
Short or Long Interval between Priming and Boosting: Does It Impact on the Vaccine Immunogenicity?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
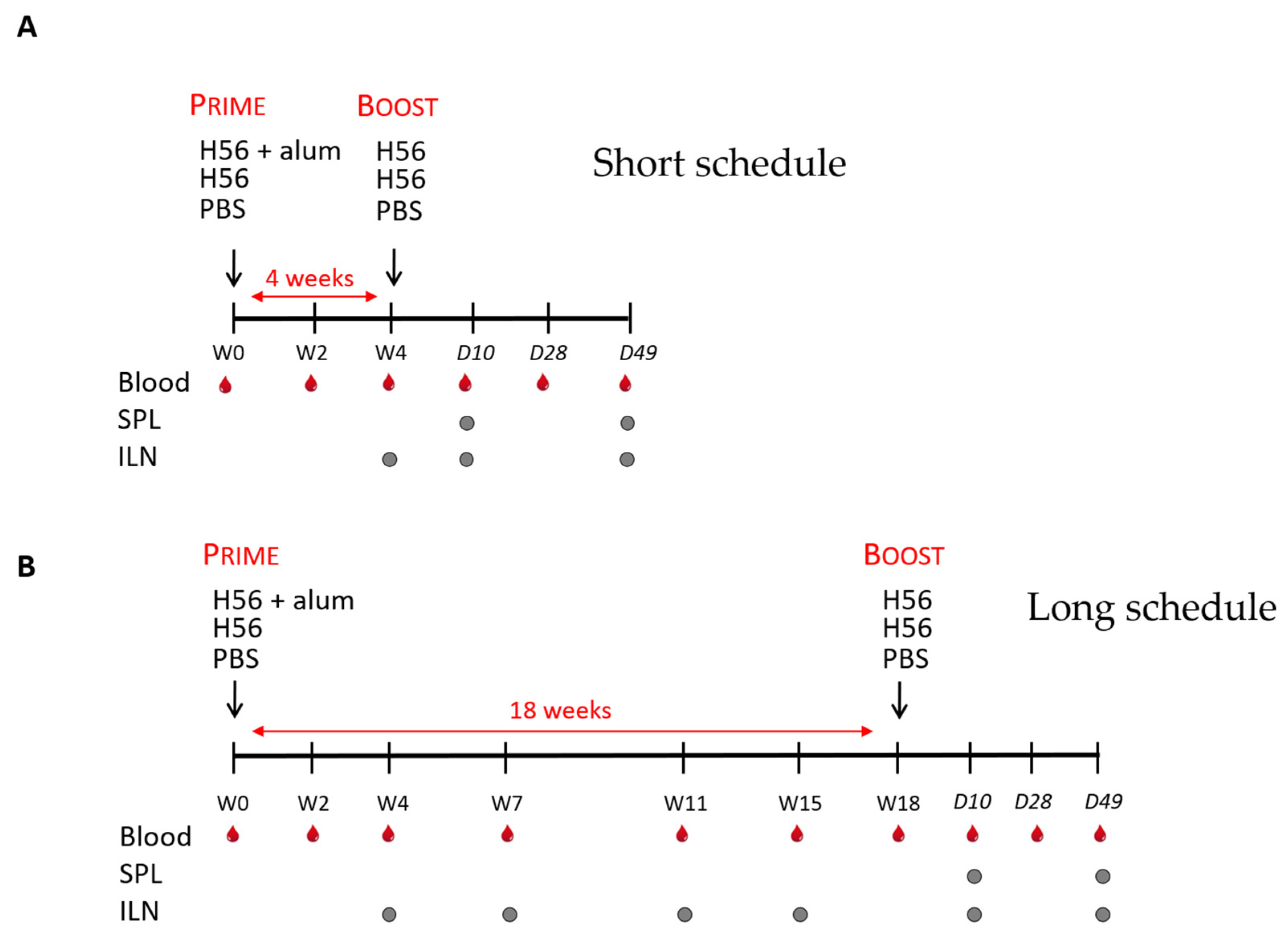
2.2. Immunizations
2.3. Sample Collection and Cell Preparation
2.4. Multiparametric Flow Cytometric Analysis
2.5. B-Cell ELISPOT
2.6. ELISA
2.7. Statistical Analysis
3. Results
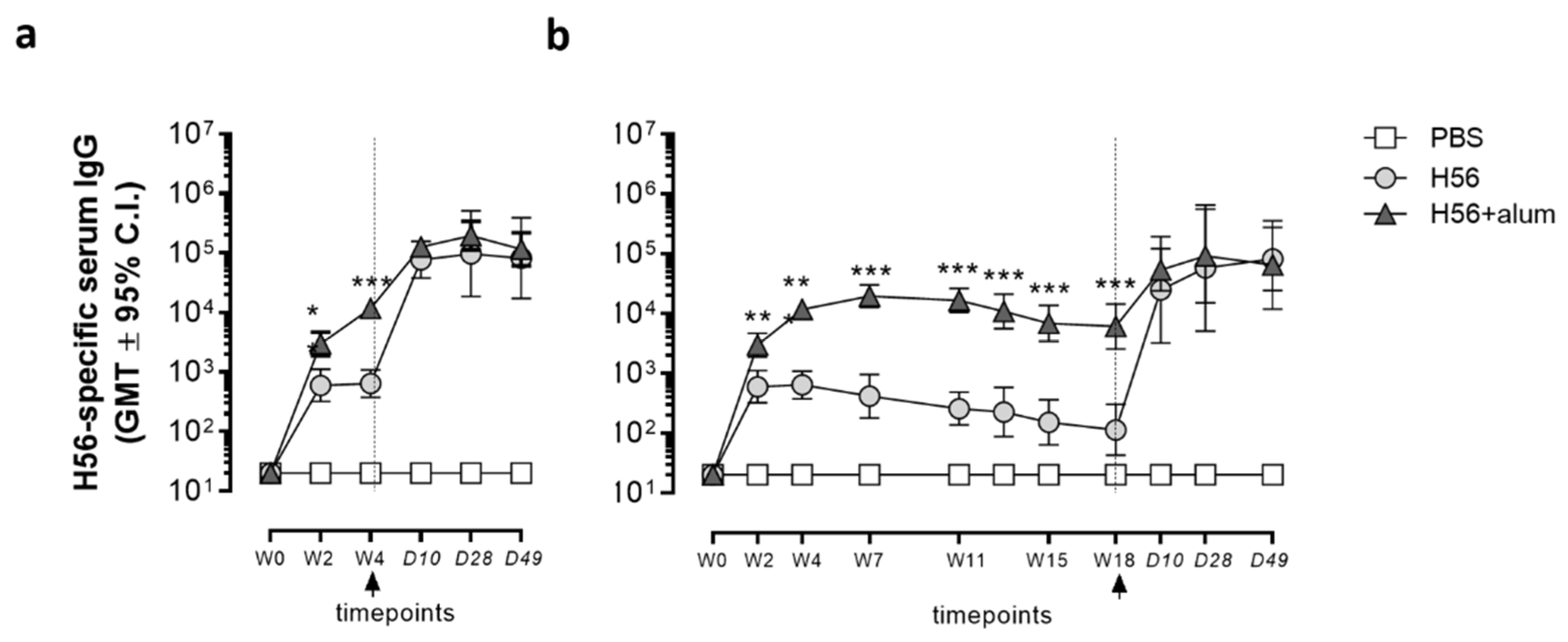
3.1. Humoral Immune Response
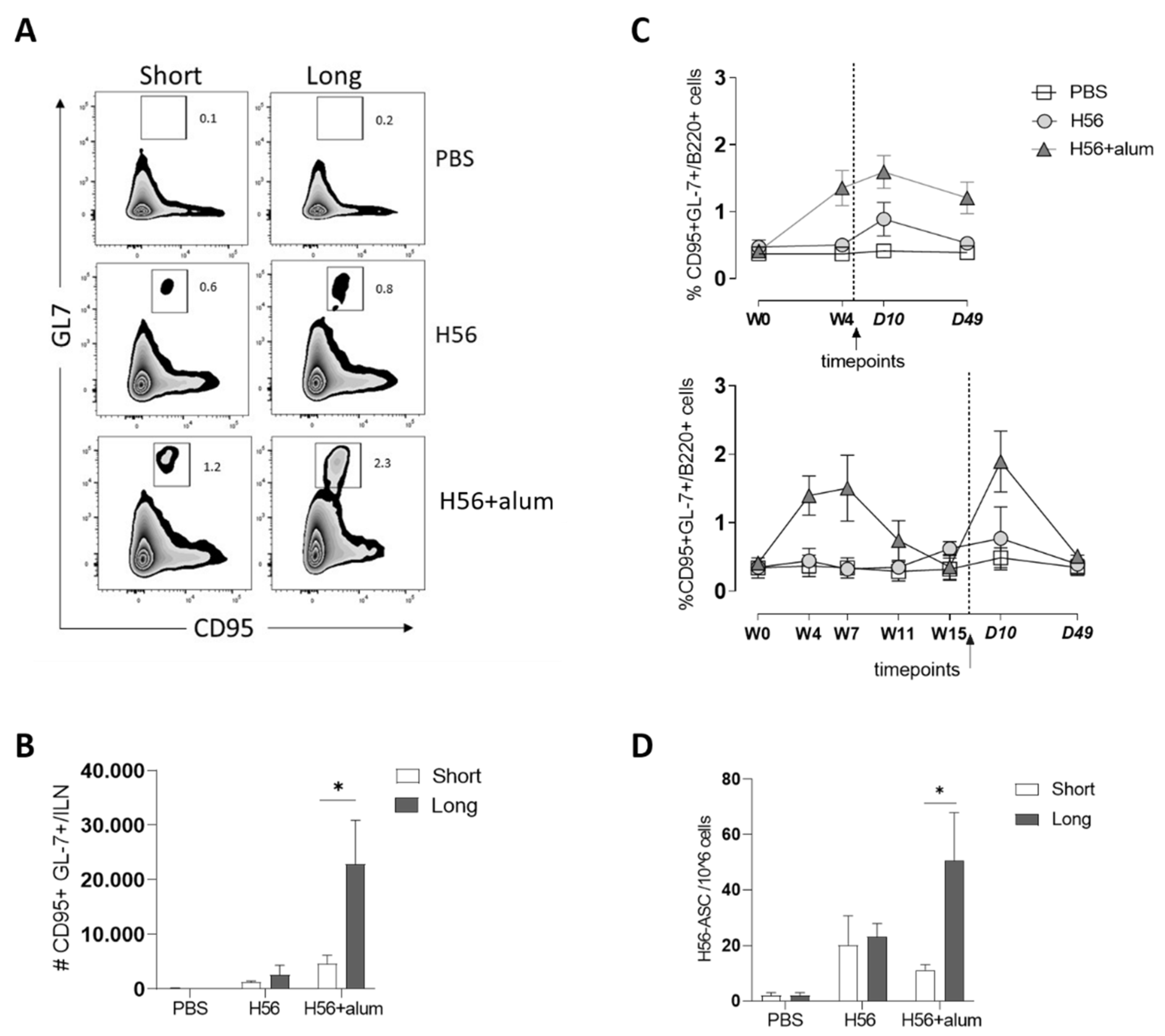
3.2. B-Cell Response
3.3. Ag-Specific CD4+ T Cell Response
3.4. CD4+ T Cell Multifunctional Profile
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Ciabattini, A.; Pettini, E.; Medaglini, D. CD4(+) T Cell Priming as Biomarker to Study Immune Response to Preventive Vaccines. Front. Immunol. 2013, 4, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciabattini, A.; Pettini, E.; Fiorino, F.; Pastore, G.; Andersen, P.; Pozzi, G.; Medaglini, D. Modulation of Primary Immune Response by Different Vaccine Adjuvants. Front. Immunol. 2016, 7, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciabattini, A.; Pettini, E.; Fiorino, F.; Lucchesi, S.; Pastore, G.; Brunetti, J.; Santoro, F.; Andersen, P.; Bracci, L.; Pozzi, G.; et al. Heterologous Prime-Boost Combinations Highlight the Crucial Role of Adjuvant in Priming the Immune System. Front. Immunol. 2018, 9, 380. [Google Scholar] [CrossRef] [PubMed]
- Fiorino, F.; Pettini, E.; Pozzi, G.; Medaglini, D.; Ciabattini, A. Prime-Boost Strategies in Mucosal Immunization Affect Local IgA Production and the Type of Th Response. Front. Immunol. 2013, 4, 128. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, A.L.; Garçon, N.; Leo, O.; Friedland, L.R.; Strugnell, R.; Laupèze, B.; Doherty, M.; Stern, P. Vaccine Development: From Concept to Early Clinical Testing. Vaccine 2016, 34, 6655–6664. [Google Scholar] [CrossRef]
- Fiorino, F.; Rondini, S.; Micoli, F.; Lanzilao, L.; Alfini, R.; Mancini, F.; MacLennan, C.A.; Medaglini, D. Immunogenicity of a Bivalent Adjuvanted Glycoconjugate Vaccine against Salmonella Typhimurium and Salmonella Enteritidis. Front. Immunol. 2017, 8, 168. [Google Scholar] [CrossRef] [Green Version]
