
Effects of the RNA-Polymerase Inhibitors Remdesivir and Favipiravir on the Structure of Lipid Bilayers—An MD Study


Abstract
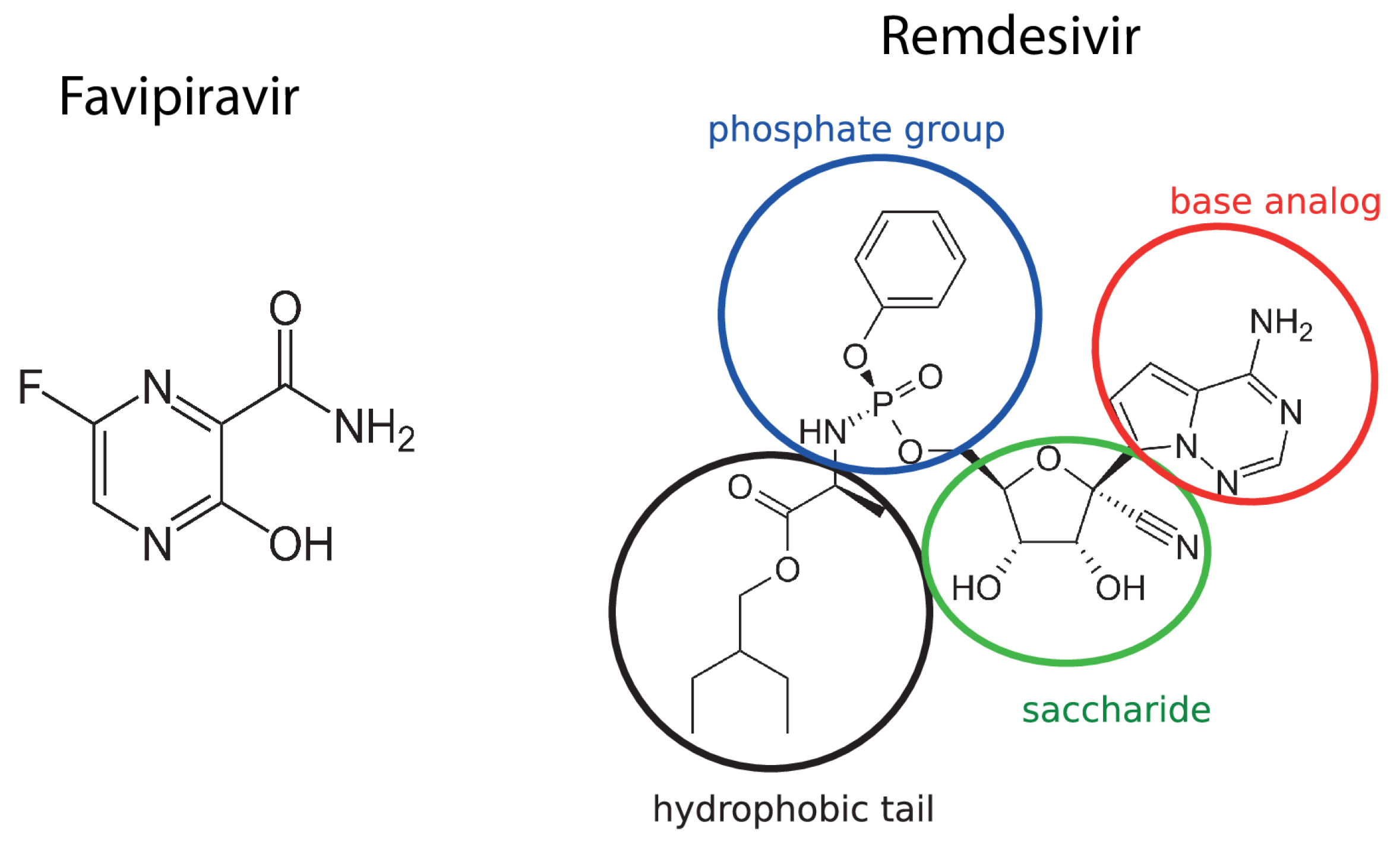
:1. Introduction
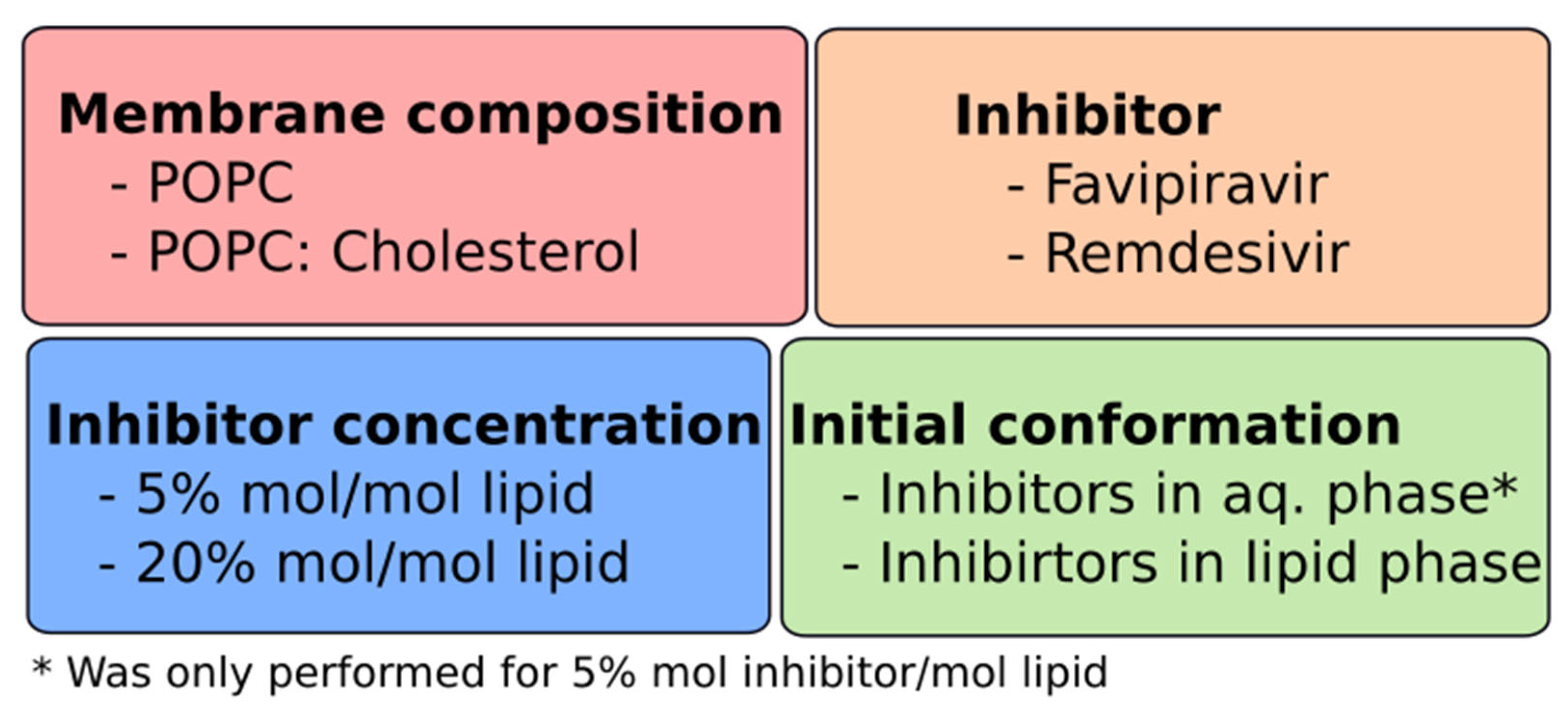
2. Computational Methods
3. Results
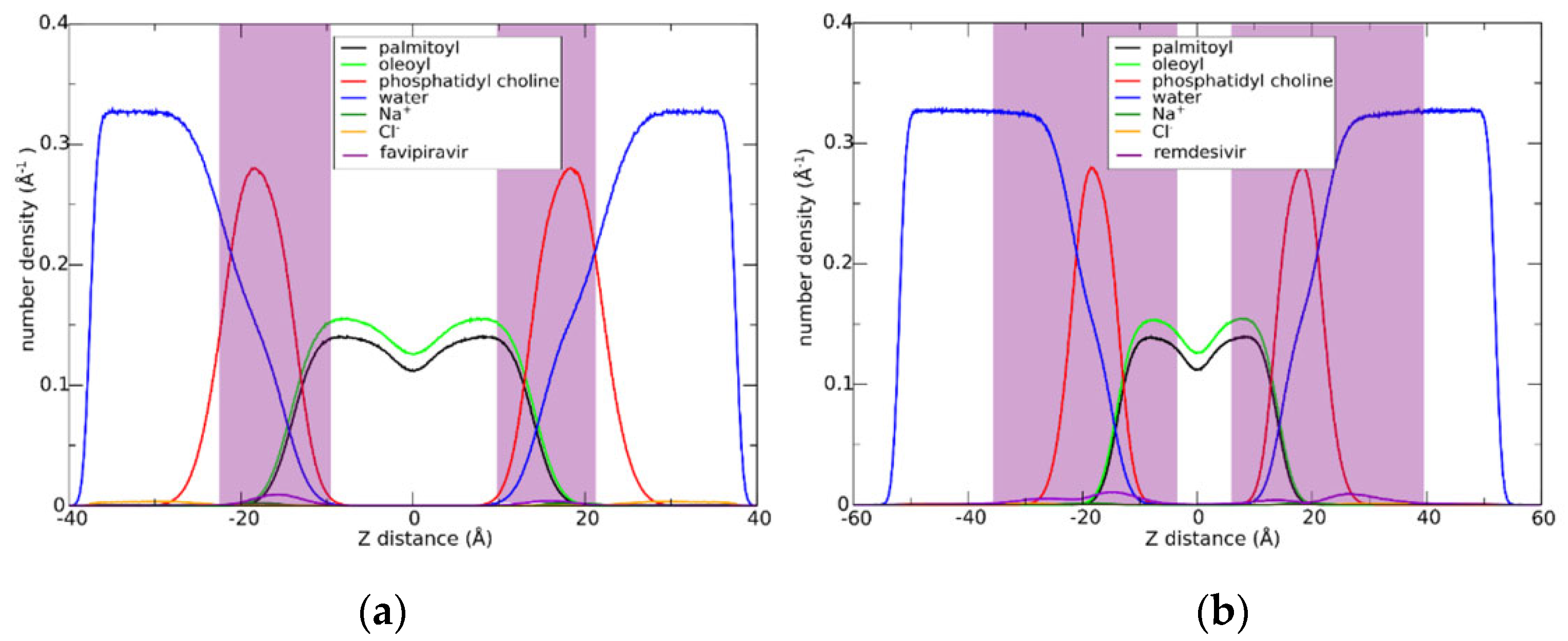
3.1. Drugs Insertion into the Membrane
3.2. Drugs Interactions with the Membranes
3.3. Effect of Drug Insertion in the Membrane Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laggner, P. Life—As a matter of fat—The emerging science of lipidomics edited by ole G. Mouritsen. Eur. J. Lipid Sci. Technol. 2007, 109, 1237. [Google Scholar] [CrossRef]
- Lopes, D.; Jakobtorweihen, S.; Nunes, C.; Sarmento, B.; Reis, S. Shedding light on the puzzle of drug-membrane interactions: Experimental techniques and molecular dynamics simulations. Prog. Lipid Res. 2017, 65, 24–44. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.I.; Saha, S.C.; Deplazes, E. Phenolic compounds alter the ion permeability of phospholipid bilayers via specific lipid interactions. Phys. Chem. Chem. Phys. 2021, 23, 22352–22366. [Google Scholar] [CrossRef] [PubMed]
- Haralampiev, I.; Scheidt, H.A.; Abel, T.; Luckner, M.; Herrmann, A.; Huster, D.; Müller, P. The interaction of sorafenib and regorafenib with membranes is modulated by their lipid composition. Biochim. Biophys. Acta 2016, 1858, 2871–2881. [Google Scholar] [CrossRef]
