
An Immunoinformatics Approach for SARS-CoV-2 in Latam Populations and Multi-Epitope Vaccine Candidate Directed towards the World’s Population

 , , , , , and
, , , , , and
Abstract
:1. Introduction
1.1. Virology of SARS-CoV-2
1.2. Medication for COVID-19
1.3. Vaccine for SARS-CoV-2
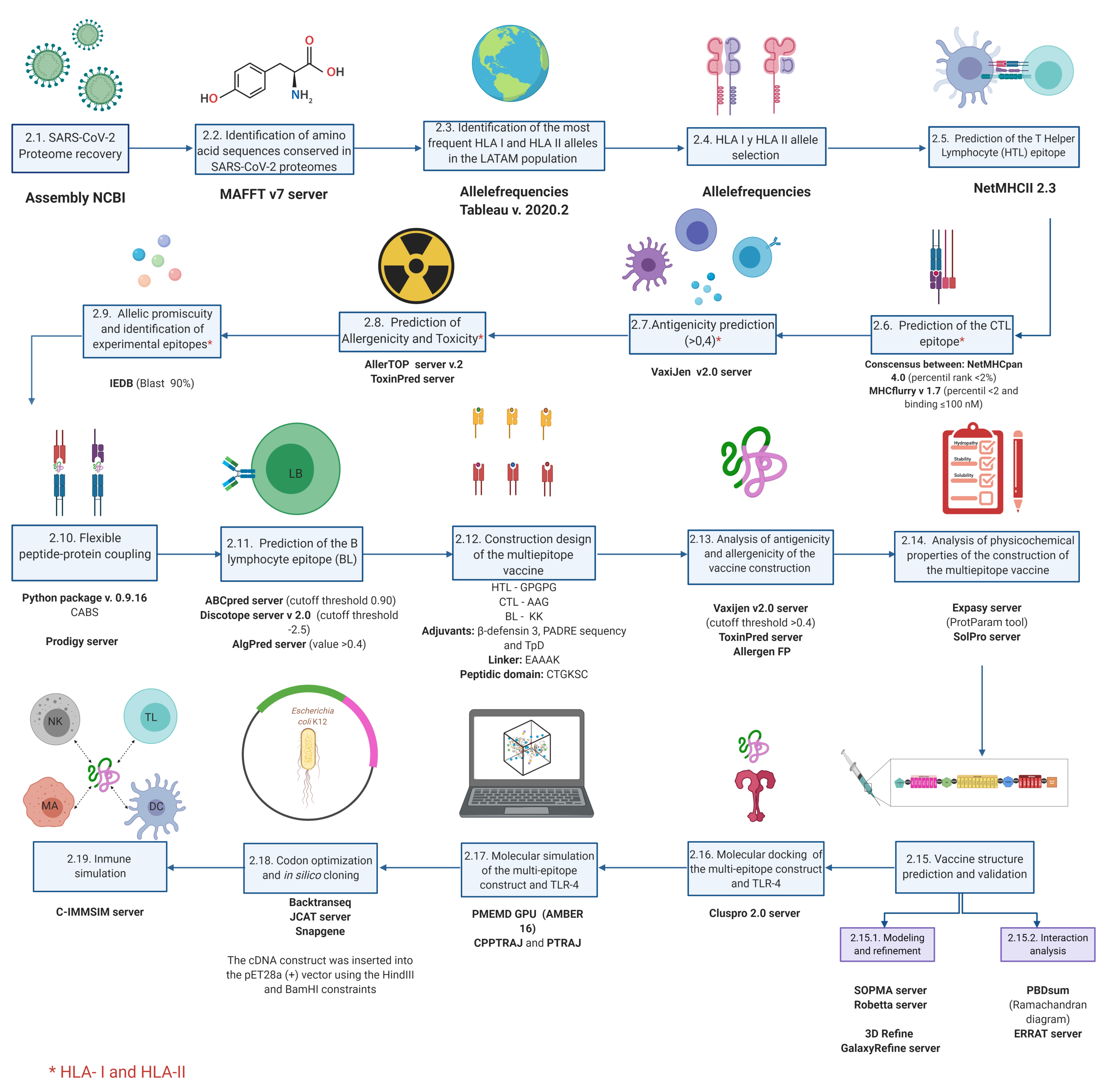
2. Materials and Methods
2.1. SARS-CoV-2 Proteome Recovery
2.2. Identification of Amino Acid Sequences Conserved in SARS-CoV-2 Proteomes
2.3. Identification of the Most Frequent HLA-I and HLA-II Alleles in LATAM Population
2.4. HLA-I and HLA-II Allele Selection
2.5. Prediction of T Helper Lymphocytes (HTL) Epitope
2.6. Prediction of the CTL Epitope
2.7. Antigenicity Prediction
2.8. Prediction of Allergenicity and Toxicity
2.9. Allelic Promiscuity and Identification of Experimental Epitopes
2.10. Flexible Peptide-Protein Docking
2.11. Prediction of the BL Epitope
2.12. Recuperation of Validated Epitopes and Identification of Post-Transcriptional Modifications
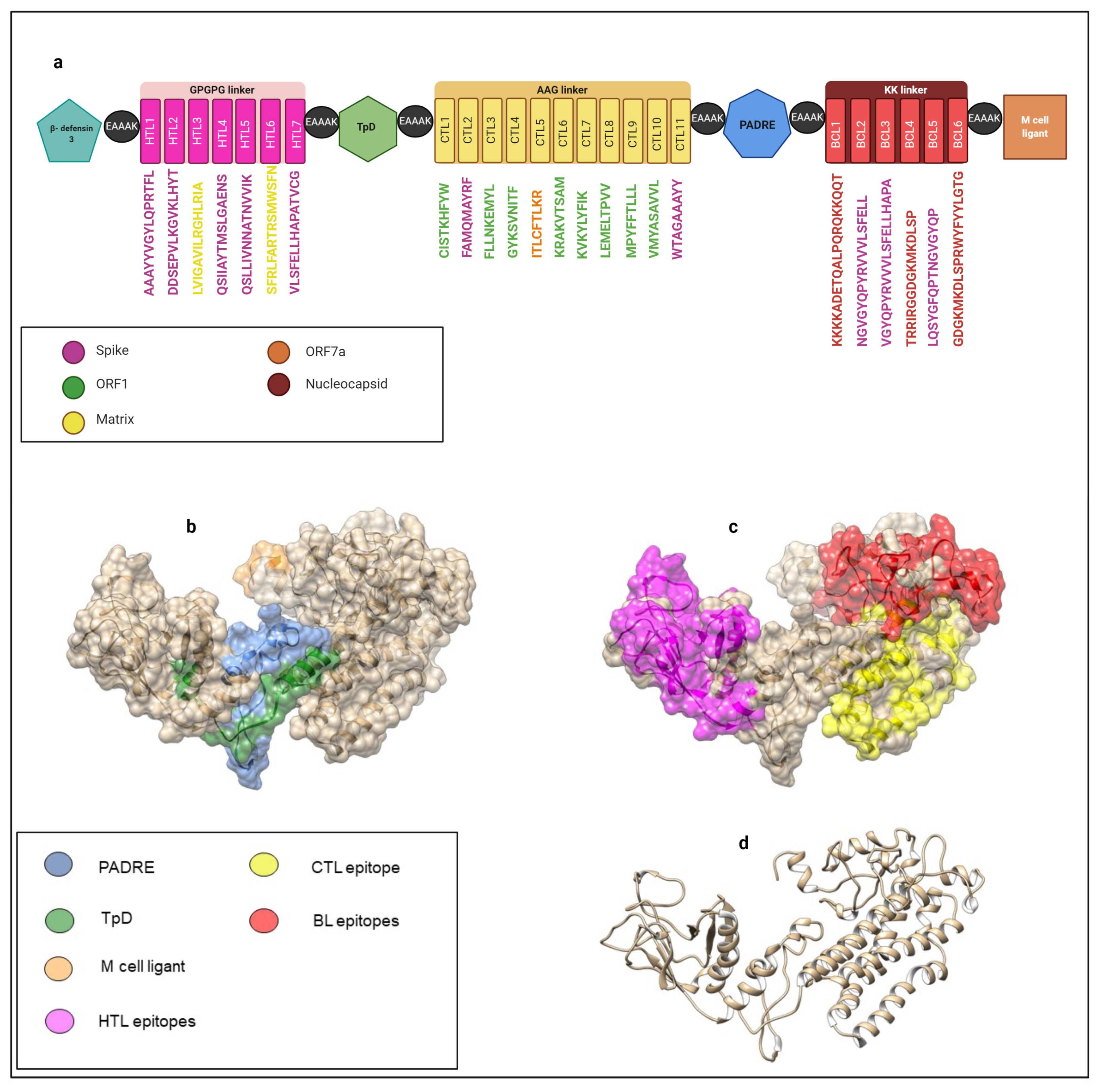
2.13. Multi-Epitope Vaccine Construct Design
2.14. Analysis of Antigenicity and Allergenicity of the Vaccine Construct
2.15. Analysis of the Physicochemical Properties of the Multi-Epitope Vaccine Construct
2.16. Vaccine Structure Prediction and Validation
2.17. Molecular Docking of the Multi-Epitope Construct and TLR-4
2.18. Molecular Dynamics Simulation of the Multi-Epitope Construct and TLR-4 Complex
2.19. Codon Optimization and In Silico Cloning
2.20. Immune Simulation
3. Results
3.1. Recovery of SARS-CoV-2 Proteomes
3.2. A Vaccine for SARS-CoV-2 Must Take into Account Non-Structural Proteins
3.3. Most Frequently Identified HLA-I and HLA-II Alleles in LATAM
3.4. Potential Epitopes of TLs, CD4+, CD8+, and BLs Are Contained in the Conserved Sequences of 92 SARS-CoV-2 Proteomes
3.4.1. HLA-II
3.4.2. HLA-I
3.4.3. BLs
3.5. SARS-CoV-2 MG and SP Contain Conserved Sequences Capable of Interacting with a Large Number of HLA-II Molecules
3.6. Most Frequent HLA-1 in LATAM and Recognition of Peptides of Conserved SARS-CoV-2 Regions
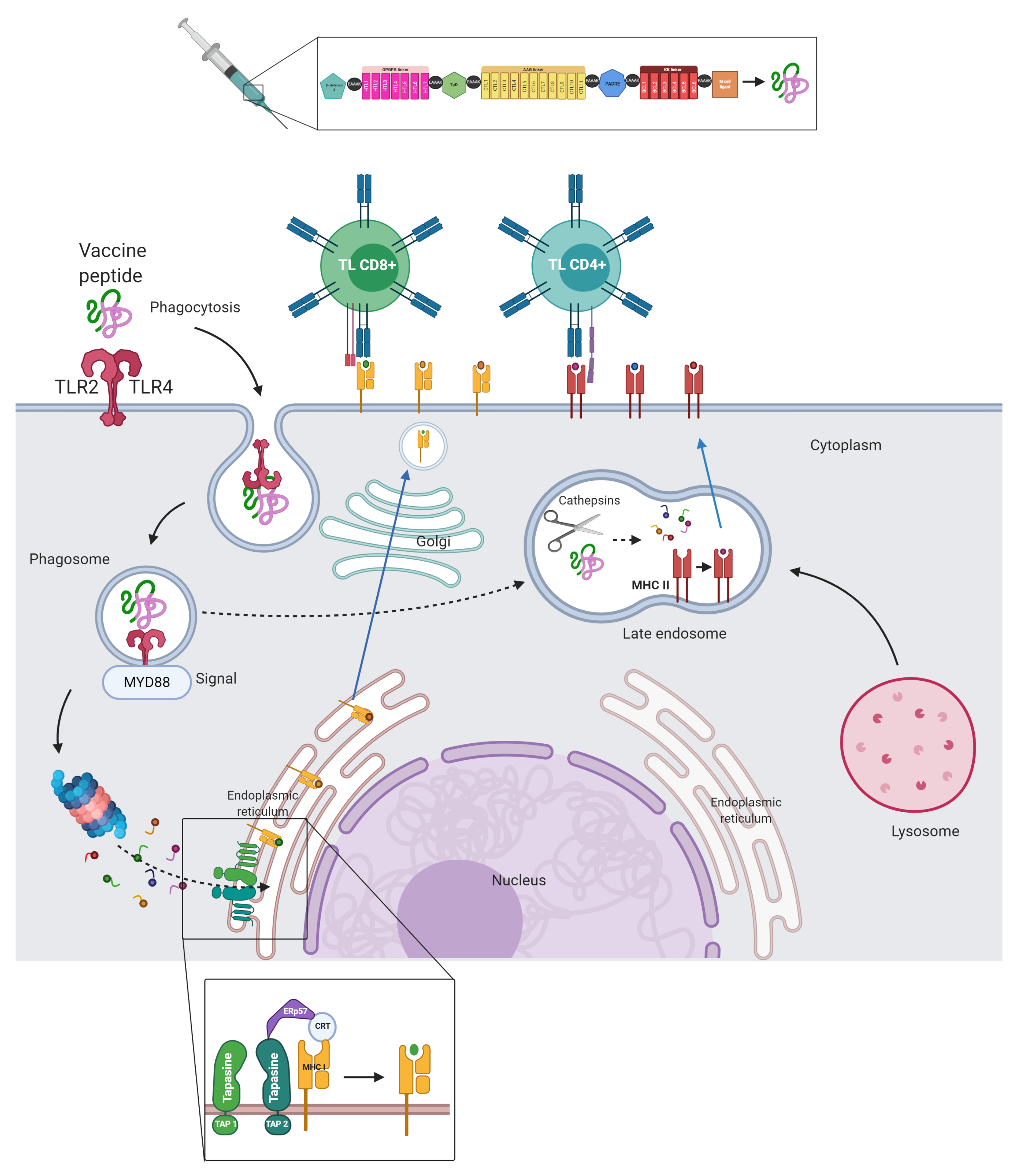
3.7. Vaccine Candidates, Post-Translational Modifications, and Experimentally Validated Peptides
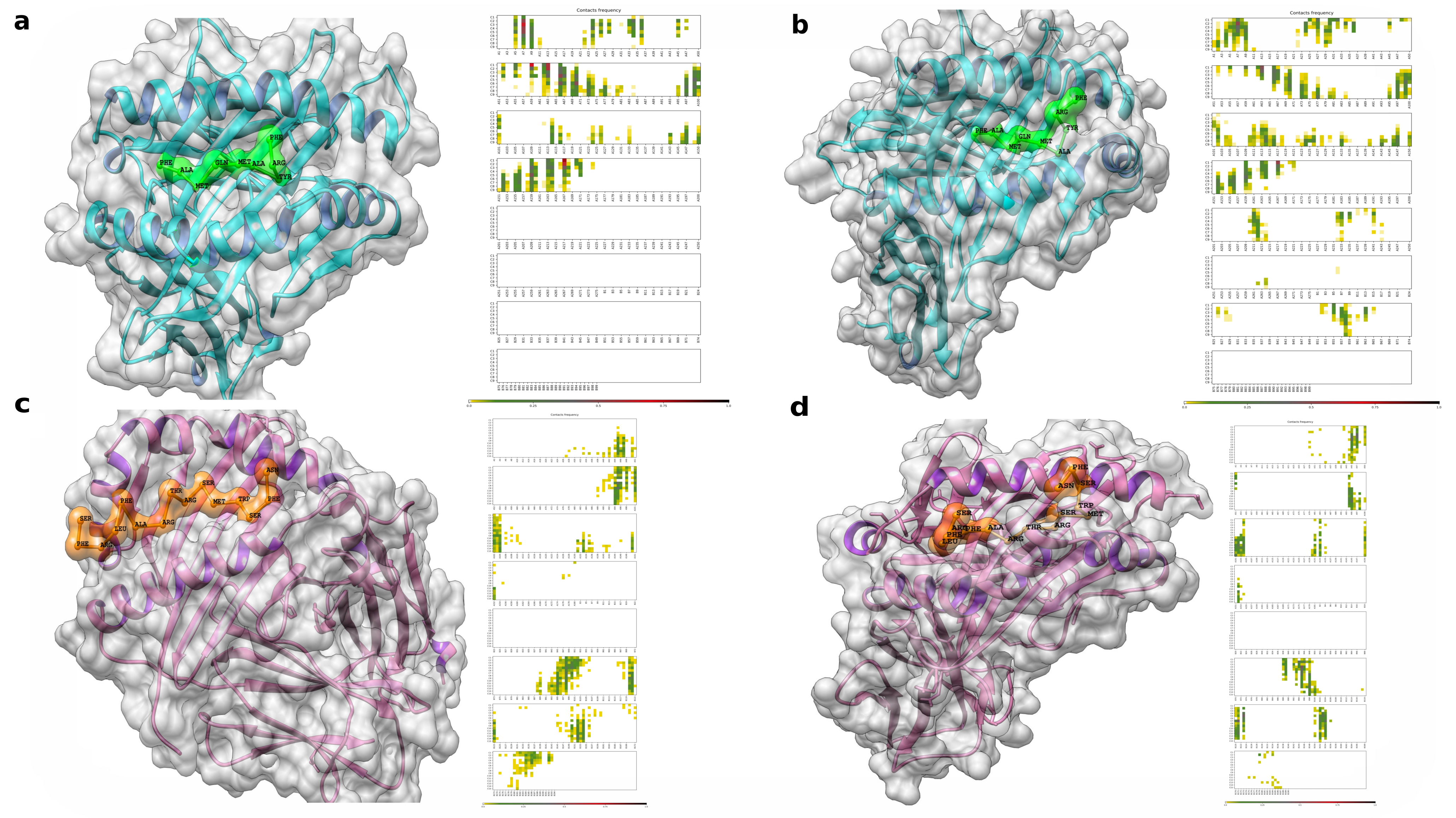
3.8. Flexible Peptide-Protein Coupling Signals Favourable Energy and Anchorage Residues to HLA Molecules
3.9. Molecular Docking, Interactions with Heterodimer TLR-4/MD-2
3.10. The Physicochemical Properties of the Multi-Epitope Construct Are Consistent with the Requirements for Generating an Immune Response in an Experimental Model
3.11. Three-Dimensional Structure Modeling and Validation of the Multi-Epitope Vaccine Construct
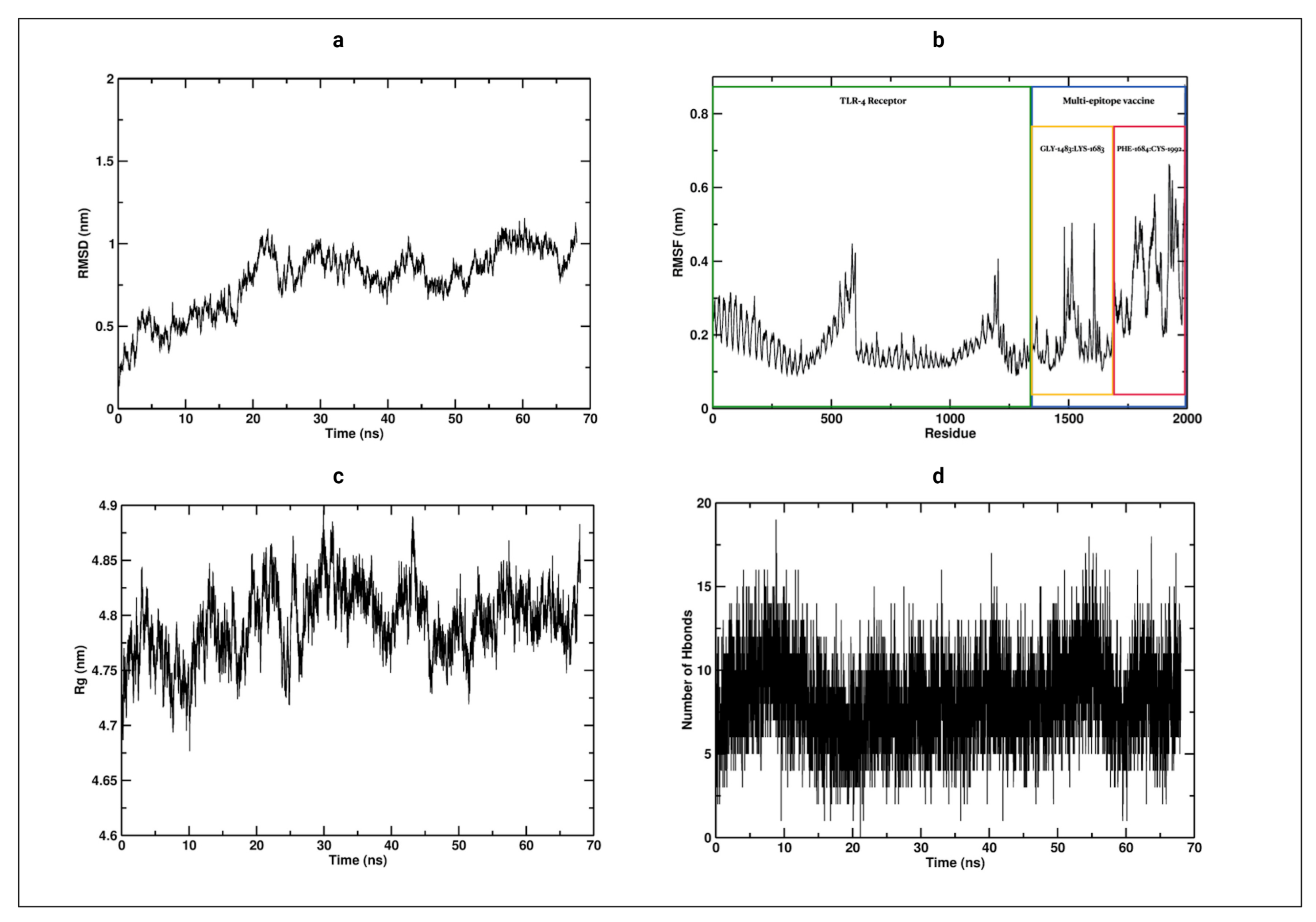
3.12. Molecular Dynamics
