
Function-Related Dynamics in Multi-Spanning Helical Membrane Proteins Revealed by Solution NMR

 , , and
, , and 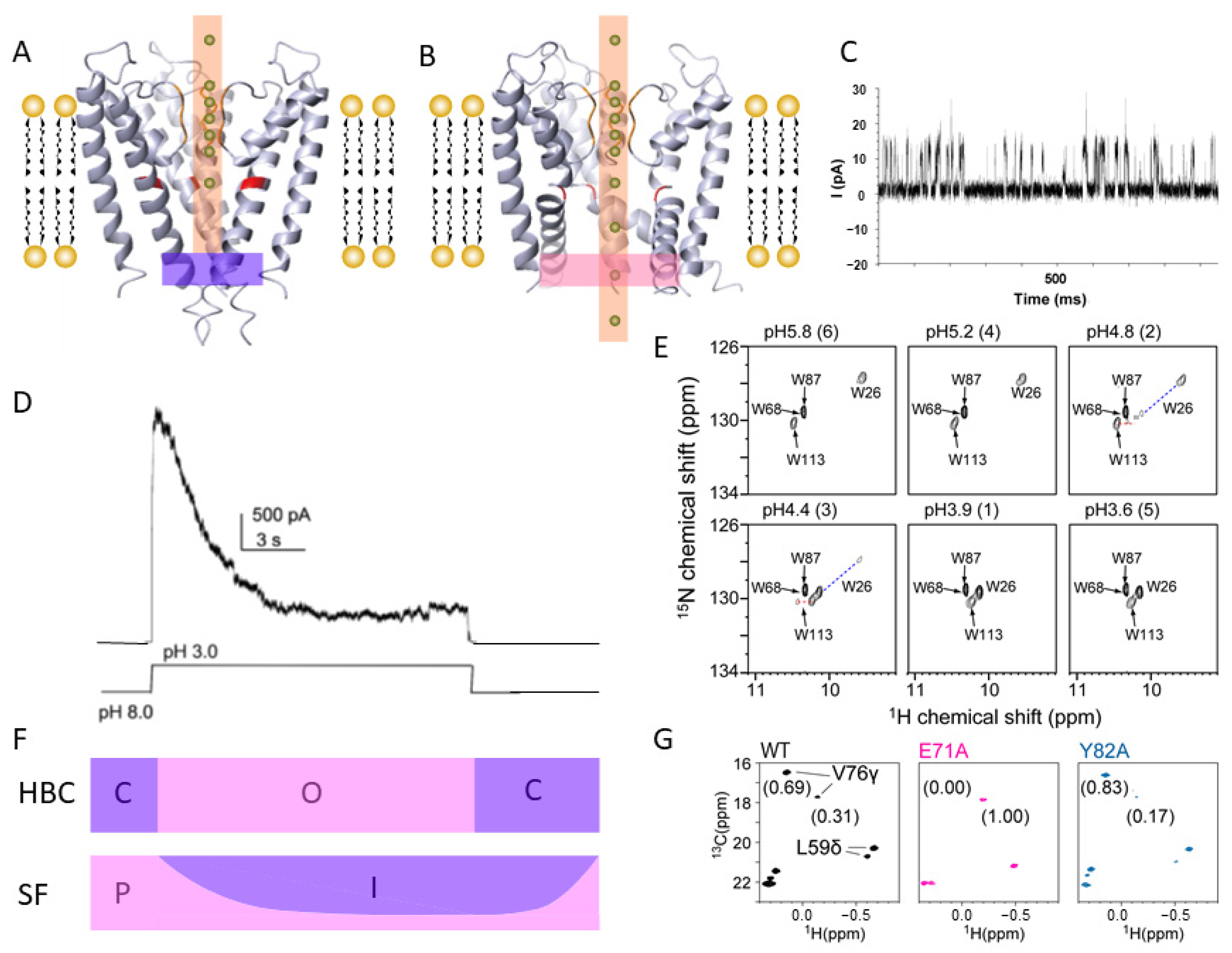{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ion Channels
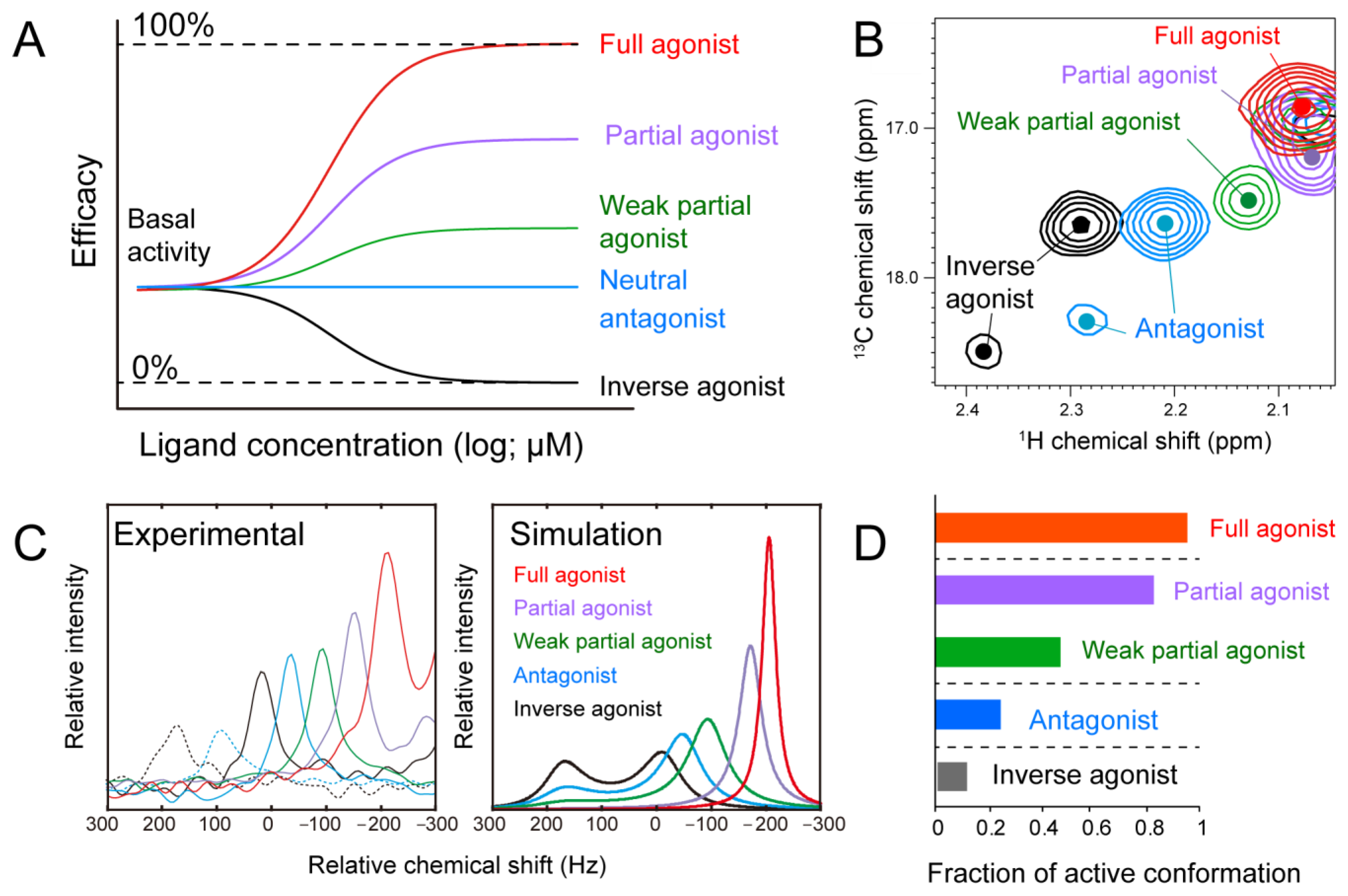
3. G-Protein Coupled Receptors (GPCRs)
4. Transporters
5. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Morais-Cabral, J.H.; Kaufman, A.; MacKinnon, R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 2001, 414, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lee, A.; Chen, J.; Cadene, M.; Chait, B.T.; MacKinnon, R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature 2002, 417, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lee, A.; Chen, J.; Cadene, M.; Chait, B.T.; MacKinnon, R. The open pore conformation of potassium channels. Nature 2002, 417, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Lü, W.; Wu, S.; Cheng, Y.; Gouaux, E. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 2015, 526, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yan, Z.; Li, Z.; Yan, C.; Lu, S.; Dong, M.; Yan, N. Structure of the voltage-gated calcium channel Cav1.1 complex. Science 2015, 350, aad2395. [Google Scholar] [CrossRef] [PubMed]
- Saotome, K.; Murthy, S.E.; Kefauver, J.M.; Whitwam, T.; Patapoutian, A.; Ward, A.B. Structure of the mechanically activated ion channel Piezo1. Nature 2018, 554, 481–486. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhou, H.; Chi, S.; Wang, Y.; Wang, J.; Geng, J.; Wu, K.; Liu, W.; Zhang, T.; Dong, M.-Q.; et al. Structure and mechanogating mechanism of the Piezo1 channel. Nature 2018, 554, 487–492. [Google Scholar] [CrossRef]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCusker, E.C.; Bagnéris, C.; Naylor, C.E.; Cole, A.R.; D’Avanzo, N.; Nichols, C.G.; Wallace, B.A. Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat. Commun 2012, 3, 1102. [Google Scholar] [CrossRef] [Green Version]
- Hite, R.K.; MacKinnon, R. Structural Titration of Slo2.2, a Na(+)-Dependent K(+) Channel. Cell 2017, 168, 390–399.e311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrapani, S.; Cordero-Morales, J.F.; Perozo, E. A Quantitative Description of KcsA Gating I: Macroscopic Currents. J. Gen. Physiol. 2007, 130, 465–478. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K.; Takahashi, H.; Kawano, S.; Shimada, I. Identification and Characterization of the Slowly Exchanging pH-dependent Conformational Rearrangement in KcsA. J. Biol. Chem. 2007, 282, 15179–15186. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Osawa, M.; Takeuchi, K.; Shimada, I. Structural basis underlying the dual gate properties of KcsA. Proc. Natl. Acad. Sci. USA 2010, 107, 6216–6221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrapani, S.; Cordero-Morales, J.F.; Perozo, E. A Quantitative Description of KcsA Gating II: Single-Channel Currents. J. Gen. Physiol. 2007, 130, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.; Osawa, M.; Mita, K.; Toyonaga, S.; Machiyama, A.; Ueda, T.; Takeuchi, K.; Oiki, S.; Shimada, I. Functional Equilibrium of the KcsA Structure Revealed by NMR. J. Biol. Chem. 2012, 287. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Xu, Y.; Zhang, D.; McDermott, A.E. Probing allosteric coupling in a constitutively open mutant of the ion channel KcsA using solid-state NMR. Proc. Natl. Acad. Sci. USA 2020, 117, 7171. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.A.; Tzitzilonis, C.; Kwiatkowski, W.; Choe, S.; Riek, R. Conformational dynamics of the KcsA potassium channel governs gating properties. Nat. Struct. Mol. Biol. 2007, 14, 1089–1095. [Google Scholar] [CrossRef]
- Bayburt, T.H.; Grinkova, Y.V.; Sligar, S.G. Self-Assembly of Discoidal Phospholipid Bilayer Nanoparticles with Membrane Scaffold Proteins. Nano Lett. 2002, 2, 853–856. [Google Scholar] [CrossRef]
- van der Cruijsen, E.A.W.; Prokofyev, A.V.; Pongs, O.; Baldus, M. Probing Conformational Changes during the Gating Cycle of a Potassium Channel in Lipid Bilayers. Biophys. J. 2017, 112, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Chill, J.H.; Louis, J.M.; Baber, J.L.; Bax, A. Measurement of 15N relaxation in the detergent-solubilized tetrameric KcsA potassium channel. J. Biomol. NMR 2006, 36, 123–136. [Google Scholar] [CrossRef]
- Chill, J.H.; Louis, J.M.; Delaglio, F.; Bax, A. Local and global structure of the monomeric subunit of the potassium channel KcsA probed by NMR. Biochim. Biophys. Acta 2007, 1768, 3260–3270. [Google Scholar] [CrossRef] [Green Version]
- Iwahashi, Y.; Toyama, Y.; Imai, S.; Itoh, H.; Osawa, M.; Inoue, M.; Shimada, I. Conformational equilibrium shift underlies altered K(+) channel gating as revealed by NMR. Nat. Commun. 2020, 11, 5168. [Google Scholar] [CrossRef]
- Minato, Y.; Suzuki, S.; Hara, T.; Kofuku, Y.; Kasuya, G.; Fujiwara, Y.; Igarashi, S.; Suzuki, E.; Nureki, O.; Hattori, M.; et al. Conductance of P2X4 purinergic receptor is determined by conformational equilibrium in the transmembrane region. Proc. Natl. Acad. Sci. USA 2016, 113, 4741–4746. [Google Scholar] [CrossRef] [Green Version]
- Mase, Y.; Yokogawa, M.; Osawa, M.; Shimada, I. Structural basis for modulation of gating property of G protein-gated inwardly rectifying potassium ion channel (GIRK) by i/o-family G protein α subunit (Gαi/o). J. Biol. Chem. 2012, 287, 19537–19549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, Y.; Kano, H.; Mase, Y.; Yokogawa, M.; Osawa, M.; Shimada, I. Structural basis for the ethanol action on G-protein-activated inwardly rectifying potassium channel 1 revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2018, 115, 3858–3863. [Google Scholar] [CrossRef] [Green Version]
