
Multi-Subunit SARS-CoV-2 Vaccine Design Using Evolutionarily Conserved T- and B- Cell Epitopes

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Nucleotide Sequence Retrieval and Phylogenetic Analysis
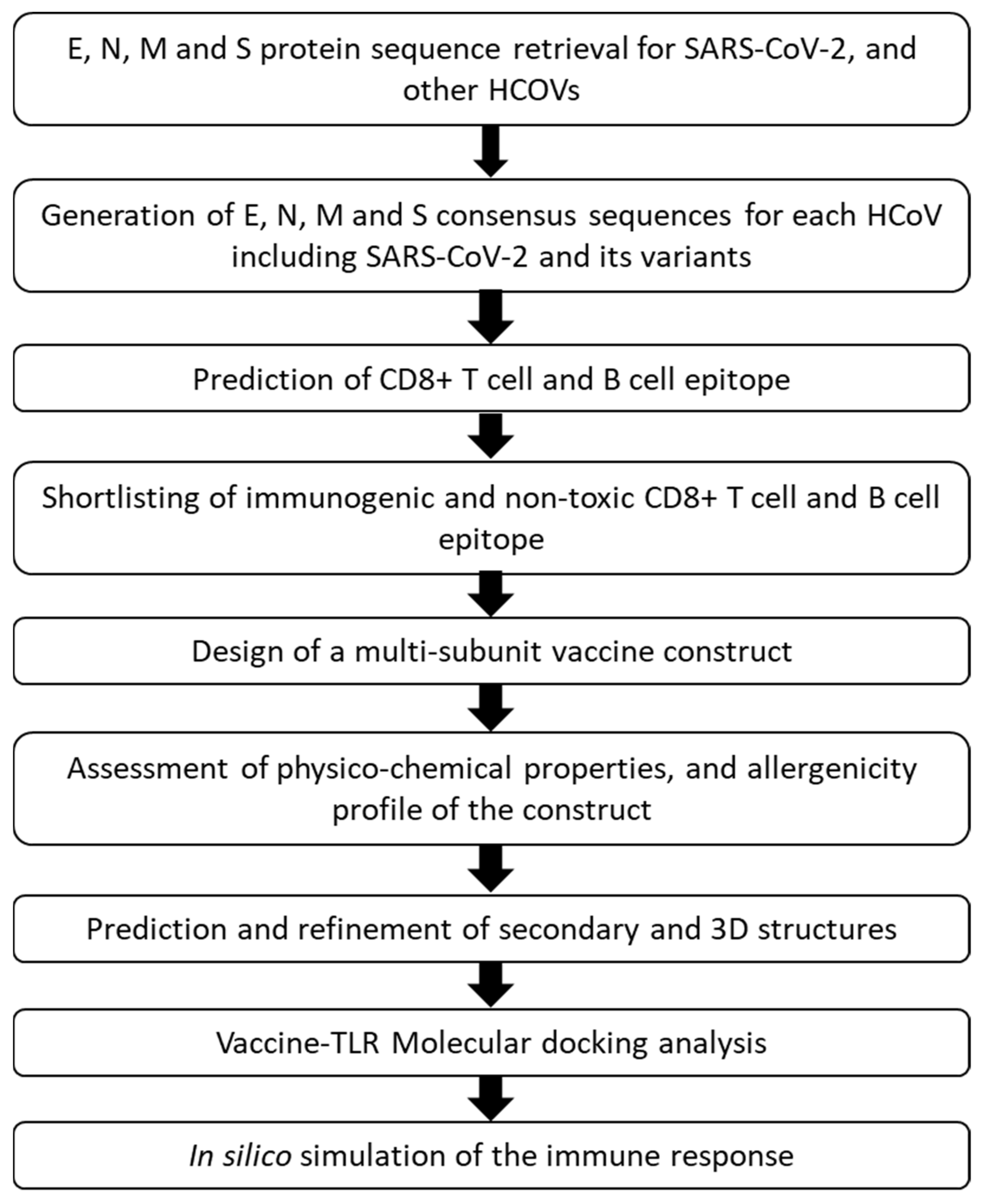
2.2. Protein Sequence Retrieval and Vaccine Design Workflow
2.3. In Silico CD8+ T Cell and B Cell Epitope Mapping, and Epitope Conservation Analysis
2.4. Selection of Immunogenic and Non-Toxic Epitopes for Multi-Subunit Vaccine Design
2.5. Design of a Multi-Subunit Vaccine
2.6. Evaluation of Physico-Chemical Properties, and Allergenicity Profile of the Multi-Subunit Vaccine
2.7. Prediction and Refinement of Secondary and 3D Structures
2.8. Molecular Docking Analysis
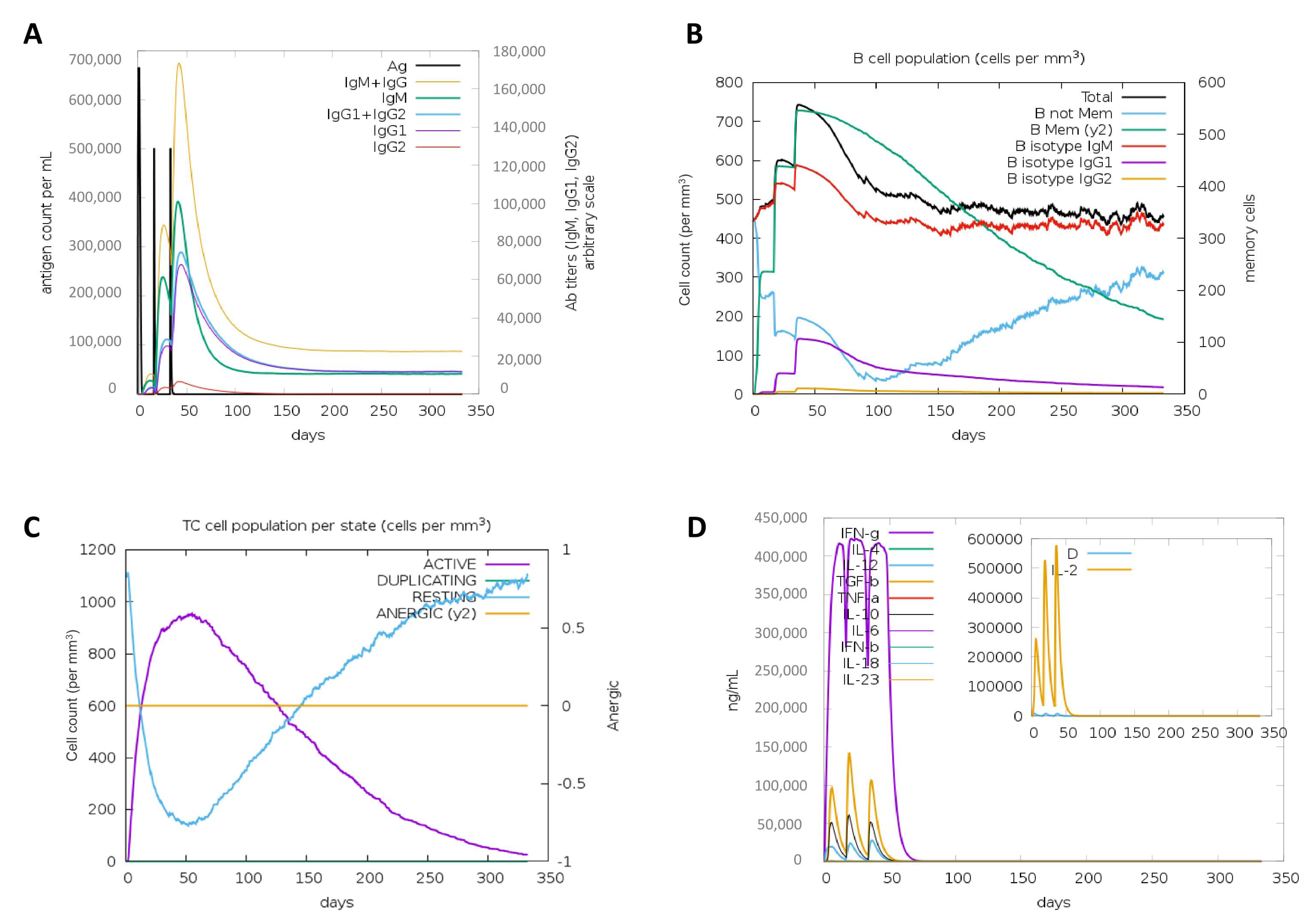
2.9. In Silico Simulation of the Immune Response
3. Results
3.1. Phylogenetic and Sequence Similarity among the Human Coronaviruses
3.2. CD8+ T Cell Epitopes Originating in Conserved Regions of E, N, M, and S Proteins
3.3. Predicted Linear B Cell Epitopes Found to Be Conserved in E, N, M, and S Protein Sequences of HCoVs
3.4. Selection of Immunogenic Non-Toxic CD8+ T and Linear B Cell Epitopes for Design of Multi-Subunit Vaccine
3.5. Design and Assessment of the Multi-Subunit Vaccine
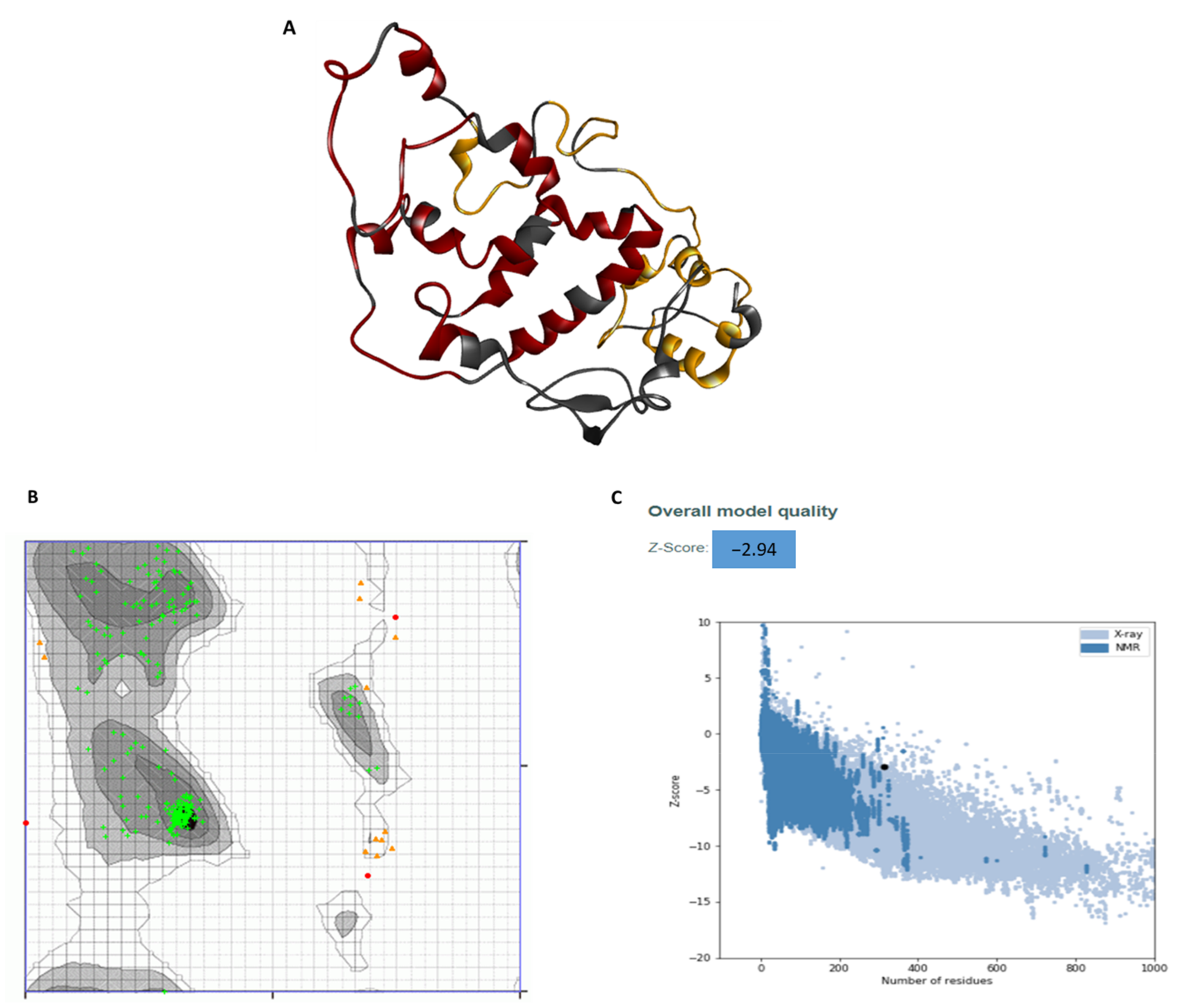
3.6. Secondary and Tertiary Structure of the Multiepitope Vaccine:
3.7. Molecular Docking Analysis of the Vaccine–TLR4 Complex
3.8. In Silico Simulation of the Immune Response against the Vaccine Construct
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273, Addendum in 2020, 588, E6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East respiratory syndrome coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef] [Green Version]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Fouchier, R.A.; Hartwig, N.G.; Bestebroer, T.M.; Niemeyer, B.; de Jong, J.C.; Simon, J.H.; Osterhaus, A.D. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. USA 2004, 101, 6212–6216. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human Coronaviruses: A Review of Virus-Host Interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280e278. [Google Scholar] [CrossRef]
- Letko, M.; Munster, V. Functional assessment of cell entry and receptor usage for lineage B beta-coronaviruses, including 2019-nCoV. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Habibzadeh, P.; Stoneman, E.K. The Novel Coronavirus: A Bird’s Eye View. Int. J. Occup. Environ. Med. 2020, 11, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Shang, W.; Yang, Y.; Rao, Y.; Rao, X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. NPJ Vaccines 2020, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Marks, F.; Clemens, J.D. Looking beyond COVID-19 vaccine phase 3 trials. Nat. Med. 2021, 27, 205–211. [Google Scholar] [CrossRef]
- Karim, S.S.A. Vaccines and SARS-CoV-2 variants: The urgent need for a correlate of protection. Lancet 2021, 397, 1263–1264. [Google Scholar] [CrossRef]
- Creech, C.B.; Walker, S.C.; Samuels, R.J. SARS-CoV-2 Vaccines. JAMA 2021, 325, 1318–1320. [Google Scholar] [CrossRef]
- Jones, I.; Roy, P. Sputnik V COVID-19 vaccine candidate appears safe and effective. Lancet 2021, 397, 642–643. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 Spike Glycoprotein Biosynthesis, Structure, Function, and Antigenicity: Implications for the Design of Spike-Based Vaccine Immunogens. Front Immunol. 2020, 11, 576622. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294e1289. [Google Scholar] [CrossRef]