- Harandi, A.M.; Medaglini, D. Mucosal Adjuvants. Curr. HIV Res. 2010, 8, 330–335. [Google Scholar] [CrossRef]
- Ciabattini, A.; Pettini, E.; Arsenijevic, S.; Pozzi, G.; Medaglini, D. Intranasal Immunization with Vaccine Vector Streptococcus Gordonii Elicits Primed CD4+ and CD8+ T Cells in the Genital and Intestinal Tracts. Vaccine 2010, 28, 1226–1233. [Google Scholar] [CrossRef]
- Ciabattini, A.; Giomarelli, B.; Parigi, R.; Chiavolini, D.; Pettini, E.; Aricò, B.; Giuliani, M.M.; Santini, L.; Medaglini, D.; Pozzi, G. Intranasal Immunization of Mice with Recombinant Streptococcus Gordonii Expressing NadA of Neisseria Meningitidis Induces Systemic Bactericidal Antibodies and Local IgA. Vaccine 2008, 26, 4244–4250. [Google Scholar] [CrossRef]
- Santoro, F.; Pettini, E.; Kazmin, D.; Ciabattini, A.; Fiorino, F.; Gilfillan, G.D.; Evenroed, I.M.; Andersen, P.; Pozzi, G.; Medaglini, D. Transcriptomics of the Vaccine Immune Response: Priming With Adjuvant Modulates Recall Innate Responses After Boosting. Front. Immunol. 2018, 9, 1248. [Google Scholar] [CrossRef]
- Lu, S. Heterologous Prime-Boost Vaccination. Curr. Opin. Immunol. 2009, 21, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Mare, A.; Lambeck, A.J.A.; Regts, J.; van Dam, G.M.; Nijman, H.W.; Snippe, H.; Wilschut, J.; Daemen, T. Viral Vector-Based Prime-Boost Immunization Regimens: A Possible Involvement of T-Cell Competition. Gene Ther. 2008, 15, 393–403. [Google Scholar] [CrossRef]
- Monath, T.P.; Vasconcelos, P.F.C. Yellow Fever. J. Clin. Virol. 2015, 64, 160–173. [Google Scholar] [CrossRef]
- Mutua, G.; Anzala, O.; Luhn, K.; Robinson, C.; Bockstal, V.; Anumendem, D.; Douoguih, M. Safety and Immunogenicity of a 2-Dose Heterologous Vaccine Regimen With Ad26.ZEBOV and MVA-BN-Filo Ebola Vaccines: 12-Month Data From a Phase 1 Randomized Clinical Trial in Nairobi, Kenya. J. Infect. Dis. 2019, 220, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Mensah, V.A.; Roetynck, S.; Kanteh, E.K.; Bowyer, G.; Ndaw, A.; Oko, F.; Bliss, C.M.; Jagne, Y.J.; Cortese, R.; Nicosia, A.; et al. Safety and Immunogenicity of Malaria Vectored Vaccines Given with Routine Expanded Program on Immunization Vaccines in Gambian Infants and Neonates: A Randomized Controlled Trial. Front. Immunol. 