3.13. Optimization of cDNA from the Vaccine Construct for Optimal Expression of the Vaccine Product
3.14. Expression of the Multi-Epitope Vaccine Construct in E. coli K 12 by In Silico Cloning
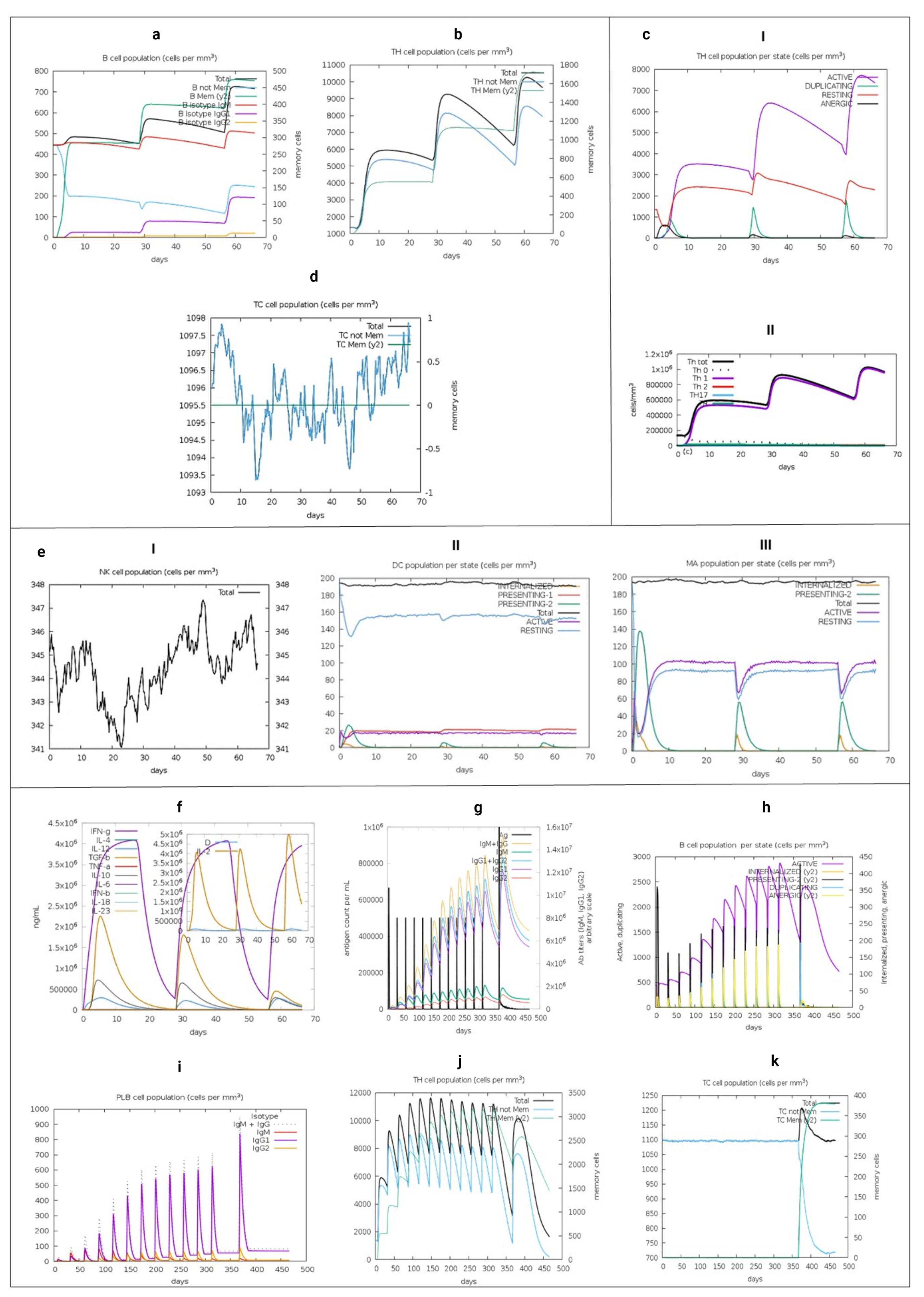
3.15. Immune System Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar] [PubMed]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, S.P.; Meng, S.; Wu, Y.J.; Mao, Y.P.; Ye, R.X.; Wang, Q.Z.; Sun, C.; Sylvia, S.; Rozelle, S.; Raat, H.; et al. Epidemiology, causes, clinical manifestation and diagnosis, prevention and control of coronavirus disease (COVID-19) during the early outbreak period: A scoping review. Infect. Dis. Poverty 2020, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Morales, A.J.; Gallego, V.; Escalera-Antezana, J.P.; Méndez, C.A.; Zambrano, L.I.; Franco-Paredes, C.; Suárez, J.A.; Rodriguez-Enciso, H.D.; Balbin-Ramon, G.J.; Savio-Larriera, E.; et al. COVID-19 in Latin America: The implications of the first confirmed case in Brazil. Travel Med. Infect. Dis. 2020, 35, 101613. [Google Scholar] [CrossRef]
- Rodriguez-Morales, A.J.; Rodriguez-Morales, A.G.; Méndez, C.A.; Hernández-Botero, S. Tracing new clinical manifestations in patients with Covid-19 in chile and its potential relationship with the SARS-CoV-2 divergence. Curr. Trop. Med. Rep. 2020, 1–4. [Google Scholar] [CrossRef]
- Millán-Oñate, J.; Millan, W.; Mendoza, L.A.; Sánchez, C.G.; Fernandez-Suarez, H.; Bonilla-Aldana, D.K.; Rodríguez-Morales, A.J. Successful recovery of COVID-19 pneumonia in a patient from Colombia after receiving chloroquine and clarithromycin. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 16. [Google Scholar] [CrossRef]
- The Lancet. The unfolding migrant crisis in Latin America. Lancet 2019, 394, 1966. [Google Scholar]
- Mellan, T.A.; Hoeltgebaum, H.H.; Mishra, S.; Whittaker, C.; Schnekenberg, R.P.; Gandy, A.; Unwin, H.J.T.; Vollmer, M.A.; Coupland, H.; Hawryluk, I.; et al. Report 21: Estimating COVID-19 cases and reproduction number in Brazil. medRxiv 2020. [Google Scholar] [CrossRef]
- Ochoa, Y.C.; Sanchez, D.E.R.; Peñaloza, M.; Motta, H.F.C.; Méndez-Fandiño, Y.R. Effective Reproductive Number estimation for initial stage of COVID-19 pandemic in Latin American Countries. Int. J. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Ong, E.; Wong, M.U.; Huffman, A.; He, Y. COVID-19 coronavirus vaccine design using reverse vaccinology and machine learning. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Renu, K.; Prasanna, P.L.; Gopalakrishnan, A.V. Coronaviruses pathogenesis, comorbidities and multi-organ damage—A review. Life Sci. 2020, 117839. [Google Scholar] [CrossRef]
