Electrospun Weak Anion-Exchange Fibrous Membranes for Protein Purification

Abstract
:1. Introduction
2. Materials and Method
2.1. Materials
2.2. Methods
2.2.1. Fabrication of Fibrous Membranes
2.2.2. UV-Initiated Polymerization of GMA
2.2.3. Membrane Morphology
2.2.4. Zeta Potential
2.3. Membrane Binding Capacity Determination
2.3.1. Static Protein Binding Capacity Measurement
2.3.2. Dynamic Binding Capacity Measurement
3. Results and Discussion
3.1. Membrane Characterization
3.1.1. Chemical Composition of Membrane Surface
3.1.2. Membrane Structure and Morphology
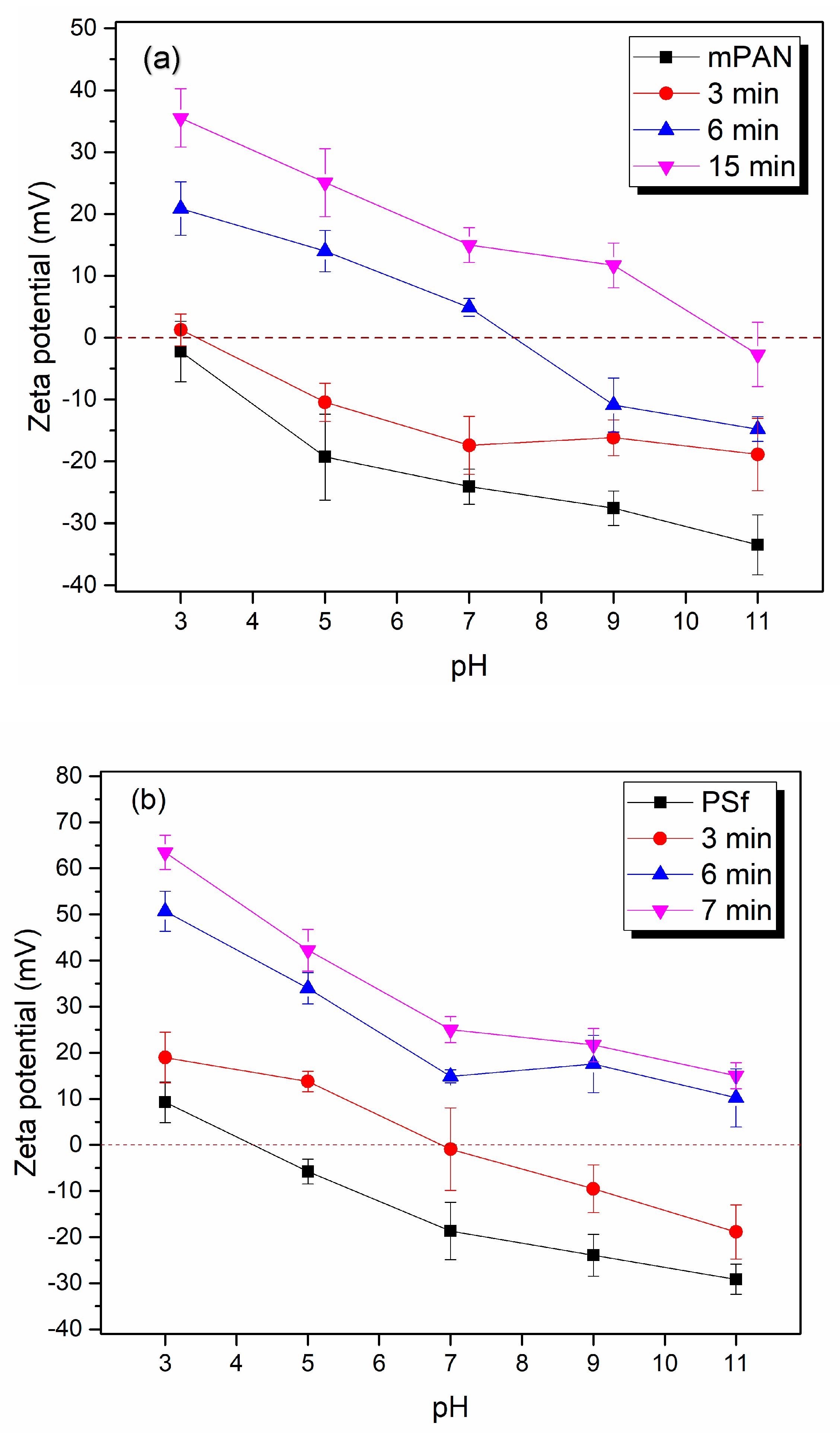
3.1.3. Zeta Potential
3.1.4. Grafting Degree
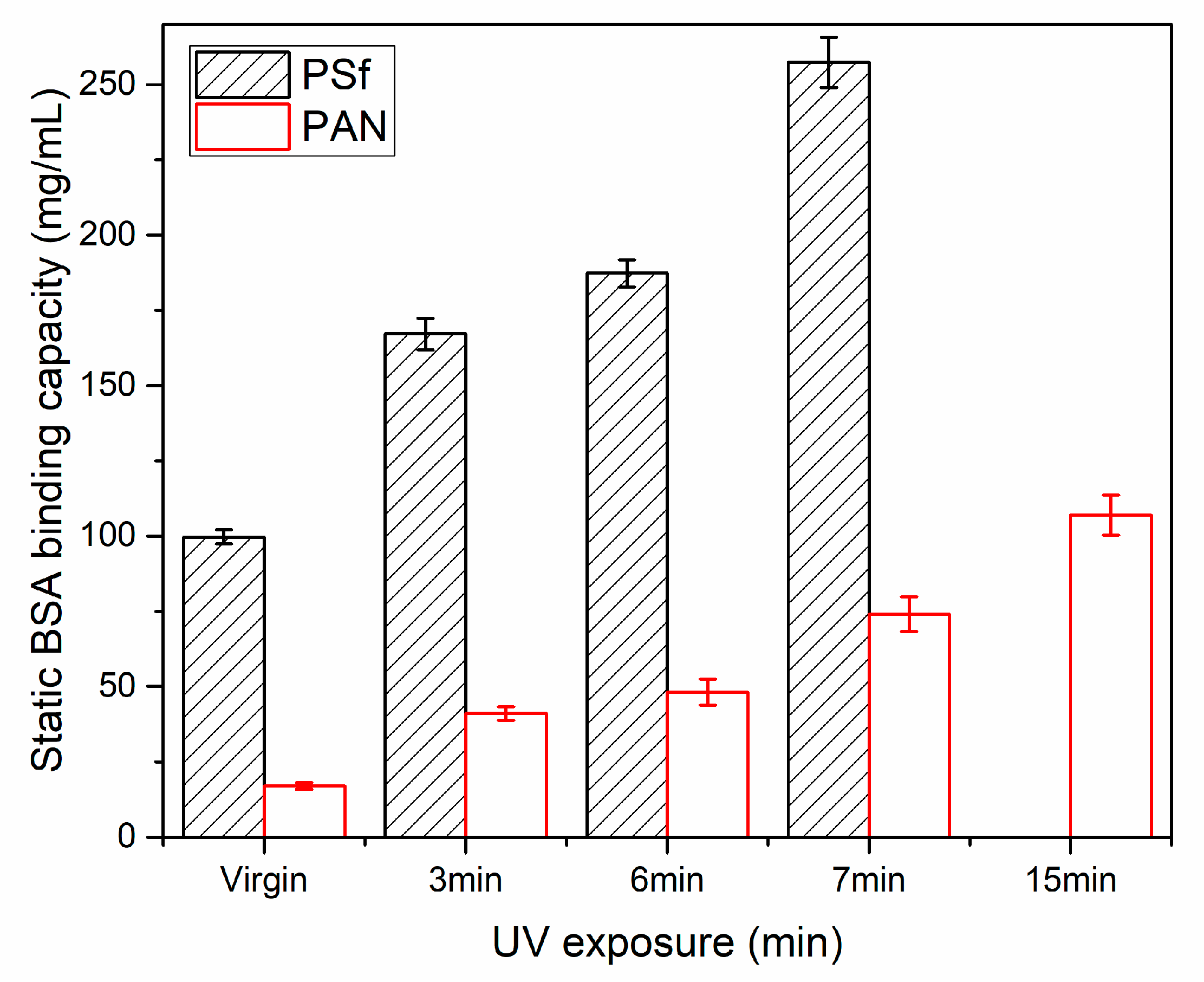
3.2. Static and Dynamic Protein Binding Capacity Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shukla, A.A.; Thömmes, J. Thömmes, Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010, 28, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs Taylor Fr. 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Pizzelli, K.; Romero, J.K.; Chrostowski, J.; Evangelist, G.; Hamzik, J.; Soice, N.; Cheng, K. Clarification of recombinant proteins from high cell density mammalian cell culture systems using new improved depth filters. Biotechnol. Bioeng. 2013, 110, 1964–1972. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, L.; E Kawkabani, E.; Jacobs, P.; Malphettes, L. Overcoming the clarification challenges of high cell density culture. BMC Proc. 2015, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Bielser, J.-M.; Wolf, M.; Souquet, J.; Broly, H.; Morbidelli, M. Perfusion mammalian cell culture for recombinant protein manufacturing—A critical review. Biotechnol. Adv. 2018, 36, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Guiochon, G.; Beaver, L.A. Separation science is the key to successful biopharmaceuticals. J. Chromatogr. A 2011, 1218, 8836–8858. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.; Husson, S.; Murphy, L.; Wickramasinghe, S.R. Anion exchange membrane adsorbers for flow-through polishing steps: Part II. Virus, host cell protein, DNA clearance, and antibody recovery. Biotechnol. Bioeng. 2012, 110, 500–510. [Google Scholar] [CrossRef]