- Brettmann, J.B.; Urusova, D.; Tonelli, M.; Silva, J.R.; Henzler-Wildman, K.A. Role of protein dynamics in ion selectivity and allosteric coupling in the NaK channel. Proc. Natl. Acad. Sci. USA 2015, 112, 15366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokogawa, M.; Osawa, M.; Takeuchi, K.; Mase, Y.; Shimada, I. NMR analyses of the Gbetagamma binding and conformational rearrangements of the cytoplasmic pore of G protein-activated inwardly rectifying potassium channel 1 (GIRK1). J. Biol. Chem. 2011, 286, 2215–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, V.S.; Loftis, A.R.; Shcherbakov, A.A.; Pentelute, B.L.; Hong, M. Atomic structures of closed and open influenza B M2 proton channel reveal the conduction mechanism. Nat. Struct. Mol. Biol. 2020, 27, 160–167. [Google Scholar] [CrossRef]
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution Structure of the Integral Human Membrane Protein VDAC-1 in Detergent Micelles. Science 2008, 321, 1206. [Google Scholar] [CrossRef] [Green Version]
- Retel, J.S.; Nieuwkoop, A.J.; Hiller, M.; Higman, V.A.; Barbet-Massin, E.; Stanek, J.; Andreas, L.B.; Franks, W.T.; van Rossum, B.J.; Vinothkumar, K.R.; et al. Structure of outer membrane protein G in lipid bilayers. Nat. Commun. 2017, 8, 2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.; Villinger, S.; Mari, S.A.; Giller, K.; Griesinger, C.; Becker, S.; Müller, D.J.; Zweckstetter, M. Molecular Plasticity of the Human Voltage-Dependent Anion Channel Embedded into a Membrane. Structure 2016, 24, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.C.; Kang, P.W.; Hou, P.; Yang, N.D.; Kuenze, G.; Smith, J.A.; Shi, J.; Huang, H.; White, K.M.; Peng, D.; et al. Structure and physiological function of the human KCNQ1 channel voltage sensor intermediate state. Elife 2020, 9. [Google Scholar] [CrossRef]
- Qasim, A.; Sher, I.; Hirschhorn, O.; Shaked, H.; Qasem, Z.; Ruthstein, S.; Chill, J.H. Investigation of a KcsA Cytoplasmic pH Gate in Lipoprotein Nanodiscs. ChemBioChem 2019, 20, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.; Akimoto, M.; Akbarizadeh, S.; Melacini, G. Free energy landscape remodeling of the cardiac pacemaker channel explains the molecular basis of familial sinus bradycardia. J. Biol. Chem. 2017, 292, 6414–6428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, S.; Kimura, T.; Nozaki, T.; Harada, H.; Shimada, I.; Osawa, M. Structural basis for the inhibition of voltage-dependent K+ channel by gating modifier toxin. Sci. Rep. 2015, 5, 14226. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K.; Yokogawa, M.; Matsuda, T.; Sugai, M.; Kawano, S.; Kohno, T.; Nakamura, H.; Takahashi, H.; Shimada, I. Structural basis of the KcsA K(+) channel and agitoxin2 pore-blocking toxin interaction by using the transferred cross-saturation method. Structure 2003, 11, 1381–1392. [Google Scholar] [CrossRef]
- Matsumura, K.; Shimomura, T.; Kubo, Y.; Oka, T.; Kobayashi, N.; Imai, S.; Yanase, N.; Akimoto, M.; Fukuda, M.; Yokogawa, M.; et al. Mechanism of hERG inhibition by gating-modifier toxin, APETx1, deduced by functional characterization. BMC Mol. Cell Biol. 2021, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Wang, S.; Cui, T.; Su, X.-C.; Chou, J.J. Ion and inhibitor binding of the double-ring ion selectivity filter of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA 2017, 114, E2846. [Google Scholar] [CrossRef] [Green Version]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Osawa, M.; Takeuchi, K.; Imai, S.; Stampoulis, P.; Kofuku, Y.; Ueda, T.; Shimada, I. Functional dynamics of cell surface membrane proteins. J. Magn. Reson. 2014, 241, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Kofuku, Y.; Ueda, T.; Okude, J.; Shiraishi, Y.; Kondo, K.; Maeda, M.; Tsujishita, H.; Shimada, I. Efficacy of the β₂-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nat. Commun. 2012, 3, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfoss, G.; DeRose, E.; Gabel, S.; Perera, L.; Krahn, J.; Mueller, G.; Zheng, X.; London, R. Conformational dependence of 13C shielding and coupling constants for methionine methyl groups. J. Biomol. NMR 2010, 48, 31–47. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The Dynamic Process of β2-Adrenergic Receptor Activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Isogai, S.; Deupi, X.; Opitz, C.; Heydenreich, F.M.; Tsai, C.-J.; Brueckner, F.; Schertler, G.F.X.; Veprintsev, D.B.; Grzesiek, S. Backbone NMR reveals allosteric signal transduction networks in the β1-adrenergic receptor. Nature 2016, 530, 237. [Google Scholar] [CrossRef]
- Solt, A.S.; Bostock, M.J.; Shrestha, B.; Kumar, P.; Warne, T.; Tate, C.G.; Nietlispach, D. Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound beta1-adrenergic receptor. Nat. Commun. 2017, 8, 1795. [Google Scholar] [CrossRef]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Biased Signaling Pathways in β2-Adrenergic Receptor Characterized by 19F-NMR. Science 2012, 335, 1106. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Kimm, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.; Yokomizo, T.; Kofuku, Y.; Shiraishi, Y.; Ueda, T.; Shimada, I. Structural equilibrium underlying ligand-dependent activation of β(2)-adrenoreceptor. Nat. Chem. Biol. 2020, 16, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Grahl, A.; Abiko, L.A.; Isogai, S.; Sharpe, T.; Grzesiek, S. A high-resolution description of β1-adrenergic receptor functional dynamics and allosteric coupling from backbone NMR. Nat. Commun. 2020, 11, 2216. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Kim, T.H.; Manglik, A.; Alvares, R.; Kobilka, B.K.; Prosser, R.S. Role of detergents in conformational exchange of a G protein-coupled receptor. J. Biol. Chem. 2012, 287, 36305–36311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Chung, K.Y.; Manglik, A.; Hansen, A.L.; Dror, R.O.; Mildorf, T.J.; Shaw, D.E.; Kobilka, B.K.; Prosser, R.S. The Role of Ligands on the Equilibria Between Functional States of a G Protein-Coupled Receptor. J. Am. Chem. Soc. 2013, 135, 9465–9474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Neale, C.; Sljoka, A.; Lyda, B.; Pichugin, D.; Tsuchimura, N.; Larda, S.T.; Pomes, R.; Garcia, A.E.; Ernst, O.P.; et al. Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 2018, 9, 1372. [Google Scholar] [CrossRef] [PubMed]
- Frei, J.N.; Broadhurst, R.W.; Bostock, M.J.; Solt, A.; Jones, A.J.Y.; Gabriel, F.; Tandale, A.; Shrestha, B.; Nietlispach, D. Conformational plasticity of ligand-bound and ternary GPCR complexes studied by 19F NMR of the β1-adrenergic receptor. Nat. Commun. 2020, 11, 669. [Google Scholar] [CrossRef]
- Clark, L.D.; Dikiy, I.; Chapman, K.; Rödström, K.E.J.; Aramini, J.; LeVine, M.V.; Khelashvili, G.; Rasmussen, S.G.F.; Gardner, K.H.; Rosenbaum, D.M. Ligand modulation of sidechain dynamics in a wild-type human GPCR. eLife 2017, 6, e28505. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, E.S.; Fuglestad, B.; Lessen, H.J.; Stetz, M.A.; Lin, D.W.; Marques, B.S.; Gupta, K.; Fleming, K.G.; Wand, A.J. Membrane Proteins Have Distinct Fast Internal Motion and Residual Conformational Entropy. Angew. Chem. Int. Ed. 2020, 59, 11108–11114. [Google Scholar] [CrossRef]
- Kooijman, L.; Schuster, M.; Baumann, C.; Jurt, S.; Löhr, F.; Fürtig, B.; Güntert, P.; Zerbe, O. Dynamics of Bacteriorhodopsin in the Dark-Adapted State from Solution Nuclear Magnetic Resonance Spectroscopy. Angew. Chem. Int. Ed. 2020, 59, 20965–20972. [Google Scholar] [CrossRef]