- Emary, K.R.W.; Golubchik, T.; Aley, P.K.; Ariani, C.V.; Angus, B.; Bibi, S.; Blane, B.; Bonsall, D.; Cicconi, P.; Charlton, S.; et al. Efficacy of ChAdOx1 nCoV-19 (AZD1222) vaccine against SARS-CoV-2 variant of concern 202012/01 (B.1.1.7): An exploratory analysis of a randomised controlled trial. Lancet 2021, 397, 1351–1362. [Google Scholar] [CrossRef]
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
- Janice Oh, H.L.; Ken-En Gan, S.; Bertoletti, A.; Tan, Y.J. Understanding the T cell immune response in SARS coronavirus infection. Emerg. Microbes Infect. 2012, 1, e23. [Google Scholar] [CrossRef] [Green Version]
- Pinotti, F.; Wikramaratna, P.S.; Obolski, U.; Paton, R.S.; Damineli, D.S.C.; Alcantara, L.C.J.; Giovanetti, M.; Gupta, S.; Lourenco, J. Potential impact of individual exposure histories to endemic human coronaviruses on age-dependent severity of COVID-19. BMC Med. 2021, 19, 19. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symposium Series 1999, 41, 95–98. [Google Scholar]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Bhasin, M.; Raghava, G.P. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine 2004, 22, 3195–3204. [Google Scholar] [CrossRef]
- Bhasin, M.; Raghava, G.P. A hybrid approach for predicting promiscuous MHC class I restricted T cell epitopes. J. Biosci. 2007, 32, 31–42. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery, C.; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Abraham Peele, K.; Srihansa, T.; Krupanidhi, S.; Ayyagari, V.S.; Venkateswarulu, T.C. Design of multi-epitope vaccine candidate against SARS-CoV-2: A in-silico study. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef]
- Ikram, A.; Zaheer, T.; Awan, F.M.; Obaid, A.; Naz, A.; Hanif, R.; Paracha, R.Z.; Ali, A.; Naveed, A.K.; Janjua, H.A. Exploring NS3/4A, NS5A and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches. Sci. Rep. 2018, 8, 16107. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein-Sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef] [Green Version]
- Biragyn, A.; Ruffini, P.A.; Leifer, C.A.; Klyushnenkova, E.; Shakhov, A.; Chertov, O.; Shirakawa, A.K.; Farber, J.M.; Segal, D.M.; Oppenheim, J.J.; et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 2002, 298, 1025–1029. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu Rev. Immunol 2015, 33, 257–290. [Google Scholar] [CrossRef] [Green Version]
- Ohto, U.; Yamakawa, N.; Akashi-Takamura, S.; Miyake, K.; Shimizu, T. Structural Analyses of Human Toll-like Receptor 4 Polymorphisms D299G and T399I*. Journal of Biological Chemistry 2012, 287, 40611–40617. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Castiglione, F. Immune system simulation online. Bioinformatics 2011, 27, 2013–2014. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 2007, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Kesmir, C.; Nussbaum, A.K.; Schild, H.; Detours, V.; Brunak, S. Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 2002, 15, 287–296. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Kesmir, C. The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005, 57, 33–41. [Google Scholar] [CrossRef]
