
Development of a Vaccine against Human Cytomegalovirus: Advances, Barriers, and Implications for the Clinical Practice


 , and
, and
Abstract
:1. Introduction
1.1. Epidemiology
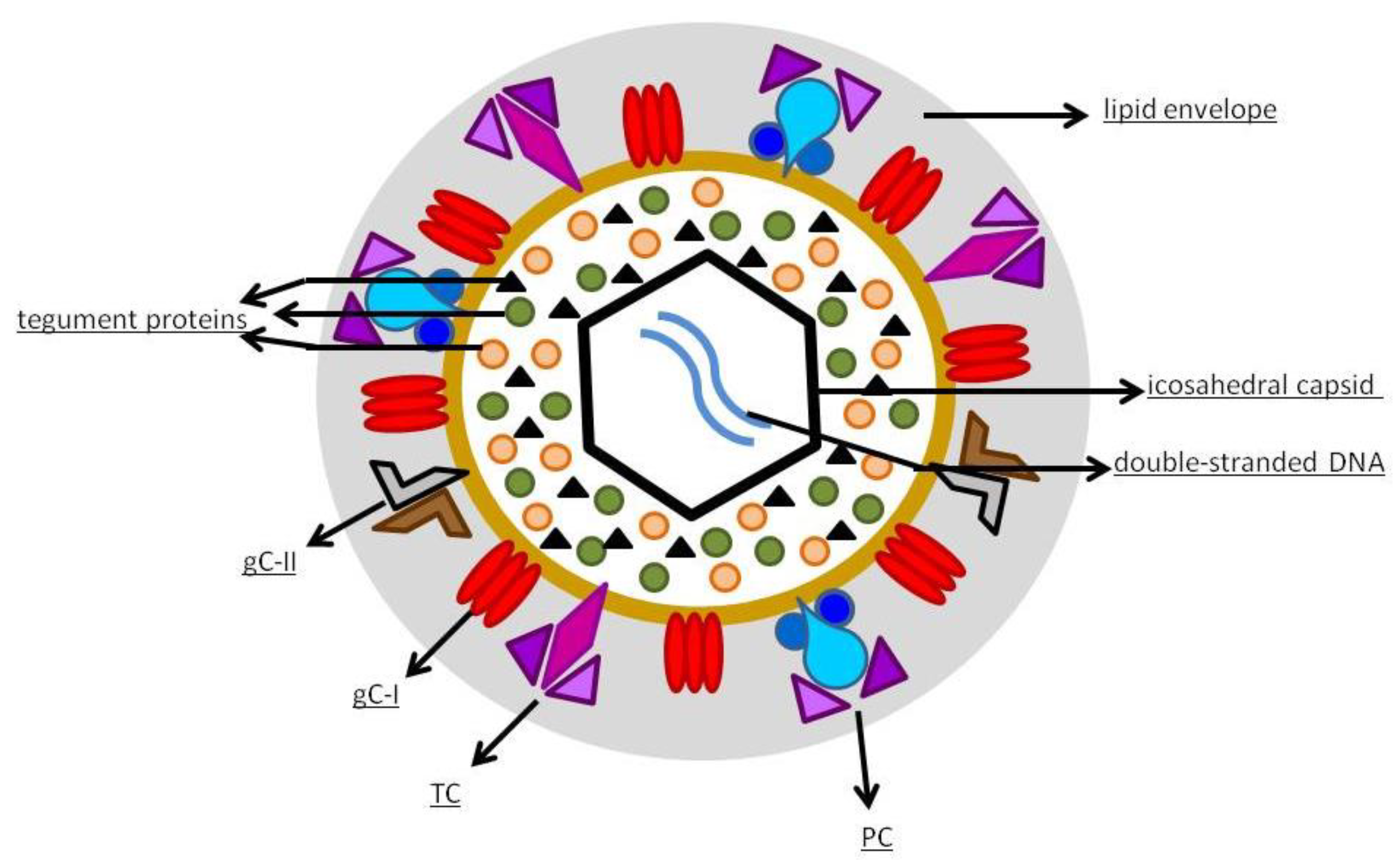
1.2. Virology
1.3. Clinical Manifestations
1.4. Diagnosis
1.4.1. cCMV
1.4.2. Allogeneic HSCT
1.4.3. SOT
1.5. Therapy
1.5.1. cCMV
1.5.2. Allogeneic HSCT
1.5.3. SOT
1.5.4. AIDS
1.6. Why Is It Necessary to Find an Effective Vaccine against hCMV?
1.7. Where Are We Now?
2. Current Candidates
2.1. Live-Attenuated Vaccines
2.2. Subunit Vaccines
2.3. Virus Vectored Vaccine
2.4. Chimeric Peptidic Vaccines
2.5. Vaccine Based on Enveloped Virus-Like Particles
2.6. Plasmid-Based DNA Vaccines
2.7. RNA-Based Vaccines
2.8. Peptide Vaccines
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bate, S.L.; Dollard, S.C.; Cannon, M.J. Cytomegalovirus Seroprevalence in the United States: The National Health and Nutrition Examination Surveys, 1988. Clin. Infect. Dis. 2010, 50, 1439–1447. [Google Scholar] [CrossRef]
- Gerna, G.; Lilleri, D. Human Cytomegalovirus (HCMV) Infection/Re-Infection: Development of a Protective HCMV Vaccine. New Microbiol. 2019, 42, 1–20. [Google Scholar] [PubMed]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef]
- Cannon, M.J.; Hyde, T.B.; Schmid, D.S. Review of cytomegalovirus shedding in bodily fluids and relevance to congenital cytomegalovirus infection. Rev. Med. Virol. 2011, 21, 240–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, T.; Mangold, J.F.; Cantrell, S.; Permar, S.R. Impact of Maternal Immunity on Congenital Cytomegalovirus Birth Prevalence and Infant Outcomes: A Systematic Review. Vaccines 2019, 7, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revello, M.G.; Fabbri, E.; Furione, M.; Zavattoni, M.; Lilleri, D.; Tassis, B.; Quarenghi, A.; Cena, C.; Arossa, A.; Montanari, L.; et al. Role of prenatal diagnosis and counseling in the management of 735 pregnancies complicated by primary human cytomegalovirus infection: A 20-year experience. J. Clin. Virol. 2011, 50, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Boppana, S.B.; Rivera, L.B.; Fowler, K.B.; Mach, M.; Britt, W.J. Intrauterine Transmission of Cytomegalovirus to Infants of Women with Preconceptional Immunity. N. Engl. J. Med. 2001, 344, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- De Vries, J.J.C.; Van Zwet, E.W.; Dekker, F.W.; Kroes, A.C.M.; Verkerk, P.H.; Vossen, A.C.T.M. The apparent paradox of maternal seropositivity as a risk factor for congenital cytomegalovirus infection: A population-based prediction model. Rev. Med. Virol. 2013, 23, 241–249. [Google Scholar] [CrossRef]
- Enders, G.; Daiminger, A.; Bäder, U.; Exler, S.; Enders, M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J. Clin. Virol. 2011, 52, 244–246. [Google Scholar] [CrossRef]
- Pass, R.F.; Fowler, K.B.; Boppana, S.B.; Britt, W.J.; Stagno, S. Congenital cytomegalovirus infection following first trimester maternal infection: Symptoms at birth and outcome. J. Clin. Virol. 2006, 35, 216–220. [Google Scholar] [CrossRef]
- Fishman, J.A. Infection in Solid-Organ Transplant Recipients. N. Engl. J. Med. 2007, 357, 2601–2614. [Google Scholar] [CrossRef] [Green Version]
- Hoover, D.R.; Saah, A.J.; Bacellar, H.; Phair, J.; Detels, R.; Anderson, R.; Kaslow, R.A. Clinical Manifestations of AIDS in the Era of Pneumocystis Prophylaxis. N. Engl. J. Med. 1993, 329, 1922–1926. [Google Scholar] [CrossRef] [PubMed]
- Sungkanuparph, S.; Chakriyanuyok, T.; Butthum, B. Antiretroviral therapy in AIDS patients with CMV disease: Impact on the survival and long-term treatment outcome. J. Infect. 2008, 56, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Cervera, C.; Gurgui, M.; Lumbreras, C. Factores de riesgo de la enfermedad por citomegalovirus en el receptor de un trasplante de órgano sólido. Enfermedades Infec. Microbiol. Clín. 2011, 29, 11–17. [Google Scholar] [CrossRef]
- Bermejo, C.L.; Manuel, O.; Len, O.; ten Berge, I.J.T.; Sgarabotto, D.; Hirsch, H.H. Cytomegalovirus infection in solid organ transplant recipients. Clin. Microbiol. Infect. 2014, 20 (Suppl. 7), 19–26. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.T.; Logan, A.C. The clinical impact of cytomegalovirus infection following allogeneic hematopoietic cell transplantation: Why the quest for meaningful prophylaxis still matters. Blood Rev. 2017, 31, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Owers, D.S.; Webster, A.C.; Strippoli, G.F.M.; Kable, K.; Hodson, E.M. Pre-emptive treatment for cytomegalovirus viraemia to prevent cytomegalovirus disease in solid organ transplant recipients. Cochrane Database Syst. Rev. 2013, CD005133. [Google Scholar] [CrossRef] [PubMed]
- Limaye, A.P.; Boeckh, M. CMV in critically ill patients: Pathogen or bystander? Rev. Med. Virol. 2010, 20, 372–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iii, J.P.T. Human cytomegalovirus tegument proteins (pp65, pp71, pp150, pp28). Virol. J. 2012, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Human Cytomegalovirus; Shenk, T.; Stinski, M. Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Sandonís, V.; García-Ríos, E.; McConnell, M.J.; Pérez-Romero, P. Role of Neutralizing Antibodies in CMV Infection: Implications for New Therapeutic Approaches. Trends Microbiol. 2020, 28, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Gerna, G.; Kabanova, A.; Lilleri, D. Human Cytomegalovirus Cell Tropism and Host Cell Receptors. Vaccines 2019, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human Cytomegalovirus UL131-128 Genes Are Indispensable for Virus Growth in Endothelial Cells and Virus Transfer to Leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [Green Version]
- Ryckman, B.J.; Jarvis, M.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human Cytomegalovirus Entry into Epithelial and Endothelial Cells Depends on Genes UL128 to UL150 and Occurs by Endocytosis and Low-pH Fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef] [Green Version]