- Sánchez-Duque, J.; Arce-Villalobos, L.; Rodríguez-Morales, A. Enfermedad por coronavirus 2019 (COVID-19) en América Latina: Papel de la atención primaria en la preparación y respuesta. Aten. Primaria Soc. Esp. Med. Fam. Comunitaria 2020, 52, 369–372. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Wang, B.; Li, R.; Lu, Z.; Huang, Y. Does comorbidity increase the risk of patients with COVID-19: Evidence from meta-analysis. Aging 2020, 12, 6049. [Google Scholar] [CrossRef]
- Totura, A.L.; Baric, R.S. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef]
- Chan, V.S.F.; Chan, K.Y.K.; Chen, Y.; Poon, L.L.M.; Cheung, A.N.Y.; Zheng, B.; Chan, K.H.; Mak, W.; Ngan, H.Y.S.; Xu, X.; et al. Homozygous L-SIGN (CLEC4M) plays a protective role in SARS coronavirus infection. Nat. Genet. 2006, 38, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.F.; Chen, K.H.; Chen, M.; Li, W.Y.; Chen, Y.J.; Tsao, C.H.; Yen, M.y.; Huang, J.C.; Chen, Y.M.A. Human-leukocyte antigen class I Cw 1502 and class II DR 0301 genotypes are associated with resistance to severe acute respiratory syndrome (SARS) infection. Viral Immunol. 2011, 24, 421–426. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, K.; Huang, C.; Makino, S. SARS coronavirus accessory proteins. Virus Res. 2008, 133, 113–121. [Google Scholar] [CrossRef] [PubMed]
- The RECOVERY Collaborative Group. Dexamethasone in hospitalized patients with Covid-19—Preliminary report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of Covid-19. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Consortium, W.S.T. Repurposed antiviral drugs for COVID-19—Interim WHO SOLIDARITY trial results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Temporary Policy for Compounding of Certain Drugs for Hospitalized Patients by Outsourcing Facilities during the COVID-19 Public Health Emergency: Guidance for Industry; Food and Drug Administration: Silver Spring, MD, USA, 2020. [Google Scholar]
- Li, L.; Zhang, W.; Hu, Y.; Tong, X.; Zheng, S.; Yang, J.; Kong, Y.; Ren, L.; Wei, Q.; Mei, H.; et al. Effect of Convalescent Plasma Therapy on Time to Clinical Improvement in Patients With Severe and Life-threatening COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 460–470. [Google Scholar] [CrossRef]
- Agarwal, A.; Mukherjee, A.; Kumar, G.; Chatterjee, P.; Bhatnagar, T.; Malhotra, P. Convalescent plasma in the management of moderate covid-19 in adults in India: Open label phase II multicentre randomised controlled trial (PLACID Trial). BMJ 2020, 371, m4232. [Google Scholar] [CrossRef]
- Italian Medicine Agency. COVID-19: Tsunami Study, Plasma Does Not Reduce the Risk of Respiratory Damage or Death; Italian Medicine Agency: Rome, Italy, 2020. [Google Scholar]
