Multitarget Approach to Cardiogenic Shock after Acute Myocardial Infarction: Extracorporeal Life Support (ECLS) and Beyond



Abstract
:1. Epidemiology of Acute Myocardial Infarction and Cardiogenic Shock (CS-AMI)
- symptoms related to ischemia;
- changes on an electrocardiogram (ECG), such as ST segment changes or new left bundle branch block;
- development of pathological Q waves on ECG;
- new regional wall motion abnormalities at imaging;
- demonstration of a coronary thrombus on angiogram or during autopsy.
2. Cardiogenic Shock after AMI: Definition and the Concept of “Spectrum of Shock”
3. The “Good Outcome” of Cardiogenic Shock after AMI
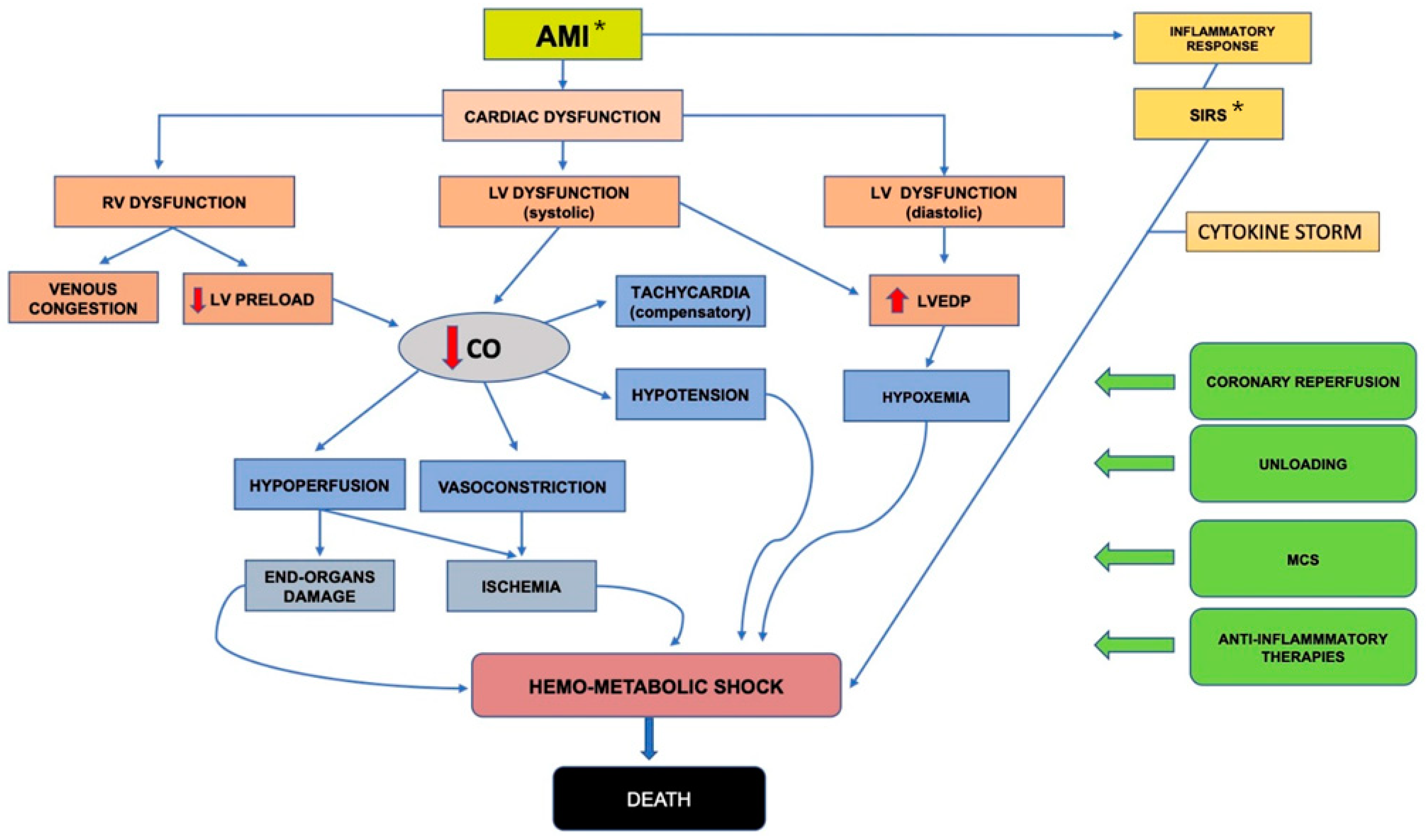
4. Pathophysiology of AMI-Related Systemic Inflammation and Therapeutic Targets
5. Extracorporeal Life Support
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction (2018). J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, E.; Wilson, L.; Wickramasinghe, K.; Bhatnagar, P.; Leal, J.; Luengo-Fernandez, R.; Burns, R.; Rayner, M.; Townsend, N. European Cardiovascular Disease Statistics Edition; European Heart Network: Brusseles, Belgium, 2017; Volume 8, pp. 94, 118, 127, 149, 162, 174. [Google Scholar]
- Vahdatpour, C.A.; Collins, D.; Goldberg, S. Cardiogenic Shock. J. Am. Heart Assoc. 2019, 8, e011991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathod, K.S.; Koganti, S.; Iqbal, M.B.; Jain, A.K.; Kalra, S.S.; Astroulakis, Z.; Lim, P.; Rakhit, R.; Dalby, M.; Lockie, T.; et al. Contemporary trends in cardiogenic shock: Incidence, intra-aortic balloon pump utilisation and outcomes from the London Heart Attack Group. Eur. Heart J. Acute Cardiovasc. Care 2017, 7, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Aissaoui, N.; Puymirat, E.; Delmas, C.; Ortuno, S.; Durand, E.; Bataille, V.; Drouet, E.; Bonello, L.; Bonnefoy-Cudraz, E.; Lesmeles, G.; et al. Trends in cardiogenic shock complicating acute myocardial infarction. Eur. J. Heart Fail. 2020, 22, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, L.; Radovanovic, D.; Jeger, R.; Pedrazzini, G.; Cuculi, F.; Urban, P.; Erne, P.; Rickli, H.; Pilgrim, T.; Hess, F.; et al. Twenty-Year Trends in the Incidence and Outcome of Cardiogenic Shock in AMIS Plus Registry. Circ. Cardiovasc. Interv. 2019, 12, e007293. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, H.R.; Hochman, J.S. Cardiogenic Shock. Circulation 2008, 117, 686–697. [Google Scholar] [CrossRef]
- Helgestad, O.K.L.; Josiassen, J.; Hassager, C.; Jensen, L.O.; Holmvang, L.; Udesen, N.L.J.; Schmidt, H.; Ravn, H.B.; Moller, J.E. Contemporary trends in use of mechanical circulatory support in patients with acute MI and cardiogenic shock. Open Hear. 2020, 7, e001214. [Google Scholar] [CrossRef]
- Baran, D.A.; Grines, C.L.; Bailey, S.; Burkhoff, D.; Hall, S.A.; Henry, T.D.; Hollenberg, S.M.; Kapur, N.K.; O’Neill, W.; Ornato, J.P.; et al. SCAI clinical expert consensus statement on the classification of cardiogenic shock. Catheter. Cardiovasc. Interv. 2019, 94, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Schrage, B.; Dabboura, S.; Yan, I.; Hilal, R.; Neumann, J.T.; Sörensen, N.A.; Goßling, A.; Becher, P.M.; Grahn, H.; Wagner, T.; et al. Application of the SCAI classification in a cohort of patients with cardiogenic shock. Catheter. Cardiovasc. Interv. 2020, 96, 213. [Google Scholar] [CrossRef]