- Ryckman, B.J.; Rainish, B.L.; Chase, M.C.; Borton, J.A.; Nelson, J.A.; Jarvis, M.; Johnson, D.C. Characterization of the Human Cytomegalovirus gH/gL/UL128-131 Complex That Mediates Entry into Epithelial and Endothelial Cells. J. Virol. 2007, 82, 60–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalejta, R.F. Tegument Proteins of Human Cytomegalovirus. Microbiol. Mol. Biol. Rev. 2008, 72, 249–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugliesi, F.; Coscia, A.; Griffante, G.; Galitska, G.; Pasquero, S.; Albano, C.; Biolatti, M. Where do we Stand after Decades of Studying Human Cytomegalovirus? Microorganisms 2020, 8, 685. [Google Scholar] [CrossRef] [PubMed]
- Stenberg, R.M. Immediate-Early Genes of Human Cytomegalovirus: Organization and Function. In Molecular Aspects of Human Cytomegalovirus Diseases; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1993; Volume 2, pp. 330–359. [Google Scholar]
- Paulus, C.; Nevels, M. The Human Cytomegalovirus Major Immediate-Early Proteins as Antagonists of Intrinsic and Innate Antiviral Host Responses. Viruses 2009, 1, 760–779. [Google Scholar] [CrossRef] [Green Version]
- Guerra, B.; Simonazzi, G.; Puccetti, C.; Lanari, M.; Farina, A.; Lazzarotto, T.; Rizzo, N. Ultrasound prediction of symptomatic congenital cytomegalovirus infection. Am. J. Obstet. Gynecol. 2008, 198, 380.e1–380.e7. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, J. Congenital Cytomegalovirus—History, Current Practice, and Future Opportunities. Neonatal Netw. 2020, 39, 293–298. [Google Scholar] [CrossRef]
- Boppana, S.B.; Ross, S.A.; Fowler, K.B. Congenital Cytomegalovirus Infection: Clinical Outcome. Clin. Infect. Dis. 2013, 57, S178–S181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kylat, R.I.; Kelly, E.N.; Ford-Jones, E.L. Clinical findings and adverse outcome in neonates with symptomatic congenital cytomegalovirus (SCCMV) infection. Eur. J. Nucl. Med. Mol. Imaging 2006, 165, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Dreher, A.M.; Arora, N.; Fowler, K.B.; Novak, Z.; Britt, W.J.; Boppana, S.B.; Ross, S.A. Spectrum of Disease and Outcome in Children with Symptomatic Congenital Cytomegalovirus Infection. J. Pediatr. 2014, 164, 855–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goderis, J.; De Leenheer, E.; Smets, K.; Van Hoecke, H.; Keymeulen, A.; Dhooge, I. Hearing Loss and Congenital CMV Infection: A Systematic Review. Pediatry 2014, 134, 972–982. [Google Scholar] [CrossRef] [Green Version]
- Cannon, M.J.; Griffiths, P.D.; Aston, V.; Rawlinson, W.D. Universal newborn screening for congenital CMV infection: What is the evidence of potential benefit? Rev. Med. Virol. 2014, 24, 291–307. [Google Scholar] [CrossRef] [Green Version]
- Bilavsky, E.; Shahar-Nissan, K.; Pardo, J.; Attias, J.; Amir, J. Hearing outcome of infants with congenital cytomegalovirus and hearing impairment. Arch. Dis. Child. 2016, 101, 433–438. [Google Scholar] [CrossRef]
- Grosse, S.D.; Ross, D.S.; Dollard, S.C. Congenital cytomegalovirus (CMV) infection as a cause of permanent bilateral hearing loss: A quantitative assessment. J. Clin. Virol. 2008, 41, 57–62. [Google Scholar] [CrossRef]
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef]
- Dahle, A.J.; Fowler, K.B.; Wright, J.D.; Boppana, S.B.; Britt, W.J.; Pass, R. Longitudinal investigation of hearing disorders in children with congenital cytomegalovirus. J. Am. Acad. Audiol. 2000, 11, 283–290. [Google Scholar] [PubMed]
- Foulon, I.; Naessens, A.; Foulon, W.; Casteels, A.; Gordts, F. A 10-Year Prospective Study of Sensorineural Hearing Loss in Children with Congenital Cytomegalovirus Infection. J. Pediatr. 2008, 153, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-N.; Zhou, Y.-P.; Jiang, X.; Yang, B.; Cheng, H.; Luo, M.-H. Hearing Loss Caused by HCMV Infection through Regulating the Wnt and Notch Signaling Pathways. Viruses 2021, 13, 623. [Google Scholar] [CrossRef]
- Dioverti, M.V.; Razonable, R.R. Cytomegalovirus. Microbiol. Spectr. 2016, 4, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R.; Humar, A.; the AST Infectious Diseases Community of Practice. Cytomegalovirus in Solid Organ Transplantation. Arab. Archaeol. Epigr. 2013, 13, 93–106. [Google Scholar] [CrossRef]
- George, B.; Pati, N.; Gilroy, N.; Ratnamohan, M.; Huang, G.; Kerridge, I.; Hertzberg, M.; Gottlieb, D.; Bradstock, K. Pre-transplant cytomegalovirus (CMV) serostatus remains the most important determinant of CMV reactivation after allogeneic hematopoietic stem cell transplantation in the era of surveillance and preemptive therapy. Transpl. Infect. Dis. 2010, 12, 322–329. [Google Scholar] [CrossRef]
- Travi, G.; Pergam, S.A. Cytomegalovirus Pneumonia in Hematopoietic Stem Cell Recipients. J. Intensive Care Med. 2014, 29, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Prince, H.E.; Lapé-Nixon, M. Role of Cytomegalovirus (CMV) IgG Avidity Testing in Diagnosing Primary CMV Infection during Pregnancy. Clin. Vaccine Immunol. 2014, 21, 1377–1384. [Google Scholar] [CrossRef] [Green Version]
- Lazzarotto, T.; Varani, S.; Spezzacatena, P.; Gabrielli, L.; Pradelli, P.; Guerra, B.; Landini, M.P. Maternal IgG Avidity and IgM Detected by Blot as Diagnostic Tools to Identify Pregnant Women at Risk of Transmitting Cytomegalovirus. Viral Immunol. 2000, 13, 137–141. [Google Scholar] [CrossRef]
- Lazzarotto, T.; Blázquez-Gamero, D.; Delforge, M.-L.; Foulon, I.; Luck, S.; Modrow, S.; Leruez-Ville, M. Congenital Cytomegalovirus Infection: A Narrative Review of the Issues in Screening and Management from a Panel of European Experts. Front. Pediatr. 2020, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Rawlinson, W.D.; Boppana, S.B.; Fowler, K.B.; Kimberlin, D.W.; Lazzarotto, T.; Alain, S.; Daly, K.; Doutré, S.; Gibson, L.; Giles, M.L.; et al. Congenital cytomegalovirus infection in pregnancy and the neonate: Consensus recommendations for prevention, diagnosis, and therapy. Lancet Infect. Dis. 2017, 17, e177–e188. [Google Scholar] [CrossRef]