- Landi, L.; Ravaglia, C.; Russo, E.; Cataleta, P.; Fusari, M.; Boschi, A.; Giannarelli, D.; Facondini, F.; Valentini, I.; Panzini, I.; et al. Blockage of interleukin-1β with canakinumab in patients with Covid-19. Sci. Rep. 2020, 10, 21775. [Google Scholar] [CrossRef]
- Lescure, F.X.; Honda, H.; Fowler, R.A.; Lazar, J.S.; Shi, G.; Wung, P.; Patel, N.; Hagino, O.; Bazzalo, I.J.; Casas, M.M.; et al. Sarilumab in patients admitted to hospital with severe or critical COVID-19: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Rilinger, J.; Kern, W.V.; Duerschmied, D.; Supady, A.; Bode, C.; Staudacher, D.L.; Wengenmayer, T. A prospective, randomised, double blind placebo-controlled trial to evaluate the efficacy and safety of tocilizumab in patients with severe COVID-19 pneumonia (TOC-COVID): A structured summary of a study protocol for a randomised controlled trial. Trials 2020, 21, 470. [Google Scholar] [CrossRef]
- Meng, F.; Xu, R.; Wang, S.; Xu, Z.; Zhang, C.; Li, Y.; Yang, T.; Shi, L.; Fu, J.; Jiang, T.; et al. Human umbilical cord-derived mesenchymal stem cell therapy in patients with COVID-19: A phase 1 clinical trial. Signal Transduct. Target. Ther. 2020, 5, 172. [Google Scholar] [CrossRef]
- Ahn, D.G.; Shin, H.J.; Kim, M.H.; Lee, S.; Kim, H.S.; Myoung, J.; Kim, B.T.; Kim, S.J. Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID-19). J. Microbiol. Biotechnol. 2020, 30, 313–324. [Google Scholar] [CrossRef]
- World Health Organization. DRAFT Landscape of COVID-19 Candidate Vaccines; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Dhama, K.; Sharun, K.; Tiwari, R.; Dadar, M.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W. COVID-19, an emerging coronavirus infection: Advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Hum. Vaccines Immunother. 2020, 16, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Le, T.T.; Andreadakis, Z.; Kumar, A.; Roman, R.G.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef]
- Ng, O.W.; Chia, A.; Tan, A.T.; Jadi, R.S.; Leong, H.N.; Bertoletti, A.; Tan, Y.J. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine 2016, 34, 2008–2014. [Google Scholar] [CrossRef]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. MedRxiv 2020. [Google Scholar] [CrossRef]
- Beltrame, M.H.; Boldt, A.B.; Catarino, S.J.; Mendes, H.C.; Boschmann, S.E.; Goeldner, I.; Messias-Reason, I. MBL-associated serine proteases (MASPs) and infectious diseases. Mol. Immunol. 2015, 67, 85–100. [Google Scholar] [CrossRef]
- Yasui, F.; Kai, C.; Kitabatake, M.; Inoue, S.; Yoneda, M.; Yokochi, S.; Kase, R.; Sekiguchi, S.; Morita, K.; Hishima, T.; et al. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J. Immunol. 2008, 181, 6337–6348. [Google Scholar] [CrossRef] [Green Version]
