartashova, N.V.; Antipov, A.E.; Modestov, A.D.; Glazkov ATAbunaeva, L.Z. Luggin Capillary Device for Membrane-Electrode Assemblies of Flow Electrochemical Reactors and Power Sources. Russia Patent 198483, 13 July 2020. [Google Scholar]
- Mussini, T.; Longhi, P. The Halogens. Bromine. In Standard Potentials in Aqueous Solutions, 1st ed.; Bard, A.J., Parsons, R., Jordan, J., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1985. [Google Scholar]
- Toth, Z.; Fabian, I. Oxidation of Chlorine (III) by Hypobromous Acid: Kinetics and Mechanism. Inorg. Chem. 2004, 43, 2717. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.X.; Kelley, M.D.; Cooper, J.N.; Beckwith, R.C.; Margerum, D.W. Equilibrium, Kinetic, and UV-Spectral Characteristics of Aqueous Bromine Chloride, Bromine, and Chlorine Species. Inorg. Chem. 1994, 33, 5872. [Google Scholar] [CrossRef]
- Betts, R.H.; Mackenzie, A.N. Formation and stability of hydrobromous acid in perchloric acid solutions of bromine and bromate ions. Can. J. Chem. 1951, 29, 666–677. [Google Scholar] [CrossRef]
- Petrov, M.M.; Loktionov, P.A.; Konev, D.V.; Antipov, A.E.; Astafiev, E.A.; Vorotyntsev, M.A. Evolution of Anolyte Composition in the Oxidative Electrolysis of Sodium Bromide in a Sulfuric Acid Medium. Russ. J. Electrochem. 2019, 55, 95–105. [Google Scholar] [CrossRef]
- Li, H.; Tang, Y.; Wang, Z.; Shi, Z.; Wu, S.; Song, D.; Zhang, J.; Fatih, K.; Zhang, J.; Wang, H.; et al. A review of water flooding issues in the proton exchange membrane fuel cell. J. Power Sources 2008, 178, 103–117. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Qcharge, ×103 Kл | Qdischarge, ×103 C | Ucharge, V | Udischarge, V | ηQ, % | ηU, % | ηE, % | Qdischarge/Qtot, % |
|---|---|---|---|---|---|---|---|---|
| 1 | 3.1 | 1.9 | 1.6 | 1.2 | 61 | 75 | 46 | 90 |
| 2 | 2.3 | 1.6 | 1.6 | 1.1 | 70 | 69 | 48 | 70 |
| 3 | 1.1 | 0.9 | 1.6 | 1 | 82 | 62 | 51 | 40 |
| 4 | 0.5 | 0.4 | 1.5 | 1 | 80 | 67 | 54 | 20 |
| Amount of Bromine Atoms Before Cycling, mmol | Amount of Bromine Atoms After Cycling (Figure 5), mmol | Relative Change in Bromine Content, % | |
|---|---|---|---|
| Catholyte | 3.98 | 3.07 | 77 |
| Escaping hydrogen trap | 0 | 0.06 | 1.6 |
| Anodic half-cell | 0 | 0.06 | 1.6 |
| Total amount of bromine atoms | 3.98 | 3.19 | 80.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kartashova, N.V.; Konev, D.V.; Loktionov, P.A.; Glazkov, A.T.; Goncharova, O.A.; Petrov, M.M.; Antipov, A.E.; Vorotyntsev, M.A. A Hydrogen-Bromate Flow Battery as a Rechargeable Chemical Power Source. Membranes 2022, 12, 1228. https://doi.org/10.3390/membranes12121228
Kartashova NV, Konev DV, Loktionov PA, Glazkov AT, Goncharova OA, Petrov MM, Antipov AE, Vorotyntsev MA. A Hydrogen-Bromate Flow Battery as a Rechargeable Chemical Power Source. Membranes. 2022; 12(12):1228. https://doi.org/10.3390/membranes12121228
Chicago/Turabian StyleKartashova, Natalia V., Dmitry V. Konev, Pavel A. Loktionov, Artem T. Glazkov, Olga A. Goncharova, Mikhail M. Petrov, Anatoly E. Antipov, and Mikhail A. Vorotyntsev. 2022. "A Hydrogen-Bromate Flow Battery as a Rechargeable Chemical Power Source" Membranes 12, no. 12: 1228. https://doi.org/10.3390/membranes12121228