- Luck, S.E.; Wieringa, J.W.; Blázquez-Gamero, D.; Henneke, P.; Schuster, K.; Butler, K.; Capretti, M.G.; Cilleruelo, M.J.; Curtis, N.; Garofoli, F.; et al. Congenital Cytomegalovirus. Pediatr. Infect. Dis. J. 2017, 36, 1205–1213. [Google Scholar] [CrossRef]
- Hughes, B.L.; Gyamfi-Bannerman, C. Diagnosis and antenatal management of congenital cytomegalovirus infection. Am. J. Obstet. Gynecol. 2016, 214, B5–B11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enders, M.; Daiminger, A.; Exler, S.; Ertan, K.; Enders, G.; Bald, R. Prenatal diagnosis of congenital cytomegalovirus infection in 115 cases: A 5 years’ single center experience. Prenat. Diagn. 2017, 37, 389–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boppana, S.B.; Ross, S.A.; Shimamura, M.; Palmer, A.L.; Ahmed, A.; Michaels, M.G.; Sánchez, P.J.; Bernstein, D.I.; Tolan, R.W.; Novak, Z.; et al. Saliva Polymerase-Chain-Reaction Assay for Cytomegalovirus Screening in Newborns. N. Engl. J. Med. 2011, 364, 2111–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, J.J.; van der Eijk, A.A.; Wolthers, K.C.; Rusman, L.G.; Pas, S.D.; Molenkamp, R.; Claas, E.C.; Kroes, A.C.; Vossen, A.C. Real-time PCR vs. viral culture on urine as a gold standard in the diagnosis of congenital cytomegalovirus infection. J. Clin. Virol. 2012, 53, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Leruez-Ville, M.; Magny, J.-F.; Couderc, S.; Pichon, C.; Parodi, M.; Bussières, L.; Guilleminot, T.; Ghout, I.; Ville, Y. Risk Factors for Congenital Cytomegalovirus Infection Following Primary and Nonprimary Maternal Infection. Clin. Infect. Dis. 2017, 65, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.A.; Ahmed, A.; Palmer, A.L.; Michaels, M.G.; Sánchez, P.J.; Stewart, A.; Bernstein, D.I.; Feja, K.; Novak, Z.; Fowler, K.B.; et al. Urine Collection Method for the Diagnosis of Congenital Cytomegalovirus Infection. Pediatr. Infect. Dis. J. 2015, 34, 903–905. [Google Scholar] [CrossRef] [Green Version]
- Vancor, E.; Shapiro, E.D.; Loyal, J. Results of a Targeted Screening Program for Congenital Cytomegalovirus Infection in Infants Who Fail Newborn Hearing Screening. J. Pediatr. Infect. Dis. Soc. 2018, 8, 55–59. [Google Scholar] [CrossRef]
- Diener, M.L.; Zick, C.D.; McVicar, S.B.; Boettger, J.; Park, A.H. Outcomes from a Hearing-Targeted Cytomegalovirus Screening Program. Pediatry 2017, 139, e20160789. [Google Scholar] [CrossRef] [Green Version]
- Marsico, C.; Kimberlin, D.W. Congenital Cytomegalovirus infection: Advances and challenges in diagnosis, prevention and treatment. Ital. J. Pediatr. 2017, 43, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Drew, W.L. Laboratory diagnosis of cytomegalovirus infection and disease in immunocompromised patients. Curr. Opin. Infect. Dis. 2007, 20, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jin, N.; Chen, B. Human cytomegalovirus infection: A considerable issue following allogeneic hematopoietic stem cell transplantation (Review). Oncol. Lett. 2021, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Huprikar, S.; Chou, S.; Danziger-Isakov, L.; Humar, A. The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-organ Transplantation. Transplant 2018, 102, 900–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimberlin, D.W.; Acosta, E.P.; Sánchez, P.J.; Sood, S.; Agrawal, V.; Homans, J.; Jacobs, R.F.; Lang, D.; Romero, J.R.; Griffin, J.; et al. Pharmacokinetic and Pharmacodynamic Assessment of Oral Valganciclovir in the Treatment of Symptomatic Congenital Cytomegalovirus Disease. J. Infect. Dis. 2008, 197, 836–845. [Google Scholar] [CrossRef]
- Kimberlin, D.W.; Lin, C.-Y.; Sánchez, P.J.; Demmler, G.J.; Dankner, W.; Shelton, M.; Jacobs, R.F.; Vaudry, W.; Pass, R.; Kiell, J.M.; et al. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: A randomized, controlled trial. J. Pediatr. 2003, 143, 16–25. [Google Scholar] [CrossRef]
- Kimberlin, D.W.; Jester, P.M.; Sánchez, P.J.; Ahmed, A.; Arav-Boger, R.; Michaels, M.G.; Ashouri, N.; Englund, J.A.; Estrada, B.; Jacobs, R.F.; et al. Valganciclovir for Symptomatic Congenital Cytomegalovirus Disease. N. Engl. J. Med. 2015, 372, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Oliver, S.E.; Cloud, G.A.; Sánchez, P.J.; Demmler, G.J.; Dankner, W.; Shelton, M.; Jacobs, R.F.; Vaudry, W.; Pass, R.; Soong, S.-J.; et al. Neurodevelopmental outcomes following ganciclovir therapy in symptomatic congenital cytomegalovirus infections involving the central nervous system. J. Clin. Virol. 2009, 46, S22–S26. [Google Scholar] [CrossRef] [Green Version]
- Gwee, A.; Curtis, N.; Connell, T.G.; Garland, S.; Daley, A.J. Ganciclovir for the Treatment of Congenital Cytomegalovirus. Pediatr. Infect. Dis. J. 2014, 33, 115. [Google Scholar] [CrossRef]
- Lombardi, G.; Garofoli, F.; Villani, P.; Tizzoni, M.; Angelini, M.; Cusato, M.; Bollani, L.; De Silvestri, A.; Regazzi, M.; Stronati, M. Oral valganciclovir treatment in newborns with symptomatic congenital cytomegalovirus infection. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1465–1470. [Google Scholar] [CrossRef]
