
Metabolic Rewiring in the Tumor Microenvironment to Support Immunotherapy: A Focus on Neutrophils, Polymorphonuclear Myeloid-Derived Suppressor Cells and Natural Killer Cells

 , and
, and 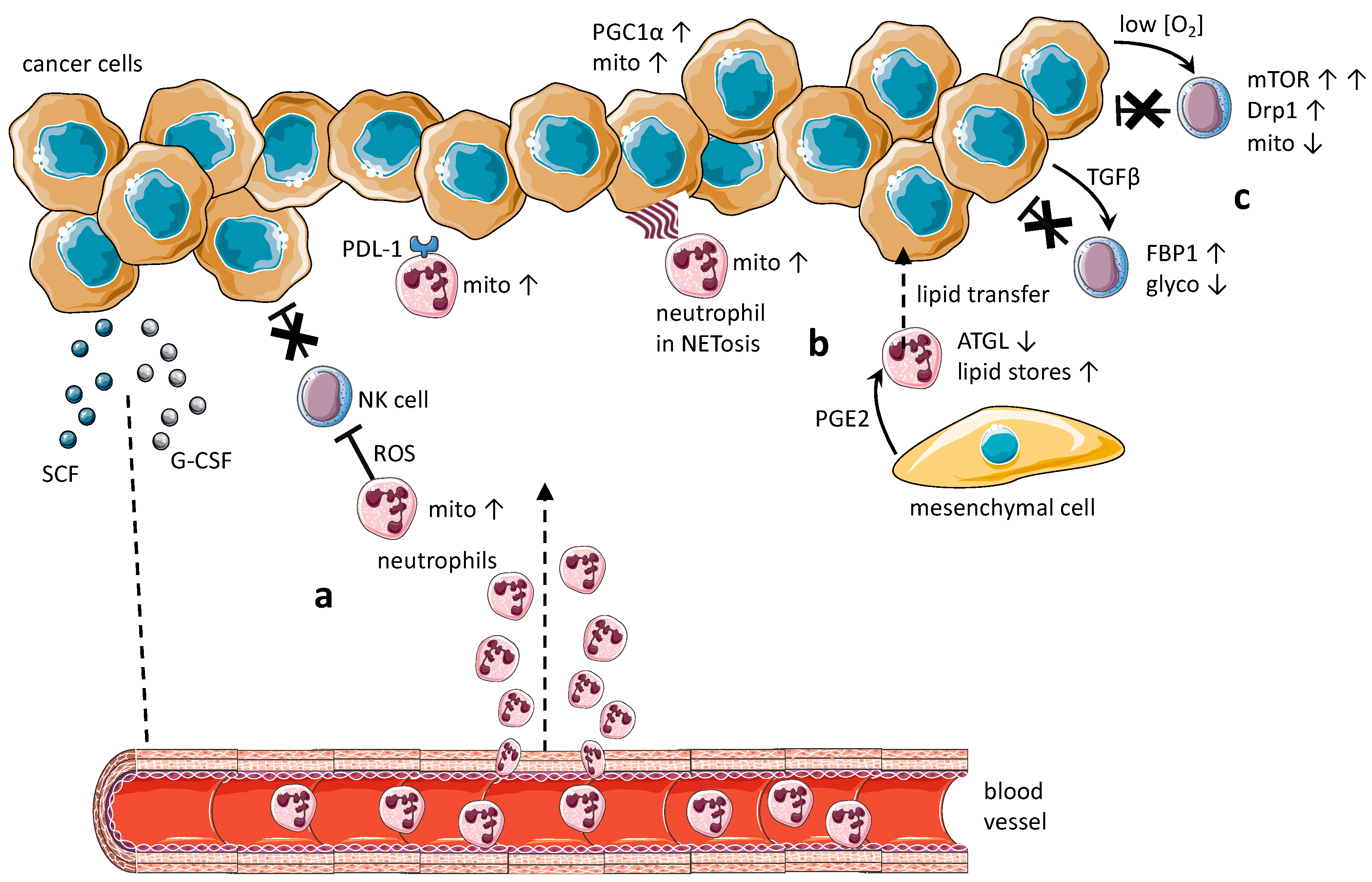{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neutrophils and Polymorphonuclear MDSCs in Health and in Cancer