- Gagnon, P. Technology trends in antibody purification. J. Chromatogr. A 2012, 1221, 57–70. [Google Scholar] [CrossRef]
- Sorci, M.; Gu, M.; Heldt, C.; Grafeld, E.; Belfort, G. A multi-dimensional approach for fractionating proteins using charged membranes. Biotechnol. Bioeng. 2013, 110, 1704–1713. [Google Scholar] [CrossRef]
- Liu, Z.; Du, H.; Wickramasinghe, S.R.; Qian, X. Membrane Surface Engineering for Protein Separations: Experiments and Simulations. Langmuir 2014, 30, 10651–10660. [Google Scholar] [CrossRef]
- Liu, Z.; Wickramasinghe, S.R.; Qian, X. The architecture of responsive polymeric ligands on protein binding and recovery. RSC Adv. 2017, 7, 27823–27832. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wickramasinghe, S.R.; Qian, X. Ion-specificity in protein binding and recovery for the responsive hydrophobic poly(vinylcaprolactam) ligand. RSC Adv. 2017, 7, 36351–36360. [Google Scholar] [CrossRef] [Green Version]
- Vu, A.; Qian, X.; Wickramasinghe, S.R. Membrane-based hydrophobic interaction chromatography. Sep. Sci. Technol. 2016, 52, 287–298. [Google Scholar] [CrossRef]
- Bhut, B.V.; Husson, S. Dramatic performance improvement of weak anion-exchange membranes for chromatographic bioseparations. J. Membr. Sci. 2009, 337, 215–223. [Google Scholar] [CrossRef]
- Wandera, D.; Wickramasinghe, S.R.; Husson, S. Stimuli-responsive membranes. J. Membr. Sci. 2010, 357, 6–35. [Google Scholar] [CrossRef]
- Ulbricht, M. Advanced functional polymer membranes. Polymer 2006, 47, 2217–2262. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Wang, J.; Ulbricht, M.; Wickramasinghe, S.R.; Husson, S. Surface-initiated atom transfer radical polymerization: A new method for preparation of polymeric membrane adsorbers. J. Membr. Sci. 2008, 309, 64–72. [Google Scholar] [CrossRef]
- Yusof, A.H.M.; Ulbricht, M. Polypropylene-based membrane adsorbers via photo-initiated graft copolymerization: Optimizing separation performance by preparation conditions. J. Membr. Sci. 2008, 311, 294–305. [Google Scholar] [CrossRef]