- Velagaleti, R.S.; Pencina, M.J.; Murabito, J.M.; Wang, T.J.; Parikh, N.I.; D’Agostino, R.B.; Levy, D.; Kannel, W.B.; Vasan, R.S. Long-Term Trends in the Incidence of Heart Failure After Myocardial Infarction. Circulation 2008, 118, 2057–2062. [Google Scholar] [CrossRef]
- Thiele, H.; Zeymer, U.; Thelemann, N.; Neumann, F.-J.; Hausleiter, J.; Abdel-Wahab, M.; Meyer-Saraei, R.; Fuernau, G.; Eitel, I.E.; Hambrecht, R.; et al. Intraaortic Balloon Pump in Cardiogenic Shock Complicating Acute Myocardial Infarction. Circulation 2019, 139, 395–403. [Google Scholar] [CrossRef]
- Hochman, J.S.; Sleeper, L.A.; Webb, J.G.; Dzavik, V.; Buller, C.E.; Aylward, P.; Col, J.; White, H.D.; for the SHOCK Investigators. Early Revascularization and Long-term Survival in Cardiogenic Shock Complicating Acute Myocardial Infarction. JAMA 2006, 295, 2511–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, H.; Akin, I.; Sandri, M.; De Waha-Thiele, S.; Meyer-Saraei, R.; Fuernau, G.; Eitel, I.; Nordbeck, P.; Geisler, T.; Landmesser, U.; et al. One-Year Outcomes after PCI Strategies in Cardiogenic Shock. N. Engl. J. Med. 2018, 379, 1699–1710. [Google Scholar] [CrossRef]
- Bauters, C.; Dubois, E.; Porouchani, S.; Saloux, E.; Fertin, M.; De Groote, P.; Lamblin, N.; Pinet, F. Long-term prognostic impact of left ventricular remodeling after a first myocardial infarction in modern clinical practice. PLoS ONE 2017, 12, e0188884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basir, M.B.; Schreiber, T.; Dixon, S.; Alaswad, K.; Patel, K.; Almany, S.; Khandelwal, A.; Hanson, I.; George, A.; Ashbrook, M.; et al. Feasibility of early mechanical circulatory support in acute myocardial infarction complicated by cardiogenic shock: The Detroit cardiogenic shock initiative. Catheter. Cardiovasc. Interv. 2018, 91, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Brilakis, E.S.; Eckman, P. The five key “ingredients” for improving outcomes in cardiogenic shock complicating acute myocardial infarction. Catheter. Cardiovasc. Interv. 2018, 91, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Kolte, D.; Sardar, P.; Khera, S.; Zeymer, U.; Thiele, H.; Hochadel, M.; Radovanovic, D.; Erne, P.; Hambraeus, K.; James, S.; et al. Culprit Vessel–Only Versus Multivessel Percutaneous Coronary Intervention in Patients with Cardiogenic Shock Complicating ST-Segment–Elevation Myocardial Infarction. Circ. Cardiovasc. Interv. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Alonso, D.R.; Scheidt, S.; Post, M.; Killip, T. Pathophysiology of Cardiogenic Shock. Circulation 1973, 48, 588–596. [Google Scholar] [CrossRef] [Green Version]
- Chioncel, O.; Parissis, J.; Mebazaa, A.; Thiele, H.; Desch, S.; Bauersachs, J.; Harjola, V.; Antohi, E.; Arrigo, M.; Ben Gal, T.; et al. Epidemiology, pathophysiology and contemporary management of cardiogenic shock—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2020, 22, 1315–1341. [Google Scholar] [CrossRef]