- Scheidt, H.A.; Haralampiev, I.; Theisgen, S.; Schirbel, A.; Sbiera, S.; Huster, D.; Kroiss, M.; Müller, P. The adrenal specific toxicant mitotane directly interacts with lipid membranes and alters membrane properties depending on lipid composition. Mol. Cell. Endocrinol. 2016, 428, 68–81. [Google Scholar] [CrossRef]
- Heimburg, T. Thermal Biophysics of Membranes; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Moradi, S.; Nowroozi, A.; Shahlaei, M. Shedding light on the structural properties of lipid bilayers using molecular dynamics simulation: A review study. RSC Adv. 2019, 9, 4644–4658. [Google Scholar] [CrossRef]
- Zhuang, X.; Dávila-Contreras, E.M.; Beaven, A.H.; Im, W.; Klauda, J.B. An extensive simulation study of lipid bilayer properties with different head groups, acyl chain lengths, and chain saturations. Biochim. Biophys. Acta 2016, 1858, 3093–3104. [Google Scholar] [CrossRef]
- Vattulainen, I.; Róg, T. Lipid membranes: Theory and simulations bridged to experiments. Biochim. Biophys. Acta 2016, 1858, 2251–2253. [Google Scholar] [CrossRef]
- Zhuang, X.; Makover, J.R.; Im, W.; Klauda, J.B. A systematic molecular dynamics simulation study of temperature dependent bilayer structural properties. Biochim. Biophys. Acta 2014, 1838, 2520–2529. [Google Scholar] [CrossRef] [Green Version]
- Haralampiev, I.; de Armiño, D.J.A.; Luck, M.; Fischer, M.; Abel, T.; Huster, D.; Di Lella, S.; Scheidt, H.A.; Müller, P. Interaction of the small-molecule kinase inhibitors tofacitinib and lapatinib with membranes. Biochim. Biophys. Acta 2020, 1862, 183414. [Google Scholar] [CrossRef]