- Himstedt, H.H.; Qian, X.; Weaver, J.R.; Wickramasinghe, S.R. Responsive membranes for hydrophobic interaction chromatography. J. Membr. Sci. 2013, 447, 335–344. [Google Scholar] [CrossRef]
- Lalia, B.S.; Kochkodan, V.; Hashaikeh, R.; Hilal, N. A review on membrane fabrication: Structure, properties and performance relationship. Desalination 2013, 326, 77–95. [Google Scholar] [CrossRef]
- Tijing, L.D.; Choi, J.-S.; Lee, S.; Kim, S.-H.; Shon, H.K. Recent progress of membrane distillation using electrospun nanofibrous membrane. J. Membr. Sci. 2014, 453, 435–462. [Google Scholar] [CrossRef]
- Alkhudhiri, A.; Darwish, N.; Hilal, N. Membrane distillation: A comprehensive review. Desalination 2012, 287, 2–18. [Google Scholar] [CrossRef]
- Tang, Y.-H.; Ledieu, E.; Cervellere, M.R.; Millett, P.C.; Ford, D.M.; Qian, X. Formation of polyethersulfone membranes via nonsolvent induced phase separation process from dissipative particle dynamics simulations. J. Membr. Sci. 2020, 599, 117826. [Google Scholar] [CrossRef]
- Ma, Z.; Lan, Z.; Matsuura, T.; Ramakrishna, S. Electrospun polyethersulfone affinity membrane: Membrane preparation and performance evaluation. J. Chromatogr. B 2009, 877, 3686–3694. [Google Scholar] [CrossRef] [PubMed]
- Schneiderman, S.; Zhang, L.; Fong, H.; Menkhaus, T.J. Surface-functionalized electrospun carbon nanofiber mats as an innovative type of protein adsorption/purification medium with high capacity and high throughput. J. Chromatogr. A 2011, 1218, 8989–8995. [Google Scholar] [CrossRef]
- Dods, S.R.; Hardick, O.; Stevens, R.; Bracewell, D.G. Fabricating electrospun cellulose nanofibre adsorbents for ion-exchange chromatography. J. Chromatogr. A 2014, 1376, 74–83. [Google Scholar] [CrossRef]
- Fu, Q.; Wang, X.; Si, Y.; Liu, L.; Yu, J.; Ding, B. Scalable Fabrication of Electrospun Nanofibrous Membranes Functionalized with Citric Acid for High-Performance Protein Adsorption. ACS Appl. Mater. Interfaces 2016, 8, 11819–11829. [Google Scholar] [CrossRef]
- Abu Seman, M.N.; Khayet, M.; Hilal, N. Comparison of two different UV-grafted nanofiltration membranes prepared for reduction of humic acid fouling using acrylic acid and N-vinylpyrrolidone. Desalination 2012, 287, 19–29. [Google Scholar] [CrossRef]