- Park, S.-Y.; Lee, S.-O.; Choi, S.-H.; Kim, Y.S.; Woo, J.H.; Baek, S.; Sung, H.; Kim, M.-N.; Kim, D.-Y.; Lee, J.-H.; et al. Efficacy and safety of low-dose ganciclovir preemptive therapy in allogeneic haematopoietic stem cell transplant recipients compared with conventional-dose ganciclovir: A prospective observational study. J. Antimicrob. Chemother. 2012, 67, 1486–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locatelli, F.; Bertaina, A.; Bertaina, V.; Merli, P. Cytomegalovirus in hematopoietic stem cell transplant recipients—management of infection. Expert Rev. Hematol. 2016, 9, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Maffini, E.; Giaccone, L.; Festuccia, M.; Brunello, L.; Busca, A.; Bruno, B. Treatment of CMV Infection after Allogeneic Hematopoietic Stem Cell Transplantation. Expert Rev. Hematol. 2016, 9, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Moretti, S.; Zikos, P.; Van Lint, M.T.; Tedone, E.; Occhini, D.; Gualandi, F.; Lamparelli, T.; Mordini, N.; Berisso, G.; Bregante, S.; et al. Foscarnet vs ganciclovir for cytomegalovirus (CMV) antigenemia after allogeneic hemopoietic stem cell transplantation (HSCT): A randomised study. Bone Marrow Transplant. 1998, 22, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Meesing, A.; Razonable, R.R. New Developments in the Management of Cytomegalovirus Infection after Transplantation. Drugs 2018, 78, 1085–1103. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- Paya, C.; Humar, A.; Dominguez, E.; Washburn, K.; Blumberg, E.; Alexander, B.; Freeman, R.; Heaton, N.; Pescovitz, M.D.; Valganciclovir Solid Organ Transplant Study Group. Efficacy and Safety of Valganciclovir vs. Oral Ganciclovir for Prevention of Cytomegalovirus Disease in Solid Organ Transplant Recipients. Arab. Archaeol. Epigr. 2004, 4, 611–620. [Google Scholar] [CrossRef]
- Razonable, R.R.; Humar, A. Cytomegalovirus in Solid Organ Transplant Recipients—Guidelines of the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13512. [Google Scholar] [CrossRef]
- Port, A.D.; Orlin, A.; Kiss, S.; Patel, S.; D’Amico, D.J.; Gupta, M.P. Cytomegalovirus Retinitis: A Review. J. Ocul. Pharmacol. Ther. 2017, 33, 224–234. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Boppana, S.B. Vaccination against the human cytomegalovirus. Vaccine 2019, 37, 7437–7442. [Google Scholar] [CrossRef]
- Fowler, K.B.; Boppana, S.B. Congenital cytomegalovirus infection. Semin. Perinatol. 2018, 42, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retzler, J.; Hex, N.; Bartlett, C.; Webb, A.; Wood, S.; Star, C.; Griffiths, P.; Jones, C.E. Economic cost of congenital CMV in the UK. Arch. Dis. Child. 2018, 104, 559–563. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, C.; Diamond, D.J. The immune response to human CMV. Futur. Virol. 2012, 7, 279–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugliesi, F.; Pasquero, S.; Griffante, G.; Scutera, S.; Albano, C.; Pacheco, S.; Riva, G.; Dell’Oste, V.; Biolatti, M. Human Cytomegalovirus and Autoimmune Diseases: Where Are We? Viruses 2021, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Akbar, A.; Beverley, P.; Caruso, C.; Derhovanessian, E.; Fülöp, T.; Griffiths, P.; Grubeck-Loebenstein, B.; Hamprecht, K.; Jahn, G.; et al. Immunosenescence and Cytomegalovirus: Where do we stand after a decade? Immun. Ageing 2010, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilieva, E.; Gianella, S.; Freeman, M.L. Novel Strategies to Combat CMV-Related Cardiovascular Disease. Pathog. Immun. 2020, 5, 240–274. [Google Scholar] [CrossRef]
- Wilski, N.A.; Snyder, C.M. From Vaccine Vector to Oncomodulation: Understanding the Complex Interplay between CMV and Cancer. Vaccines 2019, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Schleiss, M.R.; Diamond, D.J. Exciting Times for Cytomegalovirus (CMV) Vaccine Development: Navigating the Pathways toward the Goal of Protecting Infants against Congenital CMV Infection. Vaccines 2020, 8, 526. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Committee to Study Priorities for Vaccine Development. Vaccines for the 21st Century: A Tool for Decisionmaking; Stratton, K.R., Durch, J.S., Lawrence, R.S., Eds.; The National Academies Collection: Reports Funded by National Institutes of Health; National Academies Press (US): Washington, DC, USA, 2000. [Google Scholar]
- Plotkin, S.A.; Furukawa, T.; Zygraich, N.; Huygelen, C. Candidate cytomegalovirus strain for human vaccination. Infect. Immun. 1975, 12, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Elek, S.; Stern, H. Development of a vaccine against mental retardation caused by cytomegalovirus infection in utero. Lancet 1974, 303, 1–5. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Starr, S.E.; Friedman, H.M.; Brayman, K.; Harris, S.; Jackson, S.; Tustin, N.B.; Grossman, R.; Dafoe, D.; Barker, C. Effect of Towne Live Virus Vaccine on Cytomegalovirus Disease after Renal Transplant. Ann. Intern. Med. 1991, 114, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Higgins, R.; Kurtz, J.B.; Morris, P.J.; Campbell, D.A.; Shope, T.C.; Spector, S.A.; Dankner, W.M. Multi-center Trial of Towne Strain Attenuated Virus Vaccine in Seronegative Renal Transplant Recipients. Transplantation 1994, 58, 1176–1178. [Google Scholar] [PubMed]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef]
- Sijmons, S.; Thys, K.; Ngwese, M.M.; Van Damme, E.; Dvorak, J.; Van Loock, M.; Li, G.; Tachezy, R.; Busson, L.; Aerssens, J.; et al. High-Throughput Analysis of Human Cytomegalovirus Genome Diversity Highlights the Widespread Occurrence of Gene-Disrupting Mutations and Pervasive Recombination. J. Virol. 2015, 89, 7673–7695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renzette, N.; Gibson, L.; Bhattacharjee, B.; Fisher, D.; Schleiss, M.R.; Jensen, J.D.; Kowalik, T.F. Rapid Intrahost Evolution of Human Cytomegalovirus Is Shaped by Demography and Positive Selection. PLoS Genet. 2013, 9, e1003735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, J.; Super, M.; Sweny, P.; Moorhead, J.; Lui, S.; Berry, N.; Fernando, O.; Griffiths, P. Symptomatic cytomegalovirus infection in seropositive kidney recipients: Reinfection with donor virus rather than reactivation of recipient virus. Lancet 1988, 332, 132–135. [Google Scholar] [CrossRef]
- Anderholm, K.M.; Bierle, C.J.; Schleiss, M.R. Cytomegalovirus Vaccines: Current Status and Future Prospects. Drugs 2016, 76, 1625–1645. [Google Scholar] [CrossRef]