2017, 8, 1551. [Google Scholar] [CrossRef] [Green Version]
- Jones, I.; Roy, P. Sputnik V COVID-19 Vaccine Candidate Appears Safe and Effective. Lancet 2021, 397, 642–643. [Google Scholar] [CrossRef]
- Kolibab, K.; Yang, A.; Derrick, S.C.; Waldmann, T.A.; Perera, L.P.; Morris, S.L. Highly Persistent and Effective Prime/Boost Regimens against Tuberculosis That Use a Multivalent Modified Vaccine Virus Ankara-Based Tuberculosis Vaccine with Interleukin-15 as a Molecular Adjuvant. CVI 2010, 17, 793–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boianelli, A.; Pettini, E.; Prota, G.; Medaglini, D.; Vicino, A. A Stochastic Model for CD4+ T Cell Proliferation and Dissemination Network in Primary Immune Response. PLoS ONE 2015, 10, e0135787. [Google Scholar] [CrossRef] [PubMed]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef] [Green Version]
- Castiglione, F.; Mantile, F.; De Berardinis, P.; Prisco, A. How the Interval between Prime and Boost Injection Affects the Immune Response in a Computational Model of the Immune System. Comput. Math. Methods Med. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Röst, G.; Moghadas, S.M. Delay in Booster Schedule as a Control Parameter in Vaccination Dynamics. J. Math. Biol. 2019, 79, 2157–2182. [Google Scholar] [CrossRef] [Green Version]
- Ciabattini, A.; Olivieri, R.; Lazzeri, E.; Medaglini, D. Role of the Microbiota in the Modulation of Vaccine Immune Responses. Front. Microbiol. 2019, 10, 1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciabattini, A.; Garagnani, P.; Santoro, F.; Rappuoli, R.; Franceschi, C.; Medaglini, D. Shelter from the Cytokine Storm: Pitfalls and Prospects in the Development of SARS-CoV-2 Vaccines for an Elderly Population. Semin. Immunopathol. 2020, 42, 619–634. [Google Scholar] [CrossRef]