- Tilocca, B.; Soggiu, A.; Sanguinetti, M.; Musella, V.; Britti, D.; Bonizzi, L.; Urbani, A.; Roncada, P. Comparative computational analysis of SARS-CoV-2 nucleocapsid protein epitopes in taxonomically related coronaviruses. Microbes Infect. 2020, 22, 188–194. [Google Scholar] [CrossRef]
- Wang, J.; Wen, J.; Li, J.; Yin, J.; Zhu, Q.; Wang, H.; Yang, Y.; Qin, E.; You, B.; Li, W.; et al. Assessment of immunoreactive synthetic peptides from the structural proteins of severe acute respiratory syndrome coronavirus. Clin. Chem. 2003, 49, 1989–1996. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Xu, Y.; Li, L.; Weng, L.; Wang, Q.; Zhang, S.; Jia, B.; Hu, H.; He, Y.; Jacob, Y.; et al. Inhibition of influenza virus replication by constrained peptides targeting nucleoprotein. Antivir. Chem. Chemother. 2011, 22, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Li, Z.; Yang, Y.; Ma, G.; Su, Z.; Zhang, S. An Apoferritin–Hemagglutinin Conjugate Vaccine with Encapsulated Nucleoprotein Antigen Peptide from Influenza Virus Confers Enhanced Cross Protection. Bioconjugate Chem. 2020, 31, 1948–1959. [Google Scholar] [CrossRef]
- Oliveira, S.C.; de Magalhães, M.T.; Homan, E.J. Immunoinformatic Analysis of SARS-CoV-2 Nucleocapsid Protein and Identification of COVID-19 Vaccine Targets. Front. Immunol. 2020, 11, 2758. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.-F.; Du, R.-L.; Liu, J.-Z.; Li, C.; Zhang, Q.-F.; Han, L.-L.; Yu, J.-S.; Duan, S.-M.; Wang, X.-F.; Wu, K.-X.; et al. SARS patients-derived human recombinant antibodies to S and M proteins efficiently neutralize SARS-coronavirus infectivity. Biomed. Environ. Sci. 2005, 18, 363. [Google Scholar] [PubMed]
- Iyer, A.S.; Jones, F.K.; Nodoushani, A.; Kelly, M.; Becker, M.; Slater, D.; Mills, R.; Teng, E.; Kamruzzaman, M.; Garcia-Beltran, W.F.; et al. Persistence and decay of human antibody responses to the receptor binding domain of SARS-CoV-2 spike protein in COVID-19 patients. Sci. Immunol. 2020, 5, eabe0367. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Ye, F.; Cheng, M.L.; Feng, Y.; Deng, Y.Q.; Zhao, H.; Wei, P.; Ge, J.; Gou, M.; Li, X.; et al. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity 2020, 52, 971–977.e3. [Google Scholar] [CrossRef]
- Buchholz, U.J.; Bukreyev, A.; Yang, L.; Lamirande, E.W.; Murphy, B.R.; Subbarao, K.; Collins, P.L. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc. Natl. Acad. Sci. USA 2004, 101, 9804–9809. [Google Scholar] [CrossRef] [Green Version]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef]
- Sariol, A.; Perlman, S. Lessons for COVID-19 immunity from other coronavirus infections. Immunity 2020, 53, 248–263. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Tableu. Academic Programs | Tableau Software. 2020. Available online: https://www.tableau.com/community/academic (accessed on 27 March 2021).