- Diamond, D.J.; La Rosa, C.; Chiuppesi, F.; Contreras, H.; Dadwal, S.; Wussow, F.; Bautista, S.; Nakamura, R.; Zaia, J.A. A fifty-year odyssey: Prospects for a cytomegalovirus vaccine in transplant and congenital infection. Expert Rev. Vaccines 2018, 17, 889–911. [Google Scholar] [CrossRef]
- Nelson, C.S.; Herold, B.C.; Permar, S.R. A new era in cytomegalovirus vaccinology: Considerations for rational design of next-generation vaccines to prevent congenital cytomegalovirus infection. Vaccines 2018, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kagan, K.O.; Enders, M.; Schampera, M.S.; Baeumel, E.; Hoopmann, M.; Geipel, A.; Berg, C.; Goelz, R.; De Catte, L.; Wallwiener, D.; et al. Prevention of maternal-fetal transmission of cytomegalovirus after primary maternal infection in the first trimester by biweekly hyperimmunoglobulin administration. Ultrasound Obstet. Gynecol. 2019, 53, 383–389. [Google Scholar] [CrossRef]
- Lilleri, D.; Fornara, C.; Furione, M.; Zavattoni, M.; Revello, M.G.; Gerna, G. Development of Human Cytomegalovirus–Specific T Cell Immunity during Primary Infection of Pregnant Women and Its Correlation with Virus Transmission to the Fetus. J. Infect. Dis. 2007, 195, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Fornara, C.; Furione, M.; Arossa, A.; Gerna, G.; Lilleri, D. Comparative magnitude and kinetics of human cytomegalovirus-specific CD4+ and CD8+ T-cell responses in pregnant women with primary vs. remote infection and in transmitting vs. non-transmitting mothers: Its utility for dating primary infection in pre. J. Med. Virol. 2016, 88, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Tabata, T.; Petitt, M.; Fang-Hoover, J.; Freed, D.C.; Li, F.; An, Z.; Wang, D.; Fu, T.-M.; Pereira, L. Neutralizing Monoclonal Antibodies Reduce Human Cytomegalovirus Infection and Spread in Developing Placentas. Vaccines 2019, 7, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouts, A.E.; Chan, P.; Stephan, J.-P.; Vandlen, R.; Feierbach, B. Antibodies against the gH/gL/UL128/UL130/UL131 Complex Comprise the Majority of the Anti-Cytomegalovirus (Anti-CMV) Neutralizing Antibody Response in CMV Hyperimmune Globulin. J. Virol. 2012, 86, 7444–7447. [Google Scholar] [CrossRef] [Green Version]
- Freed, D.C.; Tang, Q.; Tang, A.; Li, F.; He, X.; Huang, Z.; Meng, W.; Xia, L.; Finnefrock, A.C.; Durr, E.; et al. Pentameric complex of viral glycoprotein H is the primary target for potent neutralization by a human cytomegalovirus vaccine. Proc. Natl. Acad. Sci. USA 2013, 110, E4997–E5005. [Google Scholar] [CrossRef] [Green Version]
- Ciferri, C.; Chandramouli, S.; Leitner, A.; Donnarumma, D.; Cianfrocco, M.A.; Gerrein, R.; Friedrich, K.; Aggarwal, Y.; Palladino, G.; Aebersold, R.; et al. Antigenic Characterization of the HCMV gH/gL/gO and Pentamer Cell Entry Complexes Reveals Binding Sites for Potently Neutralizing Human Antibodies. PLoS Pathog. 2015, 11, e1005230. [Google Scholar] [CrossRef]
- Chauhan, V.; Singh, M.P. Immuno-informatics approach to design a multi-epitope vaccine to combat cytomegalovirus infection. Eur. J. Pharm. Sci. 2020, 147, 105279. [Google Scholar] [CrossRef]
- Adler, S.P.; Lewis, N.; Conlon, A.; Christiansen, M.P.; Al-Ibrahim, M.; Rupp, R.; Fu, T.-M.; Bautista, O.; Tang, H.; Wang, D.; et al. Phase 1 Clinical Trial of a Conditionally Replication-Defective Human Cytomegalovirus (CMV) Vaccine in CMV-Seronegative Subjects. J. Infect. Dis. 2019, 220, 411–419. [Google Scholar] [CrossRef]
- Merck, S.; Dohme, C. A Phase I Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Safety, Tolerability and Immunogenicity of V160 (Human Cytomegalovirus Vaccine) in Healthy Japanese Men; Clinical Trial Registration NCT03840174. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03840174 (accessed on 24 May 2021).
- Merck, S.; Dohme, C. Double-Blind, Randomized, Placebo-Controlled Phase 2b, Multi-Center Study to Evaluate the Safety, Tolerability, Efficacy and Immunogenicity of a 2-Dose and a 3—Dose Regimen of V160 (Cytomegalovirus [CMV] Vaccine) in Healthy Seronegative Women, 16 to 35 Years of Age; Clinical Trial Registration NCT03486834. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03486834 (accessed on 24 May 2021).
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M.-L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, C.; et al. Vaccine Prevention of Maternal Cytomegalovirus Infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Munoz, F.M.; Callahan, S.T.; Rupp, R.; Wootton, S.H.; Edwards, K.M.; Turley, C.B.; Stanberry, L.R.; Patel, S.M.; Mcneal, M.M.; et al. Safety and efficacy of a cytomegalovirus glycoprotein B (gB) vaccine in adolescent girls: A randomized clinical trial. Vaccine 2016, 34, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, P.D.; Stanton, A.; McCarrell, E.; Smith, C.; Osman, M.; Harber, M.; Davenport, A.; Jones, G.; Wheeler, D.C.; O’Beirne, J.; et al. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: A phase 2 randomised placebo-controlled trial. Lancet 2011, 377, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, C.; Longmate, J.; Martinez, J.; Zhou, Q.; Kaltcheva, T.I.; Tsai, W.; Drake, J.; Carroll, M.; Wussow, F.; Chiuppesi, F.; et al. MVA vaccine encoding CMV antigens safely induces durable expansion of CMV-specific T cells in healthy adults. Blood 2017, 129, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; La Rosa, C.; Baden, L.R.; Longmate, J.; Ariza-Heredia, E.J.; Rida, W.N.; Lingaraju, C.R.; Zhou, Q.; Martinez, J.; Kaltcheva, T.; et al. Poxvirus Vectored Cytomegalovirus Vaccine to Prevent Cytomegalovirus Viremia in Transplant Recipients. Ann. Intern. Med. 2020, 172, 306. [Google Scholar] [CrossRef] [PubMed]
- City of Hope Medical Center. CMV-MVA Triplex Vaccination of Stem Cell Donors to Enhance CMV Specific Immunity and Prevent CMV Viremia in Recipients after Stem Cell Transplant; Clinical Trial Registration NCT03560752. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03560752 (accessed on 24 May 2021).