- Esposito, M.L.; Kapur, N.K. Acute mechanical circulatory support for cardiogenic shock: The “door to support” time. F1000Research 2017, 6, 737. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair after Myocardial Infarction. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. NLRP3 Inflammasome in Acute Myocardial Infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cell biological mechanisms in regulation of the post-infarction inflammatory response. Curr. Opin. Physiol. 2018, 1, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Prondzinsky, R.; Unverzagt, S.; Lemm, H.; Wegener, N.; Heinroth, K.; Buerke, U.; Fiedler, M.; Thiery, J.; Haerting, J.; Werdan, K.; et al. Acute myocardial infarction and cardiogenic shock. Med. Klin. Intensiv. Notfallmedizin 2012, 107, 476–484. [Google Scholar] [CrossRef]
- Cuinet, J.; Garbagnati, A.; Rusca, M.; Yerly, P.; Schneider, A.G.; Kirsch, M.; Liaudet, L. Cardiogenic shock elicits acute inflammation, delayed eosinophilia, and depletion of immune cells in most severe cases. Sci. Rep. 2020, 10, 7639. [Google Scholar] [CrossRef]
- Debrunner, M.; Schuiki, E.; Minder, E.; Straumann, E.; Naegeli, B.; Mury, R.; Bertel, O.; Frielingsdorf, J. Proinflammatory cytokines in acute myocardial infarction with and without cardiogenic shock. Clin. Res. Cardiol. 2008, 97, 298–305. [Google Scholar] [CrossRef]
- Anselmi, A.; Abbate, A.; Girola, F.; Nasso, G.; Biondi-Zoccai, G.; Possati, G.; Gaudino, S. Myocardial ischemia, stunning, inflammation, and apoptosis during cardiac surgery: A review of evidence. Eur. J. Cardio-Thoracic Surg. 2004, 25, 304–311. [Google Scholar] [CrossRef]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Biondi-Zoccai, G.G.; Van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.A.; Roberts, C.S.; Varma, A.; et al. Interleukin-1 Blockade With Anakinra to Prevent Adverse Cardiac Remodeling After Acute Myocardial Infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot Study). Am. J. Cardiol. 2010, 105, 1371–1377.e1. [Google Scholar] [CrossRef] [Green Version]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.G.L.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of Interleukin-1 Blockade With Anakinra on Adverse Cardiac Remodeling and Heart Failure After Acute Myocardial Infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) Pilot Study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Everett, B.M.; MacFadyen, J.G.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Inhibition of Interleukin-1β and Reduction in Atherothrombotic Cardiovascular Events in the CANTOS Trial. J. Am. Coll. Cardiol. 2020, 76, 1660–1670. [Google Scholar] [CrossRef]
- Silvain, J.; Kerneis, M.; Zeitouni, M.; Lattuca, B.; Galier, S.; Brugier, D.; Mertens, E.; Procopi, N.; Suc, G.; Salloum, T.; et al. Interleukin-1Beta and Risk of Premature Death in Patients with Myocardial Infarction. J. Am. Coll. Cardiol. 2020, 76, 1763–1773. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Thompson, P.L. Why Colchicine Should Be Considered for Secondary Prevention of Atherosclerosis: An Overview. Clin. Ther. 2019, 41, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deftereos, S.; Giannopoulos, G.; Angelidis, C.; Alexopoulos, N.; Filippatos, G.; Papoutsidakis, N.; Sianos, G.; Goudevenos, J.; Alexopoulos, D.; Pyrgakis, V.; et al. Anti-Inflammatory Treatment With Colchicine in Acute Myocardial Infarction. Circulation 2015, 132, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.C.; Quinn, S.; Nasis, A.; Hiew, C.; Roberts-Thomson, P.; Adams, H.; Sriamareswaran, R.; Htun, N.M.; Wilson, W.; Stub, D.; et al. Colchicine in Patients With Acute Coronary Syndrome. Circulation 2020, 142, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Jentzer, J.C.; Lawler, P.R.; Van Diepen, S.; Henry, T.D.; Menon, V.; Baran, D.A.; Džavík, V.; Barsness, G.W.; Holmes, D.R.; Kashani, K.B. Systemic Inflammatory Response Syndrome Is Associated With Increased Mortality Across the Spectrum of Shock Severity in Cardiac Intensive Care Patients. Circ. Cardiovasc. Qual. Outcomes 2020, 13, e006956. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.E.; Fanning, J.P.; McDonald, C.; McAuley, D.F.; Fraser, J.F. The inflammatory response to extracorporeal membrane oxygenation (ECMO): A review of the pathophysiology. Crit. Care 2016, 20, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Thiele, H.; Allam, B.; Chatellier, G.; Schuler, G.; Lafont, A. Shock in acute myocardial infarction: The Cape Horn for trials? Eur. Heart J. 2010, 31, 1828–1835. [Google Scholar] [CrossRef]
- Calabrò, M.G.; Febres, D.; Recca, G.; Lembo, R.; Fominskiy, E.; Scandroglio, A.M.; Zangrillo, A.; Pappalardo, F. Blood Purification With CytoSorb in Critically Ill Patients: Single-Center Preliminary Experience. Artif. Organs 2019, 43, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Datzmann, T.; Träger, K. Extracorporeal membrane oxygenation and cytokine adsorption. J. Thorac. Dis. 2018, 10, S653–S660. [Google Scholar] [CrossRef]
- Pappalardo, F.; Schulte, C.; Pieri, M.; Schrage, B.; Contri, R.; Soeffker, G.; Greco, T.; Lembo, R.; Müllerleile, K.; Colombo, A.; et al. Concomitant implantation of Impella® on top of veno-arterial extracorporeal membrane oxygenation may improve survival of patients with cardiogenic shock. Eur. J. Heart Fail. 2017, 19, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Schrage, B.; Becher, P.M.; Bernhardt, A.M.; Bezerra, H.; Blankenberg, S.; Brunner, S.; Colson, P.H.; Deseda, G.C.; Dabboura, S.; Eckner, D.; et al. Left Ventricular Unloading Is Associated With Lower Mortality in Patients With Cardiogenic Shock Treated With Venoarterial Extracorporeal Membrane Oxygenation. Circulation 2020, 142, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.S.; Adriaenssens, T.; Berry, C.; Bogaerts, K.; Erglis, A.; Distelmaier, K.; Guagliumi, G.; Haine, S.; Kastrati, A.; Massberg, S.; et al. The EURO SHOCK Trial: Design, Aims and Objectives. EuroIntervention 2020. [Google Scholar] [CrossRef]
- Thiele, H. Prospective Randomized Multicenter Study Comparing Extracorporeal Life Support Plus Optimal Medical Care Versus Optimal Medical Care Alone in Patients With Acute Myocardial Infarction Complicated by Cardiogenic Shock Undergoing Revascularization. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03637205 (accessed on 20 January 2021).