3. NK Cells in Health and in Cancer
4. Metabolism and Cancer, a Focus on Neutrophils and NK Cells
4.1. Metabolism in Immune Cells
4.2. Neutrophil Metabolism
4.3. Neutrophil Metabolism in Cancer
4.4. NK Cell Metabolism
4.5. NK Cell Metabolism in Cancer
4.6. Metabolic Cooperation in the TME between Neutrophils and Cancer Cells
4.7. Do NK Cells Exert an Editing Function on Cancer Cell Metabolism through Cytotoxic Activity?
4.8. Crosstalk between Neutrophils or PMN-MDSCs and NK Cells and Cancer Metabolism
5. Targeting Tumor Metabolism: A Focus on Neutrophils and NK Cells
5.1. Boosting Metabolism in Anti-Cancer NK Cells
5.2. Targeting Neutrophil/MDSC Metabolism
6. Considerations for Targeting Tumor Metabolism: A Focus on Neutrophils, PMN-MDSCs and NK Cells
6.1. Coexistence of Different Metabolic Pathways in the Same Cell
6.2. Cellular Heterogeneity in the TME
6.3. Pathophysiological MDSCs
6.4. Immunosuppressive Activity of MDSCs on T Cell Responses
6.5. Modulation of Lipid Metabolism, Similar Outcomes in NK Cells and MDSCs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADCC | Antibody-dependent cellular cytotoxicity |
| ATC | Adoptive cell transfer |
| ATGL | Adipose triglyceride lipase |
| ATP | Adenosine triphosphate |
| CAR | Chimeric antigen receptors |
| CeO2 | Cesium oxide |
| CPT1 | Carnitine palmitoyl transferase 1 |
| DCA | Dichloroacetate |
| dNK | Decidual natural killer |
| Drp1 | Dynamin-related protein1 |
| ECM | Extracellular matrix |
| FAO | Fatty acid oxidation |
| FATP2 | Fatty acid transport protein 2 |
| FBP-1 | Fructose-1,6-bisphosphatase 1 |
| GM-CSF | Granulocyte–macrophage colony-stimulating factor |
| IFNγ | Interferon gamma |
| IL | Interleukin |
| IL-15R | Interleukin-15 receptor |
| iLDNs | Immature low-density neutrophils |
| iPSC | Induced pluripotent stem cells |
| LDH-A | Lactate deydrogenase-A |
| M1/2 | M1/2 macrophage |
| MDSCs | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| MPO | Myeloperoxidase |
| mTOR | Mammalian target of rapamycin |
| N1/2 | N1/2 neutrophils |
| NADPH | Nicotinamide adenine dinucleotide phosphate hydrogen |
| NETs | Neutrophil extracellular traps |
| NK | Natural killer |
| OXPHO | Oxidative phosphorylation |
| P2RX1 | Purinergic receptor P2X 1 |
| PaD4-KO | Peptidyl arginine deiminase 4 knock out |
| PD-1 | Programmed cell death protein 1 |
| PDK | Pyruvate dehydrogenase kinase |
| PDL-1 | Programmed death ligand 1 |
| PGC-1a | Peroxisome proliferator-activated receptor gamma coactivator 1 alpha |
| PGE2 | Prostaglandin E2 |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| PUFA | Poly-unsaturated fatty acids |
| ROS | Reactive oxygen species |
| SCF | Stem cell factor |
| SREBP | Sterol regulatory element-binding protein |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAM | Tumor-associated macrophages |
| TAN | Tumor-associated neutrophil |
| TCA | Tricarboxylic acid |
| TGFβ | Transforming growth factor beta |
| Tigit | T cell immunoreceptor with Ig and ITIM domains |
| TIM3 | T-cell immunoglobulin and mucin domain-3 |
| TLR4 | Toll-like receptor 4 |
| TME | Tumor microenvironment |
| TNFα | Tumor necrosis factor alpha |
| TORC1/2 | Target of rapamycin complex1/2 |