- City of Hope Medical Center. A Phase 1/2 Clinical Study to Evaluate the Optimal Dose and the Protective Effect of CMV-MVA Triplex Vaccine in Pediatric Patients Receiving an Allogeneic Hematopoietic Stem Cell Transplant; Clinical Trial Registration NCT03354728. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03354728 (accessed on 24 May 2021).
- Hookipa Biotech GmbH. Randomized, Placebo-Controlled, Double-Blind Phase I Dose-Escalating Trial to Evaluate the Safety and Immunogenicity of a Vaccine against Human Cytomegalovirus; Clinical Trial Registration NCT02798692. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT02798692 (accessed on 24 May 2021).
- Hookipa Biotech GmbH. A Randomized, Placebo-Controlled, Phase 2 Study of HB-101, a Bivalent Cytomegalovirus (CMV) Vaccine, in CMV-Seronegative Recipient (R-) Patients Awaiting Kidney Transplantation from Living CMV-Seropositive Donors (D+).; Clinical Trial Registration NCT03629080. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03629080 (accessed on 24 May 2021).
- La Rosa, C.; Longmate, J.; Lacey, S.F.; Kaltcheva, T.; Sharan, R.; Marsano, D.; Kwon, P.; Drake, J.; Williams, B.; Denison, S.; et al. Clinical Evaluation of Safety and Immunogenicity of PADRE-Cytomegalovirus (CMV) and Tetanus-CMV Fusion Peptide Vaccines with or Without PF03512676 Adjuvant. J. Infect. Dis. 2012, 205, 1294–1304. [Google Scholar] [CrossRef]
- Nakamura, R.; La Rosa, C.; Longmate, J.; Drake, J.; Slape, C.; Zhou, Q.; Lampa, M.G.; O’Donnell, M.; Cai, J.-L.; Farol, L.; et al. Viraemia, immunogenicity, and survival outcomes of cytomegalovirus chimeric epitope vaccine supplemented with PF03512676 (CMVPepVax) in allogeneic haemopoietic stem-cell transplantation: Randomised phase 1b trial. Lancet Haematol. 2016, 3, e87–e98. [Google Scholar] [CrossRef] [Green Version]
- City of Hope Medical Center. A Phase II Randomized, Placebo-Controlled, Multicenter Trial to Evaluate Protective Function of an Optimized Dose of CMVPepVax in Recipients of an Allogeneic Hematopoietic Stem Cell Transplant; Clinical Trial Registration NCT02396134. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT02396134 (accessed on 24 May 2021).
- VBI Vaccines Inc. A Phase 1 Randomized, Observer-Blind, Placebo-Controlled, Study to Evaluate the Safety, Tolerability, and Immunogenicity of the Candidate Human Cytomegalovirus Vaccine (VBI-1501) in Healthy Adults; Clinical Trial Registration NCT02826798. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02826798 (accessed on 24 May 2021).
- Kharfan-Dabaja, M.A.; Boeckh, M.; Wilck, M.B.; Langston, A.A.; Chu, A.H.; Wloch, M.K.; Guterwill, D.F.; Smith, L.R.; Rolland, A.P.; Kenney, R.T. A novel therapeutic cytomegalovirus DNA vaccine in allogeneic haemopoietic stem-cell transplantation: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Infect. Dis. 2012, 12, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Astellas Pharma Global Development, Inc. A Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial to Evaluate the Efficacy and Safety of a Vaccine, ASP0113, in Cytomegalovirus (CMV)-Seronegative Kidney Transplant Recipients Re-ceiving an Organ From a CMV-Seropositive Donor; Clinical Trial Registration NCT01974206. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT01974206 (accessed on 24 May 2021).
- Astellas Pharma Global Development, Inc. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial to Evaluate the Protective Efficacy and Safety of a Therapeutic Vaccine, ASP0113, in Cytomegalovirus (CMV)-Seropositive Recipients Undergoing Allogeneic, Hematopoietic Cell Transplant (HCT); Clinical Trial Registration NCT01877655. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT01877655 (accessed on 24 May 2021).
- ModernaTX, Inc. A Phase 1, Randomized, Observer-Blind, Placebo-Controlled, Dose-Ranging Study to Evaluate the Safety, Reactogenicity, and Immunogenicity of Cytomegalovirus Vaccines MRNA-1647 and MRNA-1443 When Administered to Healthy Adults; Clinical Trial Registration NCT03382405. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03382405 (accessed on 24 May 2021).
- ModernaTX, Inc. A Phase 2, Randomized, Observer-Blind, Placebo-Controlled, Dose-Finding Trial to Evaluate the Safety and Immunogenicity of Cytomegalovirus Vaccine MRNA-1647 in Healthy Adults; Clinical Trial Registration NCT04232280. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04232280 (accessed on 24 May 2021).
- Fu, T.-M.; Wang, D.; Freed, D.C.; Tang, A.; Li, F.; He, X.; Cole, S.; Dubey, S.; Finnefrock, A.C.; Ter Meulen, J.; et al. Restoration of viral epithelial tropism improves immunogenicity in rabbits and rhesus macaques for a whole virion vaccine of human cytomegalovirus. Vaccine 2012, 30, 7469–7474. [Google Scholar] [CrossRef]
- Wang, D.; Freed, D.C.; He, X.; Li, F.; Tang, A.; Cox, K.S.; Dubey, S.A.; Cole, S.; Medi, M.B.; Liu, Y.; et al. A replication-defective human cytomegalovirus vaccine for prevention of congenital infection. Sci. Transl. Med. 2016, 8, 362ra145. [Google Scholar] [CrossRef]
- Gonczol, E.; Ianacone, J.; Ho, W.; Starr, S.; Meignier, B.; Plotkin, S. Isolated gA/gB glycoprotein complex of human cytomegalovirus envelope induces humoral and cellular immune-responses in human volunteers. Vaccine 1990, 8, 130–136. [Google Scholar] [CrossRef]
- Pass, R.F.; Duliegè, A.; Boppana, S.; Sekulovich, R.; Percell, S.; Britt, W.; Burke, R.L. A Subunit Cytomegalovirus Vaccine Based on Recombinant Envelope Glycoprotein B and a New Adjuvant. J. Infect. Dis. 1999, 180, 970–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, G.; Barchfeld, G.L.; Chernoff, D.; Radhakrishnan, R.; Van Hoogevest, P.; Van Nest, G. MF59 Design and Evaluation of a Safe and Potent Adjuvant for Human Vaccines. In Pharmaceutical Biotechnology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1995; Volume 6, pp. 277–296. [Google Scholar]
- Frey, S.E.; Harrison, C.; Pass, R.F.; Yang, E.; Boken, D.; Sekulovich, R.E.; Percell, S.; Izu, A.E.; Hirabayashi, S.; Burke, R.L.; et al. Effects of Antigen Dose and Immunization Regimens on Antibody Responses to a Cytomegalovirus Glycoprotein B Subunit Vaccine. J. Infect. Dis. 1999, 180, 1700–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, D.K.; Holmes, S.J.; Burke, R.L.; Duliege, A.M.; Adler, S.P. Immunogenicity of a recombinant human cytomegalovirus gB vaccine in seronegative toddlers. Pediatr. Infect. Dis. J. 2002, 21, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Sabbaj, S.; Pass, R.; Pichon, S.; Goepfert, P.A. Glycoprotein B Vaccine Is Capable of Boosting Both Antibody and CD4 T-Cell Responses to Cytomegalovirus in Chronically Infected Women. J. Infect. Dis. 2011, 203, 1534–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- City of Hope’s Triplex Vaccine Reduces Rate of CMV Complications in Transplant Recipients by 50 Percent. Available online: https://www.cityofhope.org/news/annals-of-internal-medicine-study-on-cmv-triplex-vaccine (accessed on 28 April 2021).