- Kared, H.; Redd, A.D.; Bloch, E.M.; Bonny, T.S.; Sumatoh, H.; Kairi, F.; Carbajo, D.; Abel, B.; Newell, E.W.; Bettinotti, M.P.; et al. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Snyder, T.M.; Gittelman, R.M.; Klinger, M.; May, D.H.; Osborne, E.J.; Taniguchi, R.; Zahid, H.J.; Kaplan, I.M.; Dines, J.N.; Noakes, M.N.; et al. Magnitude and Dynamics of the T-Cell Response to SARS-CoV-2 Infection at Both Individual and Population Levels. medRxiv 2020. [Google Scholar] [CrossRef]
- Bilich, T.; Nelde, A.; Heitmann, J.S.; Maringer, Y.; Roerden, M.; Bauer, J.; Rieth, J.; Wacker, M.; Peter, A.; Horber, S.; et al. T cell and antibody kinetics delineate SARS-CoV-2 peptides mediating long-term immune responses in COVID-19 convalescent individuals. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Schwarz, T.; Heiss, K.; Mahendran, Y.; Casilag, F.; Kurth, F.; Sander, L.E.; Wendtner, C.M.; Hoechstetter, M.A.; Muller, M.A.; Sekul, R.; et al. SARS-CoV-2 Proteome-Wide Analysis Revealed Significant Epitope Signatures in COVID-19 Patients. Front. Immunol. 2021, 12, 629185. [Google Scholar] [CrossRef]
- Tarke, A.; Sidney, J.; Kidd, C.K.; Dan, J.M.; Ramirez, S.I.; Yu, E.D.; Mateus, J.; da Silva Antunes, R.; Moore, E.; Rubiro, P.; et al. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep. Med. 2021, 2, 100204. [Google Scholar] [CrossRef]
- Ferretti, A.P.; Kula, T.; Wang, Y.; Nguyen, D.M.V.; Weinheimer, A.; Dunlap, G.S.; Xu, Q.; Nabilsi, N.; Perullo, C.R.; Cristofaro, A.W.; et al. Unbiased Screens Show CD8(+) T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 that Largely Reside outside the Spike Protein. Immunity 2020, 53, 1095–1107e1093. [Google Scholar] [CrossRef]
- Li, Y.; Ma, M.L.; Lei, Q.; Wang, F.; Hong, W.; Lai, D.Y.; Hou, H.; Xu, Z.W.; Zhang, B.; Chen, H.; et al. Linear epitope landscape of the SARS-CoV-2 Spike protein constructed from 1,051 COVID-19 patients. Cell Rep. 2021, 34, 108915. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef]
- Lu, S.; Xie, X.X.; Zhao, L.; Wang, B.; Zhu, J.; Yang, T.R.; Yang, G.W.; Ji, M.; Lv, C.P.; Xue, J.; et al. The immunodominant and neutralization linear epitopes for SARS-CoV-2. Cell Rep. 2021, 34, 108666. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; de Oliveira, T.; Vermeulen, M.; van der Berg, K.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar] [CrossRef]
- Sanami, S.; Zandi, M.; Pourhossein, B.; Mobini, G.R.; Safaei, M.; Abed, A.; Arvejeh, P.M.; Chermahini, F.A.; Alizadeh, M. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int. J. Biol. Macromol. 2020, 164, 871–883. [Google Scholar] [CrossRef]
- Kar, T.; Narsaria, U.; Basak, S.; Deb, D.; Castiglione, F.; Mueller, D.M.; Srivastava, A.P. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 10895. [Google Scholar] [CrossRef]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef]
- Martin, W.R.; Cheng, F. A rational design of a multi-epitope vaccine against SARS-CoV-2 which accounts for the glycan shield of the spike glycoprotein. J. Biomol. Struct. Dyn. 2021, 1–15. [Google Scholar] [CrossRef]
- Sarkar, B.; Ullah, M.A.; Johora, F.T.; Taniya, M.A.; Araf, Y. Immunoinformatics-guided designing of epitope-based subunit vaccines against the SARS Coronavirus-2 (SARS-CoV-2). Immunobiology 2020, 225, 151955. [Google Scholar] [CrossRef]
- Kalita, P.; Padhi, A.K.; Zhang, K.Y.J.; Tripathi, T. Design of a peptide-based subunit vaccine against novel coronavirus SARS-CoV-2. Microb. Pathog. 2020, 145, 104236. [Google Scholar] [CrossRef]