- Robinson, J.; Halliwell, J.A.; Hayhurst, J.D.; Flicek, P.; Parham, P.; Marsh, S.G.E. The IPD and IMGT/HLA database: Allele variant databases. Nucleic Acids Res. 2015, 43, D423–D431. [Google Scholar] [CrossRef] [Green Version]
- Manczinger, M.; Boross, G.; Kemény, L.; Müller, V.; Lenz, T.L.; Papp, B.; Pál, C. Pathogen diversity drives the evolution of generalist MHC-II alleles in human populations. PLoS Biol. 2019, 17, e3000131. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, A.; Altfeld, M. Regulation of NK-cell function by HLA class II. Front. Cell. Infect. Microbiol. 2020, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-source class I MHC binding affinity prediction. Cell Syst. 2018, 7, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Berzofsky, J.A. Intrinsic and Extrinsic Factors in Protein Antigenic Structure. Science 1985, 229, 932–940. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery Consortium; Raghava, G.P.S. In Silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Gao, G.F. Major histocompatibility complex: Interaction with peptides. eLS 2011. [Google Scholar] [CrossRef]
- Kurcinski, M.; Pawel Ciemny, M.; Oleniecki, T.; Kuriata, A.; Badaczewska-Dawid, A.E.; Kolinski, A.; Kmiecik, S. CABS- dock standalone: A toolbox for flexible protein-peptide docking. Bioinformatics 2019, 35, 4170–4172. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Kringelum, J.V.; Lundegaard, C.; Lund, O.; Nielsen, M. Reliable B cell epitope predictions: Impacts of method development and improved benchmarking. PLoS Comput. Biol. 2012, 8, e1002829. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P.S. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Jung, E.; Brunak, S. Prediction of N-glycosylation sites in human proteins. Preparation 2004, 46, 203–206. [Google Scholar]
- Fievez, V.; Plapied, L.; Plaideau, C.; Legendre, D.; des Rieux, A.; Pourcelle, V.; Freichels, H.; Jérôme, C.; Marchand, J.; Préat, V.; et al. In Vitro identification of targeting ligands of human M cells by phage display. Int. J. Pharm. 2010, 394, 35–42. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- Walker, J.M. The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Solanki, V.; Tiwari, M.; Tiwari, V. Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 5240. [Google Scholar] [CrossRef]
- Geourjon, C.; Deleage, G. SOPM: A self-optimized method for protein secondary structure prediction. Protein Eng. Des. Sel. 1994, 7, 157–164. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; DiMaio, F.; Wang, R.Y.R.; Kim, D.; Miles, C.; Brunette, T.J.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AmberTools 16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; Fabritiis, G.D. ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Kanduc, D.; Shoenfeld, Y. On the molecular determinants the SARS-CoV-2 attack. Clin. Immunol. 2020, 215, 108426. [Google Scholar] [CrossRef]
- Olejnik, J.; Hume, A.J.; Mühlberger, E. Toll-like receptor 4 in acute viral infection: Too much of a good thing. PLoS Pathog. 2018, 14, e1007390. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Usera, F.; Enjuanes, L. Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res. 2014, 194. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef]
- Neuman, B.W.; Joseph, J.S.; Saikatendu, K.S.; Serrano, P.; Chatterjee, A.; Johnson, M.A.; Liao, L.; Klaus, J.P.; Yates, J.R.; Wüthrich, K.; et al. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 2008, 82, 5279–5294. [Google Scholar] [CrossRef] [Green Version]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [Green Version]
- Blander, J.M. Coupling Toll-like receptor signaling with phagocytosis: Potentiation of antigen presentation. Trends Immunol. 2007, 28, 19–25. [Google Scholar] [CrossRef]
- Hegde, N.R.; Chevalier, M.S.; Johnson, D.C. Viral inhibition of MHC class II antigen presentation. Trends Immunol. 2003, 24, 278–285. [Google Scholar] [CrossRef]
- Vina, M.A.F.; Hollenbach, J.A.; Lyke, K.E.; Sztein, M.B.; Maiers, M.; Klitz, W.; Cano, P.; Mack, S.; Single, R.; Brautbar, C.; et al. Tracking human migrations by the analysis of the distribution of HLA alleles, lineages and haplotypes in closed and open populations. Philos. Trans. R. Soc. Biol. Sci. 2012, 367, 820–829. [Google Scholar] [CrossRef]
- Iturrieta-Zuazo, I.; Rita, C.G.; García-Soidán, A.; de Malet Pintos-Fonseca, A.; Alonso-Alarcón, N.; Pariente-Rodríguez, R.; Tejeda-Velarde, A.; Serrano-Villar, S.; Castañer-Alabau, J.L.; Nieto-Gañán, I. Possible role of HLA class-I genotype in SARS-CoV-2 infection and progression: A pilot study in a cohort of Covid-19 Spanish patients. Clin. Immunol. 2020, 219, 108572. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Deelen, J.; Gallina, A.M.; Caputo, M.; Citro, M.; Abate, M.; Sacchi, N.; Vecchione, C.; Martinelli, R. Correlation of the two most frequent HLA haplotypes in the Italian population to the differential regional incidence of Covid-19. J. Transl. Med. 2020, 18, 352. [Google Scholar] [CrossRef] [PubMed]
- Addetia, A.; Crawford, K.H.; Dingens, A.; Zhu, H.; Roychoudhury, P.; Huang, M.; Jerome, K.R.; Bloom, J.D.; Greninger, A. Neutralizing antibodies correlate with protection from SARS-CoV-2 in humans during a fishery vessel outbreak with high attack rate. medRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Ter Meulen, J.; Van Den Brink, E.N.; Poon, L.L.M.; Marissen, W.E.; Leung, C.S.W.; Cox, F.; Cheung, C.Y.; Bakker, A.Q.; Bogaards, J.A.; Van Deventer, E.; et al. Human monoclonal antibody combination against SARS coronavirus: Synergy and coverage of escape mutants. PLoS Med. 2006, 3, e237. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Wolfert, M.A.; Boons, G.J. Adaptive immune activation: Glycosylation does matter. Nat. Chem. Biol. 2013, 9, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, T.; Fitieh, A.; St Pierre, J.; Ostergaard, H.L.; Bundle, D.R.; Touret, N. Enhanced immunogenicity of a tricomponent mannan tetanus toxoid conjugate vaccine targeted to dendritic cells via Dectin-1 by incorporating β-glucan. J. Immunol. 2013, 190, 4116–4128. [Google Scholar] [CrossRef] [Green Version]
- Hanisch, F.G.; Ninkovic, T. Immunology of O-glycosylated proteins: Approaches to the design of a MUC1 glycopeptide-based tumor vaccine. Curr. Protein Pept. Sci. 2006, 7, 307–315. [Google Scholar] [CrossRef]
- Roulois, D.; Grégoire, M.; Fonteneau, J.F. MUC1-specific cytotoxic T lymphocytes in cancer therapy: Induction and challenge. BioMed Res. Int. 2013, 2013, 871936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, M.D.; Streib, J.E.; Leung, D.Y.M. Antiviral activity of human beta-defensin 3 against vaccinia virus. J. Allergy Clin. Immunol. 2007, 119, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, C.C.; Altreuter, D.H.; Ilyinskii, P.; Pittet, L.; LaMothe, R.A.; Keegan, M.; Johnston, L.; Kishimoto, T.K. Generation of a universal CD4 memory T cell recall peptide effective in humans, mice and non-human primates. Vaccine 2014, 32, 2896–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fast, E.; Chen, B. Potential T-cell and B-cell Epitopes of 2019-nCoV. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Ip, P.P.; Boerma, A.; Regts, J.; Meijerhof, T.; Wilschut, J.; Nijman, H.W.; Daemen, T. Alphavirus-based vaccines encoding nonstructural proteins of hepatitis C virus induce robust and protective T-cell responses. Mol. Ther. 2014, 22, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Dorosti, H.; Eslami, M.; Negahdaripour, M.; Ghoshoon, M.B.; Gholami, A.; Heidari, R.; Dehshahri, A.; Erfani, N.; Nezafat, N.; Ghasemi, Y. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. [Google Scholar] [CrossRef]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Idicula-Thomas, S.; Balaji, P.V. Understanding the relationship between the primary structure of proteins and its propensity to be soluble on overexpression in Escherichia coli. Protein Sci. 2005, 14, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the C lus P ro server motivated by CAPRI. Proteins Struct. Funct. Bioinform. 2017, 85, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Tamalika, K.; Utkarsh, N.; Srijita, B.; Debashrito, D.; Filippo, C.; Mueller, D.M.; Srivastava, A.P. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 10895. [Google Scholar]