- Schwendinger, M.; Thiry, G.; De Vos, B.; Leroux-Roels, G.; Bruhwyler, J.; Huygens, A.; Ganeff, C.; Buchinger, H.; Orlinger, K.K.; Pinschewer, D.D.; et al. A Randomized Dose-Escalating Phase I Trial of a Replication-Deficient Lymphocytic Choriomeningitis Virus Vector-Based Vaccine Against Human Cytomegalovirus. J. Infect. Dis. 2020, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleiss, M.R.; Permar, S.R.; Plotkin, S.A. Progress toward Development of a Vaccine against Congenital Cytomegalovirus Infection. Clin. Vaccine Immunol. 2017, 24, e00268-17. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, C.; Krishnan, R.; Markel, S.; Schneck, J.P.; Houghten, R.; Pinilla, C.; Diamond, D. Enhanced immune activity of cytotoxic T-lymphocyte epitope analogs derived from positional scanning synthetic combinatorial libraries. Blood 2001, 97, 1776–1786. [Google Scholar] [CrossRef] [Green Version]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Zaia, J.A.; Gallez-Hawkins, G.; Li, X.; Yao, Z.-Q.; Lomeli, N.A.; Molinder, K.M.; La Rosa, C.; Diamond, D. Infrequent Occurrence of Natural Mutations in the pp65495–503 Epitope Sequence Presented by the HLA A*0201 Allele among Human Cytomegalovirus Isolates. J. Virol. 2001, 75, 2472–2474. [Google Scholar] [CrossRef] [Green Version]
- Wloch, M.K.; Smith, L.R.; Boutsaboualoy, S.; Reyes, L.; Han, C.; Kehler, J.; Smith, H.D.; Selk, L.; Nakamura, R.; Brown, J.M.; et al. Safety and Immunogenicity of a Bivalent Cytomegalovirus DNA Vaccine in Healthy Adult Subjects. J. Infect. Dis. 2008, 197, 1634–1642. [Google Scholar] [CrossRef]
- La Rosa, C.; Wang, Z.; Brewer, J.C.; Lacey, S.F.; Villacres, M.C.; Sharan, R.; Krishnan, R.; Crooks, M.; Markel, S.; Maas, R.; et al. Preclinical development of an adjuvant-free peptide vaccine with activity against CMV pp65 in HLA transgenic mice. Blood 2002, 100, 3681–3689. [Google Scholar] [CrossRef] [Green Version]
- Gratama, J.W.; Boeckh, M.; Nakamura, R.; Cornelissen, J.J.; Brooimans, R.A.; Zaia, J.A.; Forman, S.J.; Gaal, K.; Bray, K.R.; Gasior, G.H.; et al. Immune monitoring with iTAg MHC Tetramers for prediction of recurrent or persistent cytomegalovirus infection or disease in allogeneic hematopoietic stem cell transplant recipients: A prospective multicenter study. Blood 2010, 116, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.Y.H.; Chow, H.C.H.; Kwok, J.S.Y.; Lui, C.K.H.; Cheng, V.C.C.; Yuen, K.-Y.; Lie, A.K.W.; Liang, R. Safety of vaccinating sibling donors with live-attenuated varicella zoster vaccine before hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007, 39, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, M.; Barsegian, V.; Runde, V.; Fiedler, M.; Heermann, K.-H.; Schaefer, U.W.; Roggendorf, M.; Grosse-Wilde, H. Transfer of humoral and cellular hepatitis B immunity by allogeneic hematopoietic cell transplantation. Transplantology 2003, 75, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Teira, P.; Battiwalla, M.; Ramanathan, M.; Barrett, A.J.; Ahn, K.W.; Chen, M.; Green, J.S.; Saad, A.; Antin, J.H.; Savani, B.N.; et al. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: A CIBMTR analysis. Blood 2016, 127, 2427–2438. [Google Scholar] [CrossRef]
- Zhou, W.; Longmate, J.; Lacey, S.F.; Palmer, J.M.; Gallez-Hawkins, G.; Thao, L.; Spielberger, R.; Nakamura, R.; Forman, S.J.; Zaia, J.A.; et al. Impact of donor CMV status on viral infection and reconstitution of multifunction CMV-specific T cells in CMV-positive transplant recipients. Blood 2009, 113, 6465–6476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchmeier, M.; Fluckiger, A.-C.; Soare, C.; Bozic, J.; Ontsouka, B.; Ahmed, T.; Diress, A.; Pereira, L.; Schödel, F.; Plotkin, S.; et al. Enveloped Virus-Like Particle Expression of Human Cytomegalovirus Glycoprotein B Antigen Induces Antibodies with Potent and Broad Neutralizing Activity. Clin. Vaccine Immunol. 2014, 21, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Selinsky, C.; Luke, C.; Wloch, M.; Geall, A.; Hermanson, G.; Kaslow, D.; Evans, T. A DNA-based vaccine for the prevention of human cytomegalovirus-associated diseases. Hum. Vaccines 2005, 1, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.R.; Wloch, M.K.; Chaplin, J.A.; Gerber, M.; Rolland, A.P. Clinical Development of a Cytomegalovirus DNA Vaccine: From Product Concept to Pivotal Phase 3 Trial. Vaccines 2013, 1, 398–414. [Google Scholar] [CrossRef] [Green Version]
- Vincenti, F.; Budde, K.; Merville, P.; Shihab, F.; Peddi, V.R.; Shah, M.; Wyburn, K.; Cassuto-Viguier, E.; Weidemann, A.; Lee, M.; et al. A randomized, phase 2 study of ASP0113, a DNA-based vaccine, for the prevention of CMV in CMV-seronegative kidney transplant recipients receiving a kidney from a CMV-seropositive donor. Arab. Archaeol. Epigr. 2018, 18, 2945–2954. [Google Scholar] [CrossRef]
- University of California, San Francisco. Randomized, Phase 1 Trial to Evaluate Safety and CMV-Specific Immune Response to a PDNA CMV Trivalent Vaccine (VCL-CT02) Followed by Towne CMV Vaccine (Towne) Challenge in Healthy, CMV—Seronegative Adults; Clinical Trial Registration NCT00373412. 2008. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00373412 (accessed on 24 May 2021).
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Leppek, K.; Byeon, G.W.; Kladwang, W.; Wayment-Steele, H.K.; Kerr, C.H.; Xu, A.F.; Kim, D.S.; Topkar, V.V.; Choe, C.; Rothschild, D.; et al. Combinatorial Optimization of MRNA Structure, Stability, and Translation for RNA-Based Therapeutics. Molec. Biol. 2021. [Google Scholar] [CrossRef]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, S.; Yuzhakov, O.; Woods, A.; Deterling, J.; Hassett, K.; Shaw, C.A.; Ciaramella, G. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 2018, 36, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Moderna Completes Enrollment of Cytomegalovirus (CMV) Vaccine (mRNA-1647) Phase 2 Study Moderna, Inc. Available online: https://investors.modernatx.com/news-releases/news-release-details/moderna-completes-enrollment-cytomegalovirus-cmv-vaccine-mrna/ (accessed on 28 April 2021).