- Rehman, I.; Kerndt, C.C.; Botelho, S. Biochemistry, Tertiary Protein Structure; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Enany, S. Structural and functional analysis of hypothetical and conserved proteins of Clostridium tetani. J. Infect. Public Health 2014, 7, 296–307. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Foroutan, M.; Ghaffarifar, F.; Sharifi, Z.; Dalimi, A. Vaccination with a novel multi-epitope ROP8 DNA vaccine against acute Toxoplasma gondii infection induces strong B and T cell responses in mice. Comp. Immunol. Microbiol. Infect. Dis. 2020, 69, 101413. [Google Scholar] [CrossRef]
- Wlodawer, A. Stereochemistry and Validation of Macromolecular Structures. Methods Mol. Biol. 2017, 1607, 595–610. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, Z.; Rafiei, A.; Yazdani, M.; Valadan, R. Design an Efficient Multi-Epitope Peptide Vaccine Candidate Against SARS-CoV-2: An in silico Analysis. Infect. Drug Resist. 2020, 13, 3007–3022. [Google Scholar] [CrossRef]
- Tso, F.Y.; Lidenge, S.J.; Pena, P.B.; Clegg, A.A.; Ngowi, J.R.; Mwaiselage, J.; Ngalamika, O.; Julius, P.; West, J.T.; Wood, C. High prevalence of pre-existing serological cross-reactivity against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) in sub-Saharan Africa. Int. J. Infect. Dis. 2021, 102, 577–583. [Google Scholar] [CrossRef]
- Saletti, G.; Gerlach, T.; Jansen, J.M.; Molle, A.; Elbahesh, H.; Ludlow, M.; Li, W.; Bosch, B.J.; Osterhaus, A.; Rimmelzwaan, G.F. Older adults lack SARS CoV-2 cross-reactive T lymphocytes directed to human coronaviruses OC43 and NL63. Sci. Rep. 2020, 10, 21447. [Google Scholar] [CrossRef]
- Ladner, J.T.; Henson, S.N.; Boyle, A.S.; Engelbrektson, A.L.; Fink, Z.W.; Rahee, F.; D’Ambrozio, J.; Schaecher, K.E.; Stone, M.; Dong, W.; et al. Epitope-resolved profiling of the SARS-CoV-2 antibody response identifies cross-reactivity with an endemic human CoV. bioRxiv 2020. [Google Scholar] [CrossRef]
- Khan, S.; Nakajima, R.; Jain, A.; de Assis, R.R.; Jasinskas, A.; Obiero, J.M.; Adenaiye, O.; Tai, S.; Hong, F.; Milton, D.K.; et al. Analysis of Serologic Cross-Reactivity Between Common Human Coronaviruses and SARS-CoV-2 Using Coronavirus Antigen Microarray. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Che, X.Y.; Qiu, L.W.; Liao, Z.Y.; Wang, Y.D.; Wen, K.; Pan, Y.X.; Hao, W.; Mei, Y.B.; Cheng, V.C.; Yuen, K.Y. Antigenic cross-reactivity between severe acute respiratory syndrome-associated coronavirus and human coronaviruses 229E and OC43. J. Infect. Dis 2005, 191, 2033–2037. [Google Scholar] [CrossRef] [Green Version]
- Ilinskaya, A.N.; Dobrovolskaia, M.A. Understanding the immunogenicity and antigenicity of nanomaterials: Past, present and future. Toxicol. Appl. Pharmacol. 2016, 299, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Yang, X.M.; Xie, Q.M.; Ma, J.Y.; Luo, Y.N.; Cao, Y.C.; Chen, F.; Bi, Y.Z. The potent adjuvant effects of chicken beta-defensin-1 when genetically fused with infectious bursal disease virus VP2 gene. Vet. Immunol. Immunopathol. 2010, 136, 92–97. [Google Scholar] [CrossRef]
- Lam, L.K.M.; Watson, A.M.; Ryman, K.D.; Klimstra, W.B. Gamma-interferon exerts a critical early restriction on replication and dissemination of yellow fever virus vaccine strain 17D-204. NPJ Vaccines 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Ewer, K.J.; Barrett, J.R.; Belij-Rammerstorfer, S.; Sharpe, H.; Makinson, R.; Morter, R.; Flaxman, A.; Wright, D.; Bellamy, D.; Bittaye, M.; et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat. Med. 2021, 27, 270–278. [Google Scholar] [CrossRef]