- Chenette, H.C.; Robinson, J.R.; Hobley, E.; Husson, S.M. Development of high-productivity, strong cation-exchange adsorbers for protein capture by graft polymerization from membranes with different pore sizes. J. Membr. Sci. 2012, 423, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zheng, Y.; Gurgel, P.V.; Carbonell, R.G. Affinity membrane development from PBT nonwoven by photo-induced graft polymerization, hydrophilization and ligand attachment. J. Membr. Sci. 2013, 428, 562–575. [Google Scholar] [CrossRef]
- Meng, H.; Cheng, Q.; Wang, H.; Li, C. Improving Anti-Protein-Fouling Property of Polyacrylonitrile Ultrafiltration Membrane by Grafting Sulfobetaine Zwitterions. J. Chem. 2014. [Google Scholar] [CrossRef] [Green Version]
- Ahmadiannamini, P.; Eswaranandam, S.; Wickramasinghe, R.; Qian, X. Mixed-matrix membranes for efficient ammonium removal from wastewaters. J. Membr. Sci. 2017, 526, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Gurgel, P.V.; Carbonell, R.G. Preparation and characterization of anion exchange adsorptive nonwoven membranes with high protein binding capacity. J. Membr. Sci. 2015, 493, 349–359. [Google Scholar] [CrossRef]
- Chiao, Y.-H.; Sengupta, A.; Chen, S.-T.; Huang, S.-H.; Hu, C.-C.; Hung, W.-S.; Chang, Y.; Qian, X.; Wickramasinghe, R.; Lee, K.-R. Zwitterion augmented polyamide membrane for improved forward osmosis performance with significant antifouling characteristics. Sep. Purif. Technol. 2019, 212, 316–325. [Google Scholar] [CrossRef]
- Carbajal, M.L.; Smolko, E.E.; Grasselli, M. Oriented immobilization of proteins on grafted porous polymers. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2003, 208, 416–423. [Google Scholar] [CrossRef]
- Bayramoglu, G.; Arıca, M.Y. Immobilization of laccase onto poly(glycidylmethacrylate) brush grafted poly(hydroxyethylmethacrylate) films: Enzymatic oxidation of phenolic compounds. Mater. Sci. Eng. C 2009, 29, 1990–1997. [Google Scholar] [CrossRef]