- Kofuku, Y.; Ueda, T.; Okude, J.; Shiraishi, Y.; Kondo, K.; Mizumura, T.; Suzuki, S.; Shimada, I. Functional dynamics of deuterated β2 -adrenergic receptor in lipid bilayers revealed by NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2014, 53, 13376–13379. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, M.; Damian, M.; Lescop, E.; Point, E.; Moncoq, K.; Morellet, N.; Levy, D.; Marie, J.; Guittet, E.; Banères, J.-L.; et al. Functional Modulation of a G Protein-Coupled Receptor Conformational Landscape in a Lipid Bilayer. J. Am. Chem. Soc. 2016, 138, 11170–11175. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Rajagopal, K.; Lefkowitz, R.J. Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010, 9, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharm. Toxicol. 2012, 52, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Standfuss, J.; Edwards, P.C.; D’Antona, A.; Fransen, M.; Xie, G.; Oprian, D.D.; Schertler, G.F. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 2011, 471, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Okude, J.; Ueda, T.; Kofuku, Y.; Sato, M.; Nobuyama, N.; Kondo, K.; Shiraishi, Y.; Mizumura, T.; Onishi, K.; Natsume, M.; et al. Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the μ-Opioid Receptor. Angew. Chem. Int. Ed. 2015, 54, 15771–15776. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, Y.; Natsume, M.; Kofuku, Y.; Imai, S.; Nakata, K.; Mizukoshi, T.; Ueda, T.; Iwaï, H.; Shimada, I. Phosphorylation-induced conformation of β2-adrenoceptor related to arrestin recruitment revealed by NMR. Nat. Commun. 2018, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yu, X.; Liu, C.; Qu, C.-X.; Gong, Z.; Liu, H.-D.; Li, F.-H.; Wang, H.-M.; He, D.-F.; Yi, F.; et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and 19F-NMR. Nat. Commun. 2015, 6, 8202. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.J.Y.; Gabriel, F.; Tandale, A.; Nietlispach, D. Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches. Molecules 2020, 25, 4729. [Google Scholar] [CrossRef]
- Ferré, G.; Eddy, M.T. Structural biology of human GPCR drugs and endogenous ligands—Insights from NMR spectroscopy. Methods 2020, 180, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Bosse, M.; Gauglitz, M.; Wistuba, S.; Schmidt, P.; Kaiser, A.; Gurevich, V.V.; Beck-Sickinger, A.G.; Hildebrand, P.W.; Huster, D. The Dynamics of the Neuropeptide Y Receptor Type 1 Investigated by Solid-State NMR and Molecular Dynamics Simulation. Molecules 2020, 25, 5489. [Google Scholar] [CrossRef]
- Schmidt, P.; Thomas, L.; Müller, P.; Scheidt, H.A.; Huster, D. The G-protein-coupled neuropeptide Y receptor type 2 is highly dynamic in lipid membranes as revealed by solid-state NMR spectroscopy. Chemistry 2014, 20, 4986–4992. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753. [Google Scholar] [CrossRef] [Green Version]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.S.; Oldham, M.L.; Zhang, Q.; Chen, J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 2012, 490, 566–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Sakiyama, K.; Hipolito, C.J.; Fujioka, A.; Hirokane, R.; Ikeguchi, K.; Watanabe, B.; Hiratake, J.; et al. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. USA 2014, 111, 4049. [Google Scholar] [CrossRef] [Green Version]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Matsuoka, K.; Kimura, Y.; Ueda, K.; Kato, H. Inward- and outward-facing X-ray crystal structures of homodimeric P-glycoprotein CmABCB1. Nat. Commun. 2019, 10, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Matsumoto, T.; Yamaguchi, A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 2006, 443, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Tam, H.K.; Foong, W.E.; Oswald, C.; Herrmann, A.; Zeng, H.; Pos, K.M. Allosteric drug transport mechanism of multidrug transporter AcrB. Nat. Commun. 2021, 12, 3889. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Wang, Z.; James, N.R.; Voss, J.E.; Klimont, E.; Ohene-Agyei, T.; Venter, H.; Chiu, W.; Luisi, B.F. Structure of the AcrAB–TolC multidrug efflux pump. Nature 2014, 509, 512–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuldiner, S. EmrE, a model for studying evolution and mechanism of ion-coupled transporters. Biochim. Et. Biophys. Acta Proteins Proteom. 2009, 1794, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Penders, B.; Horstman, K.; Vos, R. Proper science in moist biology. EMBO Rep. 2007, 8, 613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C. Pretty Structures, But What About the Data? Science 2007, 315, 459. [Google Scholar] [CrossRef]
- Tate, C.G.; Ubarretxena-Belandia, I.; Baldwin, J.M. Conformational changes in the multidrug transporter EmrE associated with substrate binding. J. Mol. Biol. 2003, 332, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Pornillos, O.; Lieu, S.; Ma, C.; Chen, A.P.; Chang, G. X-ray structure of EmrE supports dual topology model. Proc. Natl. Acad. Sci. USA 2007, 104, 18999–19004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, E.A.; DeKoster, G.T.; Dutta, S.; Vafabakhsh, R.; Clarkson, M.W.; Bahl, A.; Kern, D.; Ha, T.; Henzler-Wildman, K.A. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature 2012, 481, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, E.A.; Henzler-Wildman, K.A. Transported Substrate Determines Exchange Rate in the Multidrug Resistance Transporter EmrE. J. Biol. Chem. 2014, 289, 6825–6836. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Morrison, E.A.; Henzler-Wildman, K.A. Blocking dynamics of the SMR transporter EmrE impairs efflux activity. Biophys. J. 2014, 107, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Shcherbakov, A.A.; Hisao, G.; Mandala, V.S.; Thomas, N.E.; Soltani, M.; Salter, E.A.; Davis, J.H.; Henzler-Wildman, K.A.; Hong, M. Structure and dynamics of the drug-bound bacterial transporter EmrE in lipid bilayers. Nat. Commun. 