- Gallagher, K.M.E.; Leick, M.B.; Larson, R.C.; Berger, T.R.; Katsis, K.; Yam, J.Y.; Brini, G.; Grauwet, K.; MGH COVID-19 Collection & Processing Team; Maus, M.V. SARS -CoV-2 T-cell immunity to variants of concern following vaccination. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dong, R.; Chu, Z.; Yu, F.; Zha, Y. Contriving Multi-Epitope Subunit of Vaccine for COVID-19: Immunoinformatics Approaches. Front. Immunol 2020, 11, 1784. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021. [Google Scholar] [CrossRef]
- Tahir Ul Qamar, M.; Rehman, A.; Tusleem, K.; Ashfaq, U.A.; Qasim, M.; Zhu, X.; Fatima, I.; Shahid, F.; Chen, L.L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS ONE 2020, 15, e0244176. [Google Scholar] [CrossRef]
- Crooke, S.N.; Ovsyannikova, I.G.; Kennedy, R.B.; Poland, G.A. Immunoinformatic identification of B cell and T cell epitopes in the SARS-CoV-2 proteome. Sci. Rep. 2020, 10, 14179. [Google Scholar] [CrossRef]
- Kiyotani, K.; Toyoshima, Y.; Nemoto, K.; Nakamura, Y. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. J. Hum. Genet. 2020, 65, 569–575. [Google Scholar] [CrossRef]
- Mukherjee, S.; Tworowski, D.; Detroja, R.; Mukherjee, S.B.; Frenkel-Morgenstern, M. Immunoinformatics and Structural Analysis for Identification of Immunodominant Epitopes in SARS-CoV-2 as Potential Vaccine Targets. Vaccines 2020, 8, 290. [Google Scholar] [CrossRef]
- Wang, D.; Mai, J.; Zhou, W.; Yu, W.; Zhan, Y.; Wang, N.; Epstein, N.D.; Yang, Y. Immunoinformatic Analysis of T- and B-Cell Epitopes for SARS-CoV-2 Vaccine Design. Vaccines 2020, 8, 355. [Google Scholar] [CrossRef]
- Toledo, H.; Baly, A.; Castro, O.; Resik, S.; Laferte, J.; Rolo, F.; Navea, L.; Lobaina, L.; Cruz, O.; Miguez, J.; et al. A phase I clinical trial of a multi-epitope polypeptide TAB9 combined with Montanide ISA 720 adjuvant in non-HIV-1 infected human volunteers. Vaccine 2001, 19, 4328–4336. [Google Scholar] [CrossRef]
- Slingluff, C.L., Jr.; Lee, S.; Zhao, F.; Chianese-Bullock, K.A.; Olson, W.C.; Butterfield, L.H.; Whiteside, T.L.; Leming, P.D.; Kirkwood, J.M. A randomized phase II trial of multiepitope vaccination with melanoma peptides for cytotoxic T cells and helper T cells for patients with metastatic melanoma (E1602). Clin. Cancer Res. 2013, 19, 4228–4238. [Google Scholar] [CrossRef] [Green Version]
- Lennerz, V.; Gross, S.; Gallerani, E.; Sessa, C.; Mach, N.; Boehm, S.; Hess, D.; von Boehmer, L.; Knuth, A.; Ochsenbein, A.F.; et al. Immunologic response to the survivin-derived multi-epitope vaccine EMD640744 in patients with advanced solid tumors. Cancer Immunol. Immunother. 2014, 63, 381–394. [Google Scholar] [CrossRef]
- Guo, L.; Yin, R.; Liu, K.; Lv, X.; Li, Y.; Duan, X.; Chu, Y.; Xi, T.; Xing, Y. Immunological features and efficacy of a multi-epitope vaccine CTB-UE against H. pylori in BALB/c mice model. Appl. Microbiol. Biotechnol. 2014, 98, 3495–3507. [Google Scholar] [CrossRef]
- Cao, Y.; Li, D.; Fu, Y.; Bai, Q.; Chen, Y.; Bai, X.; Jing, Z.; Sun, P.; Bao, H.; Li, P.; et al. Rational design and efficacy of a multi-epitope recombinant protein vaccine against foot-and-mouth disease virus serotype A in pigs. Antiviral. Res. 2017, 140, 133–141. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HCoVs | Protein Sequences | |||