- Khajeh, A.; Modarress, H. The influence of cholesterol on interactions and dynamics of ibuprofen in a lipid bilayer. Biochim. Biophys. Acta 2014, 1838, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Boggara, M.B.; Mihailescu, M.; Krishnamoorti, R. Structural Association of Nonsteroidal Anti-Inflammatory Drugs with Lipid Membranes. J. Am. Chem. Soc. 2012, 134, 19669–19676. [Google Scholar] [CrossRef] [PubMed]
- Manabe, T.; Kambayashi, D.; Akatsu, H.; Kudo, K. Favipiravir for the treatment of patients with COVID-19: A systematic review and meta-analysis. BMC Infect. Dis. 2021, 21, 489. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Preliminary report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Adhikari, N.K.J.; Kwon, H.; Teo, K.; Siemieniuk, R.; Lamontagne, F.; Chan, A.; Mishra, S.; Murthy, S.; Kiiza, P.; et al. Anti-Ebola therapy for patients with Ebola virus disease: A systematic review. BMC Infect. Dis. 2019, 19, 376. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.K.; Jordan, R.; Arvey, A.; Sudhamsu, J.; Shrivastava-Ranjan, P.; Hotard, A.L.; Flint, M.; McMullan, L.; Siegel, D.; Clarke, M.O.; et al. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Sci. Rep. 2017, 7, srep43395. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B 2017, 93, 449–463. [Google Scholar] [CrossRef]