- Zimmermann, P.; Curtis, N. Factors That Influence the Immune Response to Vaccination. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [Green Version]
- Pauthner, M.; Havenar-Daughton, C.; Sok, D.; Nkolola, J.P.; Bastidas, R.; Boopathy, A.V.; Carnathan, D.G.; Chandrashekar, A.; Cirelli, K.M.; Cottrell, C.A.; et al. Elicitation of Robust Tier 2 Neutralizing Antibody Responses in Nonhuman Primates by HIV Envelope Trimer Immunization Using Optimized Approaches. Immunity 2017, 46, 1073–1088.e6. [Google Scholar] [CrossRef] [Green Version]
- Palgen, J.-L.; Tchitchek, N.; Rodriguez-Pozo, A.; Jouhault, Q.; Abdelhouahab, H.; Dereuddre-Bosquet, N.; Contreras, V.; Martinon, F.; Cosma, A.; Lévy, Y.; et al. Innate and Secondary Humoral Responses Are Improved by Increasing the Time between MVA Vaccine Immunizations. NPJ Vaccines 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belshe, R.B.; Frey, S.E.; Graham, I.; Mulligan, M.J.; Edupuganti, S.; Jackson, L.A.; Wald, A.; Poland, G.; Jacobson, R.; Keyserling, H.L.; et al. Safety and Immunogenicity of Influenza A H5 Subunit Vaccines: Effect of Vaccine Schedule and Antigenic Variant. J. Infect. Dis. 2011, 203, 666–673. [Google Scholar] [CrossRef]
- Lin, L.; Koren, M.A.; Paolino, K.M.; Eckels, K.H.; De La Barrera, R.; Friberg, H.; Currier, J.R.; Gromowski, G.D.; Aronson, N.E.; Keiser, P.B.; et al. Immunogenicity of a Live-Attenuated Dengue Vaccine Using a Heterologous Prime-Boost Strategy in a Phase I Randomized Clinical Trial. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.A.; Frey, S.E.; El Sahly, H.M.; Mulligan, M.J.; Winokur, P.L.; Kotloff, K.L.; Campbell, J.D.; Atmar, R.L.; Graham, I.; Anderson, E.J.; et al. Safety and Immunogenicity of a Modified Vaccinia Ankara Vaccine Using Three Immunization Schedules and Two Modes of Delivery: A Randomized Clinical Non-Inferiority Trial. Vaccine 2017, 35, 1675–1682. [Google Scholar] [CrossRef]