|---|---|---|---|---|
| Envelope | Membrane | Nucleocapsid | Spike | |
| HCoV-NL63 | 2 | 11 | 36 | 51 |
| HCoV-229E | 5 | 6 | 51 | 71 |
| HCoV-OC43 | 6 | 15 | 106 | 183 |
| HCoV-HKU1 | 3 | 8 | 28 | 26 |
| MERS-Cov | 18 | 35 | 87 | 250 |
| SARS-CoV | 13 | 23 | 29 | 80 |
| SARS-CoV-2 | 56 | 176 | 3082 | 1139 |
| Conserved CD8 + T Cell Epitopes in HCoVs | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Membrane | |||||||||||||||
| № | SARS-COV-2 | HLA-Resctriciton | SARS-COV | HLA-Resctriciton | MERS | HLA-Resctriciton | NL63 | HLA-Resctriciton | 229E | HLA-Resctriciton | OC43 | HLA-Resctriciton | HKU1 | HLA-Resctriciton | Conservation (%) |
| 1 | SYFIASFRL | HLA-A24,HLA-Cw*0401 | SYFVASFRL | HLA-A24, HLA-A*2402,HLA-Cw*0401 | SYFVQSIRL | HLA-Cw*0401 | MYFVNSFRL | HLA-A24,HLA-Cw*0401 | MYFANSFRL | HLA-A24,HLA-Cw*0401 | VYFVNSIRL | HLA-A24,HLA-A*2402,HLA-Cw*0401 | YFVNSIRLF | HLA-Cw*0401 | 55 |
| Nucleocapsid | |||||||||||||||
| 1 | SPRWYFYYL | HLA-Cw*0401 | SPRWYFYYL | HLA-Cw*0401 | APRWYFYYT | HLA-A*0202,HLA-A*0203,HLA-A*0205,HLA-A*0301,HLA-Cw*0401 | PPKVHFYYL | HLA-A*0301,HLA-Cw*0401 | SPKLHFYYL | HLA-B*3501,HLA-Cw*0401,HLA-B35 | LPRWYFYYL | HLA-A*0301,HLA-Cw*0401,HLA-B35 | LPRWYFYYL | HLA-A*0301,HLA-Cw*0401,HLA-B35 | 44 |
| Spike | |||||||||||||||
| 1 | YIKWPWYIW | HLA-A*0206,HLA-Cw*0401 | YIKWPWYVW | HLA-A*0206,HLA-Cw*0401 | YNKWPWYIW | HLA-A*0206,HLA-B*3501,HLA-B*5102,HLA-B*5103,HLA-B*5401,HLA-B7,HLA-Cw*0401,HLA-B35 | YIKWPWWVW | HLA-A2,HLA-A*0206,HLA-Cw*0401 | YIKWPWWVW | HLA-A2,HLA-A*0206,HLA-Cw*0401 | YVKWPWYVW | HLA-A*0206,HLA-B*51, HLA-Cw*0401 | YVKWPWYVW | HLA-A*0206,HLA-B*51,HLA-Cw*0401 | 66 |
| 2 | QSAPHGVVF | HLA-Cw*0401,HLA-A*3301 | QAAPHGVVF | HLA-Cw*0401, | VNAPNGLYF | HLA-B*5401,HLA-B*51,HLA-B*0702,HLA-Cw*0401 | NSAPDGLLF | HLA-Cw*0401 | NAAPEGLVF | HLA-Cw*0401 | QNAPYGLYF | HLA-B*5401,HLA-Cw*0401,HLA-A*3301 | QNAPYGLLF | HLA-B*5401,HLA-Cw*0401 | 44 |
| Conserved CD8 + T Cell Epitopes in SARS-CoV-2, SARS-CoV, and MERS-CoV | |||||||
|---|---|---|---|---|---|---|---|
| Envelope | |||||||
| № | SARS-CoV-2 | HLA-resctriciton | SARS-CoV | HLA-resctriciton | MERS-CoV | HLA-resctriciton | Conservation (%) |
| 1 | AILTALRLC | HLA-Cw*0401 | AILTALRLC | HLA-Cw*0401 | AFLTATRLC | HLA-Cw*0401 | 77 |
| 2 | SLVKPSFYV | HLA-Cw*0401 | SLVKPTVYV | HLA-A*0205,HLA-A*0301,HLA-B*5301,HLA-Cw*0401 | LLVQPALYL | HLA-A2,HLA-A3,HLA-A*0301,HLA-Cw*0401 | 44 |
| Membrane | |||||||
| 1 | SFNPETNIL | HLA-A*0203,HLA-Cw*0401, | SFNPETNIL | HLA-A*0203,HLA-Cw*0401, | SFNPETNCL | HLA-Cw*0401 | 88 |
| 2 | SYFIASFRL | HLA-A24,HLA-Cw*0401 | SYFVASFRL | HLA-A24,HLA-A*2402,HLA-Cw*0401 | SYFVQSIRL | HLA-Cw*0401 | 66 |
| 3 | KDLPKEITV | HLA-Cw*0401 | KDLPKEITV | HLA-Cw*0401 | DRLPNEVTV | HLA-B*51,HLA-Cw*0401 | 66 |
| 4 | AGDSGFAAY | HLA-A1,HLA-Cw*0401,HLA-B35,HLA-B*2703 | GTDSGFAAY | HLA-A1,HLA-A3,HLA-Cw*0401,HLA-B35,HLA-B*2703 | GTNSGVAIY | HLA-A3,HLA-Cw*0401,HLA-B35,HLA-B44 | 44 |
| Nucleocapsid | |||||||
| 1 | SPRWYFYYL | HLA-Cw*0401 | SPRWYFYYL | HLA-Cw*0401 | APRWYFYYT | HLA-A*0202,HLA-A*0203,HLA-A*0205,HLA-A*0301,HLA-Cw*0401 | 77 |
| 2 | ASAFFGMSR | HLA-Cw*0401,HLA-A*6801 | ASAFFGMSR | HLA-Cw*0401,HLA-A*6801 | ASAFMGMSQ | HLA-A*1101,HLA-A3,HLA-A31,HLA-Cw*0401,HLA-A*3301,HLA-A*6801 | 77 |
| 3 | ANKDGIIWV | HLA-B*3501,HLA-B*5101,HLA-B*5301,HLA-B*5401,HLA-B*51,HLA-B*0702,HLA-Cw*0401,HLA-B35 | ANKEGIVWV | HLA-B*3501,HLA-B*5101, HLA-B*5301,HLA-B*5401, HLA-B*51,HLA-B*0702,HLA-Cw*0401,HLA-B35 | AVKDGIVWV | HLA-B*5301,HLA-B*51,HLA-B8,HLA-Cw*0401 | 66 |