- Gao, B.; Zhang, D.; Li, Y. Constituting a special redox surface-initiating system and realizing graft-polymerization of GMA on polysulfone microfiltration membrane. J. Polym. Res. 2018, 25, 158. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, C.; Xie, B.; Hu, W.; Li, Y.; Yao, C. PAN ultrafiltration membranes grafted with natural amino acids for improving antifouling property. J. Coatings Technol. Res. 2017, 15, 403–414. [Google Scholar] [CrossRef]
- Liu, Z.; Wickramasinghe, S.R.; Qian, X. Membrane chromatography for protein purifications from ligand design to functionalization. Sep. Sci. Technol. 2016, 52, 299–319. [Google Scholar] [CrossRef]
- Huang, Y.-X.; Wang, Z.; Jin, J.; Lin, S. Novel Janus Membrane for Membrane Distillation with Simultaneous Fouling and Wetting Resistance. Environ. Sci. Technol. 2017, 51, 13304–13310. [Google Scholar] [CrossRef]
- Huang, L.; Arena, J.T.; Manickam, S.S.; Jiang, X.; Willis, B.G.; McCutcheon, J.R. Improved mechanical properties and hydrophilicity of electrospun nanofiber membranes for filtration applications by dopamine modification. J. Membr. Sci. 2014, 460, 241–249. [Google Scholar] [CrossRef]
- Zhao, R.; Li, Y.; Li, X.; Li, Y.; Sun, B.; Chao, S.; Wang, C. Facile hydrothermal synthesis of branched polyethylenimine grafted electrospun polyacrylonitrile fiber membrane as a highly efficient and reusable bilirubin adsorbent in hemoperfusion. J. Colloid Interface Sci. 2018, 514, 675–685. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Dynamic Binding Capacity (BSA, mg/mL Membrane Volume) | BSA Yield (%) |
|---|---|---|
| PSf | 201.3 ± 4.8 | 55.9 ± 5.1 |
| PAN | 87.2 ± 2.6 | 96.4 ± 3.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-T.; Wickramasinghe, S.R.; Qian, X. Electrospun Weak Anion-Exchange Fibrous Membranes for Protein Purification. Membranes 2020, 10, 39. https://doi.org/10.3390/membranes10030039
Chen S-T, Wickramasinghe SR, Qian X. Electrospun Weak Anion-Exchange Fibrous Membranes for Protein Purification. Membranes. 2020; 10(3):39. https://doi.org/10.3390/membranes10030039
Chicago/Turabian StyleChen, Shu-Ting, S. Ranil Wickramasinghe, and Xianghong Qian. 2020. "Electrospun Weak Anion-Exchange Fibrous Membranes for Protein Purification" Membranes 10, no. 3: 39. https://doi.org/10.3390/membranes10030039