- Sommerer, C.; Schmitt, A.; Hückelhoven-Krauss, A.; Giese, T.; Bruckner, T.; Wang, L.; Schnitzler, P.; Meuer, S.; Zeier, M.; Schmitt, M. Peptide Vaccination Against Cytomegalovirus Induces Specific T Cell Response in Responses in CMV Seronegative End-Stage Renal Disease Patients. Vaccines 2021, 9, 133. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Name | Type of Vaccine | Target Site or Antigen | Study Population | Phase, Ref. | Study ID (NCT) | Enrolment Time | Summary of Results |
|---|---|---|---|---|---|---|---|
| 190 healthy adults older than 18 years: 95 hCMV-seropositive and 95 hCMV-seronegative | Phase I [111] | NCT01986010 | November 2013–March 2017 | Acceptable safety profile. Levels of antibodies and T cell responses in hCMV-seronegative individuals were within ranges observed after natural CMV infection. | |||
| V160 Merck Sharp & Dohme Corp | Live-attenuated | AD169 genetically engineered to express PC | 18 healthy males (20–64 years aged), hCMV-seropositive and seronegative | Phase I [112] | NCT03840174 | March 2019–November 2019 | NA |
| 2220 females of childbearing age (16–35 years aged), hCMV seronegative | Phase II [113] | NCT03486834 | April 2018–ongoing | NA | |||
| 464 hCMV-seronegative women within 1 year after delivery | Phase II [114] | NCT00125502 | August 1999–January 2010 | Vaccine efficacy: 50%; more local reactions and systemic reactions in the vaccine group than in the placebo group | |||
| CMV gB/MF59 Sanofi Pasteur | Recombinant subunit | gB with MF59 | 409 hCMV seronegative adolescent females | Phase II [115] | NCT00133497 | June 2006–June 2013 | Vaccine efficacy 43%. Safe and immunogenic (although no conventional levels of significance) |
| 140 adults waiting for SOT (>18 years of age) | Phase II [116] | NCT00299260 | August 2006–September 2011 | gB antibody titers significantly increased in vaccine than the placebo group | |||
| CMV-MVA Triplex City of Hope | Virus vectored (MVA) | pp65, IE1-exon4, IE2-exon5 | 24 healthy adults (18–60 years), hCMV-seropositive and seronegative | Phase I [117] | NA | NA | Well tolerated with no dose-limiting toxicities; elicit expansions of hCMV-specific T cells, also in hCMV-seronegative subjects and in adults who previous received smallpox vaccination |
| 102 hCMV-seropositive HSCT recipients at high riskfor hCMV reactivation | Phase II [118] | NCT02506933 | July 2015–January 2021 | The risk for a significant hCMV event during the first 100 days after HSCT was reduced by half; less hCMV reactivations and higher levels of hCMV-specific T cells; no significant adverse event | |||
| 36 donors of hCMV seropositive HSCT recipient | Phase II [119] | NCT03560752 | June 2018–ongoing | NA | |||
| 80 hCMV seropositive children receiving an allogeneic HSCT | Phase I/II [120] | NCT03354728 | May 2018–ongoing | NA | |||
| HB-101 Hookipa Biotech GmbH | Virus vectored (dr) | gB and pp65 | 54 healthy adults (18–45 years) hCMV seronegative | Phase I [121] | NCT02798692 | June 2016–March 2018 | Well tolerated; induced hCVM-specific cellular responses, principally pp65 specific CD8 T cell, and neutralizing Ab production |
| 150 hCMV seronegative recipient awaiting kidney transplantation from hCMV seropositive donors | Phase II [122] | NCT03629080 | December 2018–ongoing | NA | |||
| pp65 fused to either pan DR helper T lymphocyte epitope or natural tetanus sequence | 68 healthy adults (18–55 years), HLA A*0201 subtyped, hCMV seropositive or seronegative | Phase I [123] | NCT00722839 | December 2006–April 2012 | No serious adverse events. Immune responses were detected in hCMV-seropositive subjects who received the vaccine co-administered with PF03512676. | ||
| CMVPepVax City of Hope, National Cancer Institute | Chimeric peptidic | pp65 fused to a natural tetanus sequence | 36 patients (18–75 years), HLA A*0201 subtyped, hCMV seropositive, who undergone HSCT | Phase Ib [124] | NCT01588015 | August 2012–November 2014 | Acceptable safety profile. Patients allocated the vaccine had less hCMV reactivation, lower necessity of antiviral use, and better relapse-free survival. |
| pp65 fused to a natural tetanus sequence | 133 patients (18–75 years), HLA A*0201 subtyped, hCMV seropositive, post-HSCT | Phase II [125] | NCT02396134 | May 2015–May 2019 | NA | ||
| VBI-1501 VBI Laboratories | Enveloped virus-like particles | gB | 125 healthy adults (18–40 years) hCMV seronegative | Phase I [126] | NCT02826798 | June 2016–August 2017 | Immunogenic at very low doses; amplification of neutralizing Ab titers; no safety problems; |
| 108 hCMV-positive, allogeneic HSCT adult recipients | Phase II [127] | NCT00285259 | January 2006–November 2010 | Well-tolerated, significant reduction in viral load endpoints, no significant reduction in the need of hCMV antiviral therapy | |||
| ASP0113 (VCL-CB01) Astellas | Plasmid-based | gB, pp65 with CRL1005 and benzalkonium chloride | 150 hCMV-seronegative kidney transplant recipients from hCMV-seropositive donors | Phase II [128] | NCT01974206 | November 2013–November 2020 | No statistically significant difference in the primary endpoint between the ASP0113 and placebo groups. |
| 514 hCMV-seropositive recipients undergoing allogeneic HSCT | Phase III [129] | NCT01877655 | September 2013–September 2017 | No significant improvement in overall survival and reduction in hCMV end-organ disease. Well tolerated | |||
| mRNA-1647 Moderna | mRNA | mRNA-1647:gB and PC; mRNA-1443: pp65 | 181 healthy adults (18–49 years), hCMV seropositive and seronegative | Phase I [130] | NCT03382405 | November 2017–October 2020 | Positive seven-month interim safety and immunogenicity data after the third vaccination with mRNA-1647 |
| mRNA-1647: gB and PC | 452 healthy adults (18–40 years), hCMV seropositive and seronegative | Phase II [131] | NCT04232280 | December 2019–ongoing | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpini, S.; Morigi, F.; Betti, L.; Dondi, A.; Biagi, C.; Lanari, M. Development of a Vaccine against Human Cytomegalovirus: Advances, Barriers, and Implications for the Clinical Practice. Vaccines 2021, 9, 551. https://doi.org/10.3390/vaccines9060551
Scarpini S, Morigi F, Betti L, Dondi A, Biagi C, Lanari M. Development of a Vaccine against Human Cytomegalovirus: Advances, Barriers, and Implications for the Clinical Practice. Vaccines. 2021; 9(6):551. https://doi.org/10.3390/vaccines9060551
Chicago/Turabian StyleScarpini, Sara, Francesca Morigi, Ludovica Betti, Arianna Dondi, Carlotta Biagi, and Marcello Lanari. 2021. "Development of a Vaccine against Human Cytomegalovirus: Advances, Barriers, and Implications for the Clinical Practice" Vaccines 9, no. 6: 551. https://doi.org/10.3390/vaccines9060551