| 4 | QASSRSSSR | HLA-Cw*0401 | QASSRSSSR | HLA-Cw*0401 | QSSSRASSV | HLA-B*5301,HLA-Cw*0401 | 66 |
| 5 | LLLDRLNQL | HLA-A2,HLA-A*0201,HLA-A*0206,HLA-Cw*0401 | LLLDRLNQL | HLA-A2,HLA-A*0201,HLA-A*0206,HLA-Cw*0401 | LYLDLLNRL | HLA-A2,HLA-A24,HLA-Cw*0401 | 66 |
| 6 | VPINTNSSP | HLA-Cw*0401 | VPINTNSGP | VPLNANSTP | HLA-Cw*0401 | 66 | |
| 7 | GYYRRATRR | HLA-Cw*0401 | GYYRRATRR | HLA-Cw*0401 | GYWRRQDRK | HLA-A24,HLA-A*2402,HLA-B*2705,HLA-Cw*0401,HLA-B*2902 | 55 |
| 8 | NPANNAAIV | HLA-A*2402,HLA-B*5301,HLA-Cw*0401 | NPNNNAATV | HLA-B*5301,HLA-Cw*0401 | NPNNDSAIV | HLA-Cw*0401 | 55 |
| Spike | |||||||
| 1 | ARDLICAQK | HLA-A1,HLA-Cw*0401 | ARDLICAQK | HLA-A1,HLA-Cw*0401 | ARDLICAQY | HLA-A1,HLA-Cw*0401,HLA-B*2703 | 88 |
| 2 | DLLFNKVTL | HLA-A2,HLA-A*0206,HLA-Cw*0401 | DLLFNKVTL | HLA-A2,HLA-A*0206,HLA-Cw*0401 | DLLFDKVTI | HLA-A3,HLA-Cw*0401 | 77 |
| 3 | NLNESLIDL | HLA-Cw*0401 | NLNESLIDL | HLA-Cw*0401 | ALNESYIDL | HLA-A*0203,HLA-Cw*0401 | 77 |
| 4 | YIKWPWYIW | HLA-A*0206,HLA-Cw*0401 | YIKWPWYVW | HLA-A*0206, HLA-Cw*0401 | YNKWPWYIW | HLA-A*0206,HLA-B*3501,HLA-B*5102,HLA-B*5103,HLA-B*5401, HLA-B7, HLA-Cw*0401,HLA-B35 | 77 |
| 5 | GFIAGLIAI | HLA-B8,HLA-Cw*0401 | GFIAGLIAI | HLA-B8,HLA-Cw*0401 | GFIAGLVAL | HLA-B8,HLA-Cw*0401,HLA-B35 | 77 |
| 6 | QYIKWPWYI | HLA-A*1101,HLA-A11,HLA-A24,HLA-A3,HLA-B8,HLA-Cw*0401, | QYIKWPWYV | HLA-A24, HLA-A*2402,HLA-B*5301,HLA-B8,HLA-Cw*0401 | YYNKWPWYI | HLA-A*1101,HLA-A11,HLA-B8,HLA-Cw*0401, | 66 |
| 7 | NQKLIANQF | HLA-B27,HLA-Cw*0401,HLA-B*2703 | NQKQIANQF | HLA-B14,HLA-B27,HLA-Cw*0401 | NQKLIANKF | HLA-B27,HLA-Cw*0401, | 66 |
| 8 | VVNQNAQAL | HLA-B*3501,HLA-B*51,HLA-B*0702,HLA-Cw*0401,HLA-B35 | VVNQNAQAL | HLA-B*3501,HLA-B*51,HLA-B*0702,HLA-Cw*0401,HLA-B35 | AVNNNAQAL | HLA-B*51,HLA-Cw*0401,HLA-B35 | 66 |
| 9 | AYRFNGIGV | HLA-A*2402,HLA-Cw*0401,HLA-B*2706,HLA-A*3301 | AYRFNGIGV | HLA-A*2402,HLA-Cw*0401,HLA-B*2706,HLA-A*3301 | FYRLNGVGI | HLA-A*1101,HLA-A11,HLA-Cw*0401,HLA-A*3301 | 55 |
| 10 | DFCGKGYHL | HLA-Cw*0401 | DFCGKGYHL | HLA-Cw*0401 | GFCGQGTHI | HLA-A*1101,HLA-A11,HLA-B8,HLA-Cw*0401, | 55 |
| 11 | APHGVVFLH | HLA-A*2402,HLA-A*0301,HLA-Cw*0401 | APHGVVFLH | HLA-A*2402,HLA-A*0301,HLA-Cw*0401, | APNGLYFMH | HLA-B*5102,HLA-B*5103,HLA-Cw*0401 | 55 |
| 12 | QSAPHGVVF | HLA-Cw*0401,HLA-A*3301 | QAAPHGVVF | HLA-Cw*0401, | VNAPNGLYF | HLA-B*5401,HLA-B*51,HLA-B*0702,HLA-Cw*0401 | 44 |
| 13 | VVFLHVTYV | HLA-B*51,HLA-Cw*0401 | VVFLHVTYV | HLA-B*51,HLA-Cw*0401 | LYFMHVGYY | HLA-A1,HLA-A*2402,HLA-A3,HLA-B*51,HLA-Cw*0401,HLA-A*3301 | 44 |
| 14 | QMAYRFNGI | HLA-Cw*0401,HLA-A*3301,HLA-A*6801 | QMAYRFNGI | HLA-Cw*0401,HLA-A*3301,HLA-A*6801 | SIFYRLNGV | HLA-B*51,HLA-Cw*0401 | 44 |
| Conserved B cell epitopes in SARS-CoV-2, SARS-CoV, and MERS-CoV | ||||
|---|---|---|---|---|
| Envelope | ||||
| № | SARS-CoV-2 | SARS-CoV | MERS-CoV | Conservation (%) |
| 1 | YSFVSEETGTLIVN | YSFVSEETGTLIVN | LPFVQERIGLFIVN | 50 |
| 2 | YSFVSEETGTLI | YSFVSEETGTLI | LPFVQERIGLFI | 42 |
| Membrane | ||||
| 1 | SMWSFNPETNILLN | SMWSFNPETNILLN | SWWSFNPETNCLLN | 86 |
| Nucleocapsid | ||||
| 1 | TASWFTALTQHGKEDL | TASWFTALTQHGKEEL | TVSWYTGLTQHGKVPL | 69 |
| 2 | YNVTQAFGRRGPEQTQ | YNVTQAFGRRGPEQTQ | FNMVQAFGLRGPGDLQ | 56 |
| 3 | DQVILLNKHIDAYKTF | DNVILLNKHIDAYKTF | KWLELLEQNIDAYKTF | 56 |
| 4 | TGAIKLDDKDPNFKDQ | HGAIKLDDKDPQFKDN | SGAIKLDPKNPNYNKW | 50 |