2021, 12, 172. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Viennet, T.; Arthanari, H.; Takeuchi, K. Spotlight on the Ballet of Proteins: The Structural Dynamic Properties of Proteins Illuminated by Solution NMR. Int. J. Mol. Sci. 2020, 21, 1829. [Google Scholar] [CrossRef] [Green Version]
- Arthanari, H.; Takeuchi, K.; Dubey, A.; Wagner, G. Emerging solution NMR methods to illuminate the structural and dynamic properties of proteins. Curr. Opin. Struct. Biol. 2019, 58, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M.; Takeuchi, K.; Ueda, T.; Nishida, N.; Shimada, I. Functional dynamics of proteins revealed by solution NMR. Curr. Opin. Struct. Biol. 2012, 22, 660–669. [Google Scholar] [CrossRef]
- Takeuchi, K.; Arthanari, H.; Imai, M.; Wagner, G.; Shimada, I. Nitrogen-detected TROSY yields comparable sensitivity to proton-detected TROSY for non-deuterated, large proteins under physiological salt conditions. J. Biomol. NMR 2016, 64, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeszoermenyi, A.; Chhabra, S.; Dubey, A.; Radeva, D.L.; Burdzhiev, N.T.; Chanev, C.D.; Petrov, O.I.; Gelev, V.M.; Zhang, M.; Anklin, C.; et al. Aromatic 19F-13C TROSY: A background-free approach to probe biomolecular structure, function, and dynamics. Nat. Methods 2019, 16, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, Y.; Takeuchi, K.; Okude, J.; Ori, K.; Torizawa, T.; Shimada, I. Structural Fingerprints of an Intact Monoclonal Antibody Acquired under Formulated Storage Conditions via 15N Direct Detection Nuclear Magnetic Resonance. J. Med. Chem. 2020, 63, 5360–5366. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeuchi, K.; Kofuku, Y.; Imai, S.; Ueda, T.; Tokunaga, Y.; Toyama, Y.; Shiraishi, Y.; Shimada, I. Function-Related Dynamics in Multi-Spanning Helical Membrane Proteins Revealed by Solution NMR. Membranes 2021, 11, 604. https://doi.org/10.3390/membranes11080604
Takeuchi K, Kofuku Y, Imai S, Ueda T, Tokunaga Y, Toyama Y, Shiraishi Y, Shimada I. Function-Related Dynamics in Multi-Spanning Helical Membrane Proteins Revealed by Solution NMR. Membranes. 2021; 11(8):604. https://doi.org/10.3390/membranes11080604
Chicago/Turabian StyleTakeuchi, Koh, Yutaka Kofuku, Shunsuke Imai, Takumi Ueda, Yuji Tokunaga, Yuki Toyama, Yutaro Shiraishi, and Ichio Shimada. 2021. "Function-Related Dynamics in Multi-Spanning Helical Membrane Proteins Revealed by Solution NMR" Membranes 11, no. 8: 604. https://doi.org/10.3390/membranes11080604