- Fischer, M.; Müller, P.; Scheidt, H.A.; Luck, M. Drug–Membrane Interactions: Effects of Virus-Specific RNA-Dependent RNA Polymerase Inhibitors Remdesivir and Favipiravir on the Structure of Lipid Bilayers. Biochemistry 2022, 61, 1392–1403. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D., III; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Dickson, C.J.; Walker, R.C.; Gould, I.R. Lipid21: Complex Lipid Membrane Simulations with AMBER. J. Chem. Theory Comput. 2022, 18, 1726–1736. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., 3rd. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Ide, S.; Saito, S.; Akazawa, T.; Furuya, T.; Masuda, J.; Nagashima, M.; Asai, Y.; Ogawa, T.; Yamamoto, R.; Ishioka, H.; et al. Extracorporeal membrane oxygenation may decrease the plasma concentration of remdesivir in a patient with severe coronavirus disease 2019. IDCases 2021, 26, e01343. [Google Scholar] [CrossRef]
- Fresta, M.; Guccione, S.; Beccari, A.R.; Furneri, P.M.; Puglisi, G. Combining molecular modeling with experimental methodologies: Mechanism of membrane permeation and accumulation of ofloxacin. Bioorganic Med. Chem. 2002, 10, 3871–3889. [Google Scholar] [CrossRef]
- Brien, Z.; Moghaddam, M.F. A Systematic Analysis of Physicochemical and ADME Properties of All Small Molecule Kinase Inhibitors Approved by US FDA from January 2001 to October 2015. Curr. Med. Chem. 2017, 24, 3159–3184. [Google Scholar]
- Lee, C.M.; Kang, M.-A.; Bae, J.S.; Park, K.; Yang, Y.-H.; Lee, J.; Jang, K.Y.; Park, S.-H. An in vitro study on anti-carcinogenic effect of remdesivir in human ovarian cancer cells via generation of reactive oxygen species. Hum. Exp. Toxicol. 2022, 41, 09603271221089257. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.; König, J.; Hofmann, U.; Kölz, C.; Fromm, M.; Schwab, M. Interaction of Remdesivir with Clinically Relevant Hepatic Drug Uptake Transporters. Pharmaceutics 2021, 13, 369. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Favipiravir | Remdesivir | |||
|---|---|---|---|---|
| POPC | POPC:Cholesterol | POPC | POPC:Cholesterol | |
| POPC | 34 ± 1 | 27.6 ± 0.4 | 18.4 ± 0.4 | 19.1 ± 0.3 |
| Cholesterol | - | 2.4 0.1 | - | 1.3 ± 0.1 |
| Favipiravir/Remdesivir | 0.3 ± 0.1 | 0.4 ± 0.05 | 4.8 ± 0.2 | 7.4 ± 0.2 |
| POPC (Å2) | POPC:Cholesterol (Å2) | |
|---|---|---|
| Favipiravir | 64 ± 1 | 54.0 ± 0.9 |
| Remdesivir | 65 ± 1 | 55 ± 1 |
| control | 64 ± 1 | 59.9 ± 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bringas, M.; Luck, M.; Müller, P.; Scheidt, H.A.; Di Lella, S. Effects of the RNA-Polymerase Inhibitors Remdesivir and Favipiravir on the Structure of Lipid Bilayers—An MD Study. Membranes 2022, 12, 941. https://doi.org/10.3390/membranes12100941
Bringas M, Luck M, Müller P, Scheidt HA, Di Lella S. Effects of the RNA-Polymerase Inhibitors Remdesivir and Favipiravir on the Structure of Lipid Bilayers—An MD Study. Membranes. 2022; 12(10):941. https://doi.org/10.3390/membranes12100941
Chicago/Turabian StyleBringas, Mauro, Meike Luck, Peter Müller, Holger A. Scheidt, and Santiago Di Lella. 2022. "Effects of the RNA-Polymerase Inhibitors Remdesivir and Favipiravir on the Structure of Lipid Bilayers—An MD Study" Membranes 12, no. 10: 941. https://doi.org/10.3390/membranes12100941