| Spike | ||||
| 1 | NEVAKNLNESLIDLQE | NEVAKNLNESLIDLQE | QQVVKALNESYIDLKE | 63 |
| 2 | ESLIDLQELGKYEQYI | ESLIDLQELGKYEQYI | ESYIDLKELGNYTYYN | 63 |
| 3 | CVLGQSKRVDFCGKGY | CVLGQSKRVDFCGKGY | CVKAQSKRSGFCGQGT | 56 |
| 4 | RDLICAQKFNGLTVLP | RDLICAQKFNGLTVLP | RDLICAQYVAGYKVLP | 50 |
| 5 | EAEVQIDRLITGRLQS | EAEVQIDRLITGRLQS | EQDAQIDRLINGRLTT | 50 |
| SARS-CoV-2 Antigenic Non-Toxic CD8+ T and B Cell Epitopes | |||||||
|---|---|---|---|---|---|---|---|
| T Cell Epitopes | |||||||
| Envelope | |||||||
| No | Peptide | Immunogenecity (Vaxijen) | Toxicity (ToxinPred) | Conservation in 7 HCoVs (%) | Conservation in SARS-CoV-2, SARS-CoV, MERS-CoV (%) | Conservation in B.1.1.7, B.1.351, P.1 (%) | Studies Experimentally Validating the Epitopes |
| 1 | SLVKPSFYV | 0.414 | Non-toxin | - | 44 | 100 | [58,59] |
| Membrane | |||||||
| 1 | SYFIASFRL | 0.4821 | Non-toxin | 55 | 66 | 100 | [59,60,61] |
| 2 | AGDSGFAAY | 0.9095 | Non-toxin | - | 44 | 100 | [61,62] |
| Nucleocapsid | |||||||
| 1 | SPRWYFYYL | 0.734 | Non-toxin | 44 | 77 | 100 | [62,63] |
| 2 | QASSRSSSR | 0.8294 | Non-toxin | - | 66 | 100 | [59] |
| 3 | VPINTNSSP | 0.4439 | Non-toxin | - | 66 | 88 | [62] |
| Spike | |||||||
| 1 | DLLFNKVTL | 0.68 | Non-toxin | - | 77 | 100 | [62,64,65] |
| 2 | NLNESLIDL | 0.6827 | Non-toxin | - | 77 | 100 | [58,59,64] |
| 3 | VVNQNAQAL | 0.4749 | Non-toxin | - | 66 | 100 | [64] |
| 4 | AYRFNGIGV | 1.2995 | Non-toxin | - | 55 | 100 | [62,64,65] |
| 5 | VVFLHVTYV | 1.5122 | Non-toxin | - | 44 | 100 | [62] |
| 6 | QMAYRFNGI | 0.6803 | Non-toxin | - | 44 | 100 | [62,65] |
| B cell epitopes | |||||||
| Envelope | |||||||
| № | Peptide | Immunogenecity (Vaxijen) | Toxicity (ToxinPred) | Conservation in 7 HCoVs (%) | Conservation in SARS-CoV-2, SARS-CoV, MERS-CoV (%) | Conservation in B.1.1.7, B.1.351, P.1 (%) | Experimentally Validated Epitope |
| 1 | YSFVSEETGTLIVN | 0.4532 | Non-Toxin | - | 50 | 100 | [66] |
| 2 | YSFVSEETGTLI | 0.5014 | Non-Toxin | - | 42 | 100 | [66] |
| Nucleocapsid | |||||||
| 1 | TASWFTALTQHGKEDL | 0.4149 | Non-Toxin | - | 69 | 100 | - |
| 2 | YNVTQAFGRRGPEQTQ | 0.4899 | Non-Toxin | - | 56 | 100 | - |
| 3 | TGAIKLDDKDPNFKDQ | 1.8438 | Non-Toxin | - | 50 | 100 | - |
| Spike | |||||||
| 1 | ESLIDLQELGKYEQYI | 0.6105 | Non-Toxin | - | 63 | 100 | - |
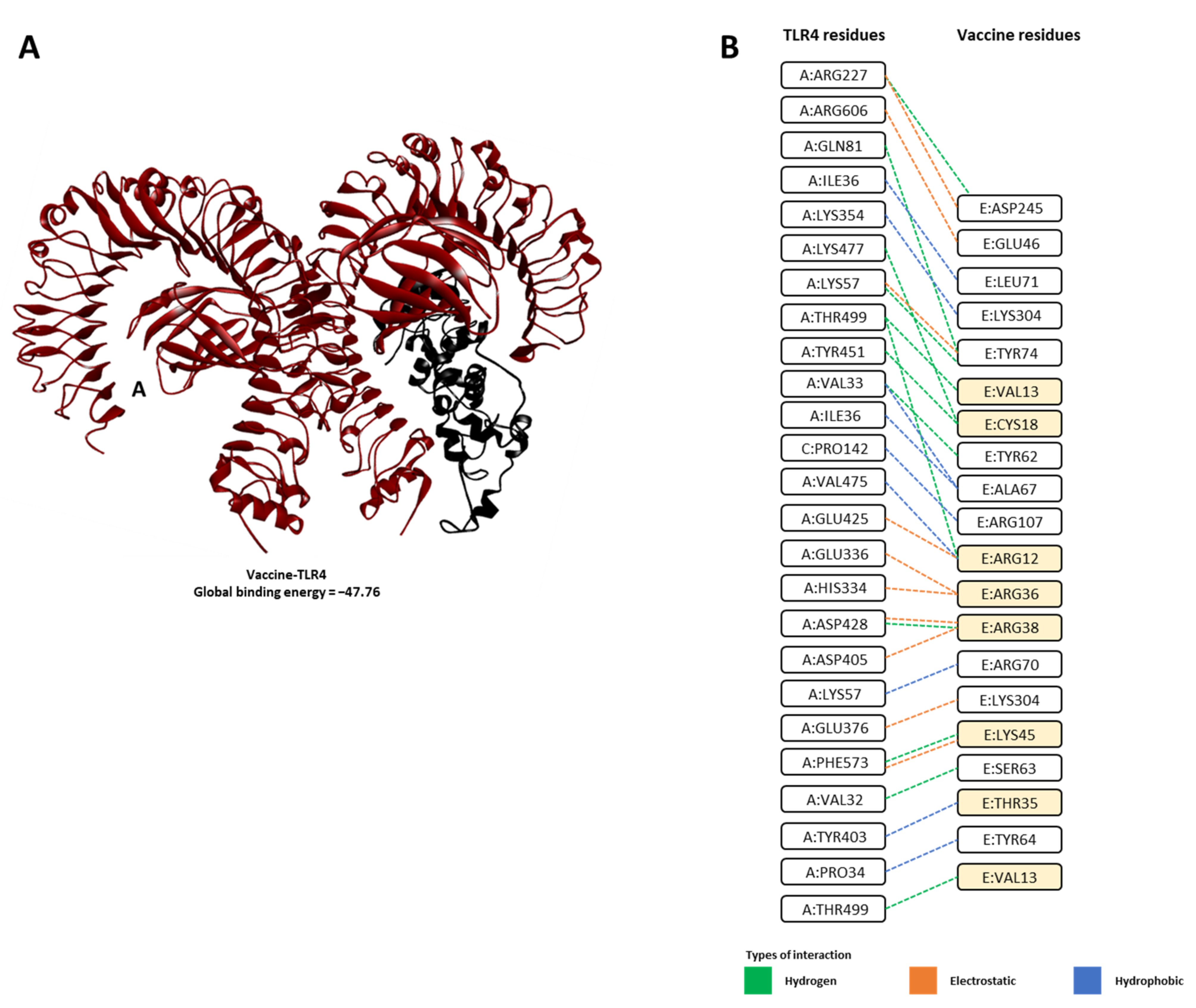
| Complex | Global Binding Energy | Energy Contributions | |||
|---|---|---|---|---|---|
| Attractive Van Der Waals (vdW) | Repulsive Van Der Waals (vdW) | Atomic Contact Energy (ACE) | Hydrogen Bond (HB) | ||
| Vaccine-TLR4 | −47.76 | −36.33 | 16.90 | 15.29 | −0.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akbay, B.; Abidi, S.H.; Ibrahim, M.A.A.; Mukhatayev, Z.; Ali, S. Multi-Subunit SARS-CoV-2 Vaccine Design Using Evolutionarily Conserved T- and B- Cell Epitopes. Vaccines 2021, 9, 702. https://doi.org/10.3390/vaccines9070702
Akbay B, Abidi SH, Ibrahim MAA, Mukhatayev Z, Ali S. Multi-Subunit SARS-CoV-2 Vaccine Design Using Evolutionarily Conserved T- and B- Cell Epitopes. Vaccines. 2021; 9(7):702. https://doi.org/10.3390/vaccines9070702
Chicago/Turabian StyleAkbay, Burkitkan, Syed Hani Abidi, Mahmoud A. A. Ibrahim, Zhussipbek Mukhatayev, and Syed Ali. 2021. "Multi-Subunit SARS-CoV-2 Vaccine Design Using Evolutionarily Conserved T- and B- Cell Epitopes" Vaccines 9, no. 7: 702. https://doi.org/10.3390/vaccines9070702