- Enayatkhani, M.; Hasaniazad, M.; Faezi, S.; Guklani, H.; Davoodian, P.; Ahmadi, N.; Einakian, M.A.; Karmostaji, A.; Ahmadi, K. Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: An in silico study. J. Biomol. Struct. Dyn. 2020, 39, 2857–2872. [Google Scholar] [CrossRef] [Green Version]
- Almofti, Y.A.; Abd-elrahman, K.A.; Gassmallah, S.A.E.; Salih, M.A. Multi Epitopes Vaccine Prediction against Severe Acute Respiratory Syndrome (SARS) Coronavirus Using Immunoinformatics Approaches. Am. J. Microbiol. Res. 2018, 6, 94–114. [Google Scholar] [CrossRef] [Green Version]
- Rakib, A.; Sami, S.A.; Mimi, N.J.; Chowdhury, M.M.; Eva, T.A.; Nainu, F.; Paul, A.; Shahriar, A.; Tareq, A.M.; Emon, N.U.; et al. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Comput. Biol. Med. 2020, 103967. [Google Scholar] [CrossRef] [PubMed]
- Abraham Peele, K.; Srihansa, T.; Krupanidhi, S.; Ayyagari, V.S.; Venkateswarulu, T.C. Design of multi-epitope vaccine candidate against SARS-CoV-2: A In-Silico study. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Su, Q.d.; Yi, Y.; Zou, Y.n.; Jia, Z.y.; Qiu, F.; Wang, F.; Yin, W.j.; Zhou, W.t.; Zhang, S.; Yu, P.c.; et al. The biological characteristics of SARS-CoV-2 Spike protein Pro330-Leu650. Vaccine 2020, 38, 5071–5075. [Google Scholar] [CrossRef]
- Mitra, D.; Shekhar, N.; Pandey, J.; Jain, A.; Swaroop, S. Multi-epitope based peptide vaccine design against SARS-CoV-2 using its spike protein. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Joshi, B.C.; Mannan, M.A.u.; Kaushik, V. Epitope based vaccine prediction for SARS-COV-2 by deploying immuno-informatics approach. Inform. Med. Unlocked 2020, 19, 100338. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.W.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- Wang, Y.D.; Sin, W.Y.F.; Xu, G.B.; Yang, H.H.; Wong, T.y.; Pang, X.W.; He, X.Y.; Zhang, H.G.; Ng, J.N.L.; Cheng, C.S.S.; et al. T-cell epitopes in severe acute respiratory syndrome (SARS) coronavirus spike protein elicit a specific T-cell immune response in patients who recover from SARS. J. Virol. 2004, 78, 5612–5618. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.C.; Burgers, W.A. SARS-CoV-2 evolution and vaccines: Cause for concern? Lancet Respir. Med. 2021, 9, 333–335. [Google Scholar] [CrossRef]
- Chakradhar, S. Updated, augmented vaccines compete with original antigenic sin. Nat. Med. 2015, 21, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Tavasolian, F.; Rashidi, M.; Hatam, G.R.; Jeddi, M.; Hosseini, A.Z.; Mosawi, S.H.; Abdollahi, E.; Inman, R.D. HLA, Immune Response, and Susceptibility to COVID-19. Front. Immunol. 2021, 11, 3581. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G.W. Characterizing SARS-CoV-2 mutations in the United States. arXiv 2020, arXiv:2007.12692. [Google Scholar]
- Elizondo, V.; Harkins, G.W.; Mabvakure, B.; Smidt, S.; Zappile, P.; Marier, C.; Maurano, M.; Perez, V.; Mazza, N.; Beloso, C.; et al. SARS-CoV-2 genomic characterization and clinical manifestation of the COVID-19 outbreak in Uruguay. medRxiv 2020, 10, 51–65. [Google Scholar]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, M.; Cong, H. Advances in the study of HLA-restricted epitope vaccines. Hum. Vaccines Immunother. 2013, 9, 2566–2577. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Guo, T.; Yu, Q.; Zhang, H.; Du, J.; Zhang, Y.; Xia, S.; Yang, H.; Li, Q. Association of human leukocyte antigen alleles and supertypes with immunogenicity of oral rotavirus vaccine given to infants in China. Medicine 2018, 97, e12706. [Google Scholar] [CrossRef]
- Wu, C.Y.; Monie, A.; Pang, X.; Hung, C.F.; Wu, T.C. Improving therapeutic HPV peptide-based vaccine potency by enhancing CD4+ T help and dendritic cell activation. J. Biomed. Sci. 2010, 17, 88. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Su, X.; Zhang, P.; Liang, J.; Wei, H.; Wan, M.; Wu, X.; Yu, Y.; Wang, L. Recombinant heat shock protein 65 carrying PADRE and HBV epitopes activates dendritic cells and elicits HBV-specific CTL responses. Vaccine 2011, 29, 2328–2335. [Google Scholar] [CrossRef]
- Funderburg, N.; Lederman, M.M.; Feng, Z.; Drage, M.G.; Jadlowsky, J.; Harding, C.V.; Weinberg, A.; Sieg, S.F. Human β-defensin-3 activates professional antigen-presenting cells via Toll-like receptors 1 and 2. Proc. Natl. Acad. Sci. USA 2007, 104, 18631–18635. [Google Scholar] [CrossRef] [Green Version]
- Ferris, L.K.; Mburu, Y.K.; Mathers, A.R.; Fluharty, E.R.; Larregina, A.T.; Ferris, R.L.; Falo, L.D., Jr. Human beta-defensin 3 induces maturation of human Langerhans cell-like dendritic cells: An antimicrobial peptide that functions as an endogenous adjuvant. J. Investig. Dermatol. 2013, 133, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie-Dyck, S.; Kovacs-Nolan, J.; Snider, M.; Babiuk, L.A.; van Drunnen Littel-van den Hurk, S. Inclusion of the bovine neutrophil beta-defensin 3 with glycoprotein D of bovine herpesvirus 1 in a DNA vaccine modulates immune responses of mice and cattle. Clin. Vaccine Immunol. 2014, 21, 463–477. [Google Scholar] [CrossRef] [Green Version]
- Pridgen, E.M.; Alexis, F.; Farokhzad, O.C. Polymeric nanoparticle drug delivery technologies for oral delivery applications. Expert Opin. Drug Deliv. 2015, 12, 1459–1473. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Gao, Z.; Zhang, Z.; Pan, L.; Zhang, Y. Roles of M cells in infection and mucosal vaccines. Hum. Vaccines Immunother. 2014, 10, 3544–3551. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Pandey, R.K.; Khatoon, N.; Narula, A.; Mishra, A.; Prajapati, V.K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 9232. [Google Scholar] [CrossRef]
- Kamthania, M.; Srivastava, S.; Desai, M.; Jain, A.; Shrivastav, A.; Sharma, D. Immunoinformatics Approach to Design T-cell Epitope-Based Vaccine Against Hendra Virus. Int. J. Pept. Res. Ther. 2019, 25, 1627–1637. [Google Scholar] [CrossRef]
- Ojha, R.; Pareek, A.; Pandey, R.K.; Prusty, D.; Prajapati, V.K. Strategic development of a next-generation multi-epitope vaccine to prevent Nipah virus zoonotic infection. ACS Omega 2019, 4, 13069–13079. [Google Scholar] [CrossRef] [Green Version]
- Dar, H.A.; Zaheer, T.; Shehroz, M.; Ullah, N.; Naz, K.; Muhammad, S.A.; Zhang, T.; Ali, A. Immunoinformatics-Aided Design and Evaluation of a Potential Multi-Epitope Vaccine against Klebsiella Pneumoniae. Vaccines 2019, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, V.; Rungta, T.; Goyal, K.; Singh, M.P. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci. Rep. 2019, 9, 2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeli, S.; Hassan, S.S.; Yap, W.B. Multi-Epitope Peptide-Based and Vaccinia-Based Universal Influenza Vaccine Candidates Subjected to Clinical Trials. Malays. J. Med. Sci. MJMS 2020, 27, 10. [Google Scholar]