- Udesen, N.J.; Møller, J.E.; Lindholm, M.G.; Eiskjær, H.; Schäfer, A.; Werner, N.; Holmvang, L.; Terkelsen, C.J.; Jensen, L.O.; Junker, A.; et al. Rationale and design of DanGer shock: Danish-German cardiogenic shock trial. Am. Heart J. 2019, 214, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Combesa, A. Assessment of ECMO in Acute Myocardial Infarction with Non-reversible Cardiogenic Shock to Halt Organ Failure and Reduce Mortality (ANCHOR). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04184635 (accessed on 20 January 2021).


{kind=link}
| Stage | Definition | Physical Exam/Bedside Findings | Biochemical Markers | Hemodynamics |
|---|---|---|---|---|
| A At risk | A patient who is not currently experiencing signs or symptoms of CS but is at risk for its development. These patients may include those with large acute myocardial infarction or prior infarction acute and/or acute on chronic heart failure symptoms. | Normal Jugular Venous Pressure Lung sounds clear Warm and well perfused • Strong distal pulses • Normal mentation | Normal labs • Normal renal function • Normal lactic acid | Normotensive (SBP ≥ 100 or normal for pt.) If hemodynamics done • cardiac index ≥ 2.5 • CVP < 10 • PA sat ≥ 65% |
| B Beginning CS | A patient who has clinical evidence of relative hypotension or tachycardia without hypoperfusion. | Elevated JVP Rales in lung fields Warm and well perfused • Strong distal pulses • Normal mentation | Normal lactate Minimal renal function impairment Elevated BNP | SBP < 90 OR MAP < 60 OR > 30 mmHg drop from baseline Pulse ≥ 100 If hemodynamics done • cardiac index ≥ 2.2 • PA sat ≥ 65% |
| C Classic CS | A patient that manifests with hypoperfusion that requires intervention (inotrope, pressor or mechanical support, including ECMO) beyond volume resuscitation to restore perfusion. These patients typically present with relative hypotension. | May Include Any of: Looks unwell Panicked Ashen, mottled, dusky Volume overload Extensive rales Killip class 3 or 4 BiPap or mechanical ventilation Cold, clammy Acute alteration in mental status Urine output < 30 mL/h | May Include Any of: Lactate ≥ 2 Creatinine doubling OR > 50% drop in GFR Increased LFTs Elevated BNP | May Include Any of: SBP < 90 OR MAP < 60 OR > 30 mmHg drop from baseline ANDdrugs/device used to maintain BP above these targets Hemodynamics • cardiac index < 2.2 • PCWP > 15 • RAP/PCWP ≥ 0.8 • PAPI < 1.85 • cardiac power output ≤ 0.6 |
| D Deteriorating/doom | A patient that is similar to category C but are getting worse. They have failure to respond to initial interventions. | Any of stage C | Any of Stage C AND: Deteriorating | Any of Stage C AND: Requiring multiple pressors ORaddition of mechanical circulatory support devices to maintain perfusion |
| E Extremis | A patient that is experiencing cardiac arrest with ongoing CPR and/or ECMO, being supported by multiple interventions. | Near Pulselessness Cardiac collapse Mechanical ventilation Defibrillator used | “Trying to die” CPR (A-modifier) pH ≤ 7.2 Lactate ≥ 5 | No SBP without resuscitation PEA or refractory VT/VF Hypotension despite maximal support |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pappalardo, F.; Malara, G.; Montisci, A. Multitarget Approach to Cardiogenic Shock after Acute Myocardial Infarction: Extracorporeal Life Support (ECLS) and Beyond. Membranes 2021, 11, 87. https://doi.org/10.3390/membranes11020087
Pappalardo F, Malara G, Montisci A. Multitarget Approach to Cardiogenic Shock after Acute Myocardial Infarction: Extracorporeal Life Support (ECLS) and Beyond. Membranes. 2021; 11(2):87. https://doi.org/10.3390/membranes11020087
Chicago/Turabian StylePappalardo, Federico, Giulia Malara, and Andrea Montisci. 2021. "Multitarget Approach to Cardiogenic Shock after Acute Myocardial Infarction: Extracorporeal Life Support (ECLS) and Beyond" Membranes 11, no. 2: 87. https://doi.org/10.3390/membranes11020087