- Venkatraman, N.; Ndiaye, B.P.; Bowyer, G.; Wade, D.; Sridhar, S.; Wright, D.; Powlson, J.; Ndiaye, I.; Dièye, S.; Thompson, C.; et al. Safety and Immunogenicity of a Heterologous Prime-Boost Ebola Virus Vaccine Regimen in Healthy Adults in the United Kingdom and Senegal. J. Infect. Dis. 2019, 219, 1187–1197. [Google Scholar] [CrossRef]
- Pitisuttithum, P.; Nitayaphan, S.; Chariyalertsak, S.; Kaewkungwal, J.; Dawson, P.; Dhitavat, J.; Phonrat, B.; Akapirat, S.; Karasavvas, N.; Wieczorek, L.; et al. Late Boosting of the RV144 Regimen with AIDSVAX B/E and ALVAC-HIV in HIV-Uninfected Thai Volunteers: A Double-Blind, Randomised Controlled Trial. Lancet HIV 2020, 7, e238–e248. [Google Scholar] [CrossRef]
- Frey, S.E.; Winokur, P.L.; Salata, R.A.; El-Kamary, S.S.; Turley, C.B.; Walter, E.B.; Hay, C.M.; Newman, F.K.; Hill, H.R.; Zhang, Y.; et al. Safety and Immunogenicity of IMVAMUNE® Smallpox Vaccine Using Different Strategies for a Post Event Scenario. Vaccine 2013, 31, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Sallusto, F.; Lanzavecchia, A.; Araki, K.; Ahmed, R. From Vaccines to Memory and Back. Immunity 2010, 33, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and Efficacy of the ChAdOx1 NCoV-19 Vaccine (AZD1222) against SARS-CoV-2: An Interim Analysis of Four Randomised Controlled Trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Aagaard, C.; Hoang, T.; Dietrich, J.; Cardona, P.-J.; Izzo, A.; Dolganov, G.; Schoolnik, G.K.; Cassidy, J.P.; Billeskov, R.; Andersen, P. A Multistage Tuberculosis Vaccine That Confers Efficient Protection before and after Exposure. Nat. Med. 2011, 17, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Prota, G.; Christensen, D.; Andersen, P.; Medaglini, D.; Ciabattini, A. Peptide-Specific T Helper Cells Identified by MHC Class II Tetramers Differentiate into Several Subtypes upon Immunization with CAF01 Adjuvanted H56 Tuberculosis Vaccine Formulation. Vaccine 2015, 33, 6823–6830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, C.J.; Griffiths, E. The Global Impact of Vaccines Containing Aluminium Adjuvants. Vaccine 2002, 20 (Suppl. 3), S24–S33. [Google Scholar] [CrossRef]