- Bazhan, S.I.; Antonets, D.V.; Karpenko, L.I.; Oreshkova, S.F.; Kaplina, O.N.; Starostina, E.V.; Dudko, S.G.; Fedotova, S.A.; Ilyichev, A.A. In silico designed ebola virus T-cell multi-epitope DNA vaccine constructions are immunogenic in mice. Vaccines 2019, 7, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, L.M.F.; Morale, M.G.; Chaves, A.A.M.; Cavalher, A.M.; Lopes, A.S.; de Oliveira Diniz, M.; Schanoski, A.S.; de Melo, R.L.; de Souza Ferreira, L.C.; de Oliveira, M.L.S.; et al. Design, immune responses and anti-tumor potential of an HPV16 E6E7 multi-epitope vaccine. PLoS ONE 2015, 10, e0138686. [Google Scholar] [CrossRef] [Green Version]
- Hasanzadeh, S.; Habibi, M.; Shokrgozar, M.A.; Cohan, R.A.; Ahmadi, K.; Karam, M.R.A.; Bouzari, S. In Silico analysis and in vivo assessment of a novel epitope-based vaccine candidate against uropathogenic Escherichia coli. Sci. Rep. 2020, 10, 16258. [Google Scholar] [CrossRef]
- Nelde, A.; Bilich, T.; Heitmann, J.S.; Maringer, Y.; Salih, H.R.; Roerden, M.; Lübke, M.; Bauer, J.; Rieth, J.; Wacker, M.; et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat. Immunol. 2020, 22, 1–12. [Google Scholar] [CrossRef]
- Ascarateil, S.; Puget, A.; Koziol, M.E. Safety data of Montanide ISA 51 VG and Montanide ISA 720 VG, two adjuvants dedicated to human therapeutic vaccines. J. Immunother. Cancer 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Gauttier, V.; Morello, A.; Girault, I.; Mary, C.; Belarif, L.; Desselle, A.; Wilhelm, E.; Bourquard, T.; Pengam, S.; Teppaz, G.; et al. Tissue-resident memory CD8 T-cell responses elicited by a single injection of a multi-target COVID-19 vaccine. bioRxiv 2020. [Google Scholar] [CrossRef]
- Khan, M.A.A.; Ami, J.Q.; Faisal, K.; Chowdhury, R.; Ghosh, P.; Hossain, F.; Abd El Wahed, A.; Mondal, D. An immunoinformatic approach driven by experimental proteomics: In silico design of a subunit candidate vaccine targeting secretory proteins of Leishmania donovani amastigotes. Parasites Vectors 2020, 13, 196. [Google Scholar] [CrossRef] [Green Version]
- Maeshima, N.; Fernandez, R. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 2013, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Haij, N.B.; Leghmari, K.; Planès, R.; Thieblemont, N.; Bahraoui, E. HIV-1 Tat protein binds to TLR4-MD2 and signals to induce TNF-α and IL-10. Retrovirology 2013, 10, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef] [Green Version]
- Rallabhandi, P.; Phillips, R.L.; Boukhvalova, M.S.; Pletneva, L.M.; Shirey, K.A.; Gioannini, T.L.; Weiss, J.P.; Chow, J.C.; Hawkins, L.D.; Vogel, S.N.; et al. Respiratory syncytial virus fusion protein-induced toll-like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E5564) and requires direct interaction with MD-2. MBio 2012, 3, e00218-12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xu, L.; Park, H.B.; Hwang, J.; Kwak, M.; Lee, P.C.; Liang, G.; Zhang, X.; Xu, J.; Jin, J.O. Escherichia coli adhesion portion FimH functions as an adjuvant for cancer immunotherapy. Nat. Commun. 2020, 11, 1187. [Google Scholar] [CrossRef] [Green Version]
- Piccinini, A.; Midwood, K. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010, 672395. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Su, L.; Morin, M.D.; Jones, B.T.; Whitby, L.R.; Surakattula, M.M.; Huang, H.; Shi, H.; Choi, J.H.; Wang, K.W.; et al. TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc. Natl. Acad. Sci. USA 2016, 113, E884–E893. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.K. Molecular evolution of elements controlling HLA-C expression: Adaptation to a role as a killer-cell immunoglobulin-like receptor ligand regulating natural killer cell function. HLA 2018, 92, 271–278. [Google Scholar] [CrossRef]
- Blais, M.E.; Dong, T.; Rowland-Jones, S. HLA-C as a mediator of natural killer and T-cell activation: Spectator or key player? Immunology 2011, 133, 1–7. [Google Scholar] [CrossRef]
- Tey, S.K.; Khanna, R. Autophagy mediates transporter associated with antigen processing-independent presentation of viral epitopes through MHC class I pathway. Blood J. Am. Soc. Hematol. 2012, 120, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Peptide | Peptide | Positions | Protein of Origin | Vaxijen | AllerTOP | HLA | IEBD |
|---|---|---|---|---|---|---|---|
| 1 | AAAYYVGYLQPRTFL | 262–276 | SP | 0.48 | Non-Allergen | n/a | Validated region |
| 2 | DDSEPVLKGVKLHYT * | 1259–1273 | SP | 1.18 | Non-Allergen | n/a | Validated peptide—Assay in BLs |
| 3 | LVIGAVILRGHLRIA | 138–152 | MG | 0.88 | Non-Allergen | n/a | n/a |
| 4 | QSIIAYTMSLGAENS | 690–704 | SP | 0.57 | Non-Allergen | n/a | Validated region |
| 5 | QSLLIVNNATNVVIK | 115–129 | SP | 0.43 | Non-Allergen | n/a | Validated region |
| 6 | SFRLFARTRSMWSFN | 99–113 | MG | 0.80 | Non-Allergen | n/a | n/a |
| 7 | VLSFELLHAPATVCG | 512–526 | SP | 0.48 | Non-Allergen | n/a | Validated region |
| 8 | CISTKHFYW | 3147–3155 | ORF1-NSP4 | 1.90 | Non-Allergen | n/a | n/a |
| 9 | FAMQMAYRF | 898–906 | SP | 1.03 | Non-Allergen | n/a | Validated region |
| 10 | FLLNKEMYL * | 3183–3191 | ORF1-NSP4 | 0.44 | Non-Allergen | HLA-A*02:01 | Validated peptide—Assay in TLs. |
| 11 | GYKSVNITF | 835–843 | ORF1-NSP3 | 2.37 | Non-Allergen | n/a | n/a |
| 12 | ITLCFTLKR | 110–118 | ORF7a | 2.02 | Non-Allergen | n/a | n/a |
| 13 | KRAKVTSAM | 4022–4030 | ORF1-NSP8 | 0.76 | Non-Allergen | n/a | n/a |
| 14 | KVKYLYFIK * | 4225–4233 | ORF1-NSP9 | 1.06 | Non-Allergen | HLA-A*11:01 | Validated peptide—Assay in TLs. |
| 15 | LEMELTPVV | 1012–1020 | ORF1-NSP3 | 1.97 | Non-Allergen | n/a | n/a |
| 16 | MPYFFTLLL | 2169–2177 | ORF1-NSP3 | 0.49 | Non-Allergen | n/a | n/a |
| 17 | VMYASAVVL | 3684–3692 | ORF1-NSP6 | 0.48 | Non-Allergen | n/a | n/a |
| 18 | WTAGAAAYY | 258–266 | SP | 0.63 | Non-Allergen | HLA-A*01:01 | Validated region |
| Peptide C Terminal | CTGKSC+ | CTGKSC− |
|---|---|---|
| Vaxijen V2.0 | 0.6419 | 0.6455 |
| AllerToP V2.0 | Non-Allergenic | Non-Allergenic |
| Allergen FP V1.0 | Non-Allergenic | Non-Allergenic |
| ToxinPred | Non-Toxic | Non-Toxic |
| Physicochemical Parameters | ||
| GRAVY | −0.038 | −0.033 |
| Molecular Weight | 55.67 kDa | 54.62 kDa |
| Stability | 33.76 (<40) | 33.47 (<40) |
| Solubility | 72% | 65% |
| Half-Life (reticulocytes) | 30 h | 30 h |
| Aliphatic index | 81.81 | 82.62 |
| Size (amino acids) | 510 | 499 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuspoca, A.F.; Díaz, L.L.; Acosta, A.F.; Peñaloza, M.K.; Méndez, Y.R.; Clavijo, D.C.; Yosa Reyes, J. An Immunoinformatics Approach for SARS-CoV-2 in Latam Populations and Multi-Epitope Vaccine Candidate Directed towards the World’s Population. Vaccines 2021, 9, 581. https://doi.org/10.3390/vaccines9060581
Cuspoca AF, Díaz LL, Acosta AF, Peñaloza MK, Méndez YR, Clavijo DC, Yosa Reyes J. An Immunoinformatics Approach for SARS-CoV-2 in Latam Populations and Multi-Epitope Vaccine Candidate Directed towards the World’s Population. Vaccines. 2021; 9(6):581. https://doi.org/10.3390/vaccines9060581
Chicago/Turabian StyleCuspoca, Andrés Felipe, Laura Lorena Díaz, Alvaro Fernando Acosta, Marcela Katherine Peñaloza, Yardany Rafael Méndez, Diana Carolina Clavijo, and Juvenal Yosa Reyes. 2021. "An Immunoinformatics Approach for SARS-CoV-2 in Latam Populations and Multi-Epitope Vaccine Candidate Directed towards the World’s Population" Vaccines 9, no. 6: 581. https://doi.org/10.3390/vaccines9060581