- Mbow, M.L.; De Gregorio, E.; Ulmer, J.B. Alum’s Adjuvant Action: Grease Is the Word. Nat. Med. 2011, 17, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Nussenzweig, M.C. Germinal Centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef]
- Huang, C. Germinal Center Reaction. Adv. Exp. Med. Biol. 2020, 1254, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; SoRelle, J.A.; Choi, J.H.; Yue, T.; Wang, K.; McAlpine, W.; Wang, J.; Liu, A.; Tabeta, K.; Turer, E.; et al. Mutual Inhibition between Prkd2 and Bcl6 Controls T Follicular Helper Cell Differentiation. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef] [PubMed]
- Ehreth, J. The Value of Vaccination: A Global Perspective. Vaccine 2003, 21, 4105–4117. [Google Scholar] [CrossRef]
- Lucchesi, S.; Nolfi, E.; Pettini, E.; Pastore, G.; Fiorino, F.; Pozzi, G.; Medaglini, D.; Ciabattini, A. Computational Analysis of Multiparametric Flow Cytometric Data to Dissect B Cell Subsets in Vaccine Studies. Cytom. A 2020, 97, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.; Carrington, E.M.; Zhang, Y.; Heinzel, S.; Lew, A.M. Life and Death of Activated T Cells: How Are They Different from Naïve T Cells? Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlach, C.; van Heijst, J.W.J.; Schumacher, T.N.M. The Descent of Memory T Cells. Ann. N. Y. Acad. Sci. 2011, 1217, 139–153. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pettini, E.; Pastore, G.; Fiorino, F.; Medaglini, D.; Ciabattini, A. Short or Long Interval between Priming and Boosting: Does It Impact on the Vaccine Immunogenicity? Vaccines 2021, 9, 289. https://doi.org/10.3390/vaccines9030289
Pettini E, Pastore G, Fiorino F, Medaglini D, Ciabattini A. Short or Long Interval between Priming and Boosting: Does It Impact on the Vaccine Immunogenicity? Vaccines. 2021; 9(3):289. https://doi.org/10.3390/vaccines9030289
Chicago/Turabian StylePettini, Elena, Gabiria Pastore, Fabio Fiorino, Donata Medaglini, and Annalisa Ciabattini. 2021. "Short or Long Interval between Priming and Boosting: Does It Impact on the Vaccine Immunogenicity?" Vaccines 9, no. 3: 289. https://doi.org/10.3390/vaccines9030289