| VEGF | Vascular endothelial growth factor |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Cassim, S.; Vucetic, M.; Zdralevic, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Querol, E.; Rosales, C. Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res. 2015, 2015, 983698. [Google Scholar] [CrossRef] [Green Version]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Bassani, B.; Baci, D.; Gallazzi, M.; Poggi, A.; Bruno, A.; Mortara, L. Natural Killer Cells as Key Players of Tumor Progression and Angiogenesis: Old and Novel Tools to Divert Their Pro-Tumor Activities into Potent Anti-Tumor Effects. Cancers 2019, 11, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Xiong, Y.; Liu, H.; Gao, C.; Su, L.; Weng, J.; Yuan, X.; Zhang, D.; Feng, J. Tumor-associated neutrophils suppress antitumor immunity of NK cells through the PD-L1/PD-1 axis. Transl. Oncol. 2020, 13, 100825. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; Cassatella, M.A. The defensive alliance between neutrophils and NK cells as a novel arm of innate immunity. J. Leukoc. Biol. 2011, 89, 221–233. [Google Scholar] [CrossRef]
- Molgora, M.; Supino, D.; Mavilio, D.; Santoni, A.; Moretta, L.; Mantovani, A.; Garlanda, C. The yin-yang of the interaction between myelomonocytic cells and NK cells. Scand. J. Immunol. 2018, 88, e12705. [Google Scholar] [CrossRef] [Green Version]
- Maurer, S.; Ferrari de Andrade, L. NK Cell Interaction With Platelets and Myeloid Cells in the Tumor Milieu. Front. Immunol. 2020, 11, 608849. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiu, L.; Li, Z.; Wang, X.Y.; Yi, H. Understanding the Multifaceted Role of Neutrophils in Cancer and Autoimmune Diseases. Front. Immunol. 2018, 9, 2456. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lu, M.; Shi, J.; Hua, L.; Gong, Z.; Li, Q.; Shultz, L.D.; Ren, G. Dual roles of neutrophils in metastatic colonization are governed by the host NK cell status. Nat. Commun. 2020, 11, 4387. [Google Scholar] [CrossRef] [PubMed]
- Piccard, H.; Muschel, R.J.; Opdenakker, G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit. Rev. Oncol. Hematol. 2012, 82, 296–309. [Google Scholar] [CrossRef]
- Cerezo-Wallis, D.; Ballesteros, I. Neutrophils in cancer, a love-hate affair. FEBS J. 2021. [Google Scholar] [CrossRef]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.F.; Gereke, M.; von Kockritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef]
- Furumaya, C.; Martinez-Sanz, P.; Bouti, P.; Kuijpers, T.W.; Matlung, H.L. Plasticity in Pro- and Anti-tumor Activity of Neutrophils: Shifting the Balance. Front. Immunol. 2020, 11, 2100. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSCs: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef]
- Mehmeti-Ajradini, M.; Bergenfelz, C.; Larsson, A.M.; Carlsson, R.; Riesbeck, K.; Ahl, J.; Janols, H.; Wullt, M.; Bredberg, A.; Kallberg, E.; et al. Human G-MDSCs are neutrophils at distinct maturation stages promoting tumor growth in breast cancer. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Hashimoto, A.; Dweep, H.; Sanseviero, E.; De Leo, A.; Tcyganov, E.; Kossenkov, A.; Mulligan, C.; Nam, B.; Masters, G.; et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J. Exp. Med. 2021, 218, e20201803. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Karre, K. NK cells, MHC class I molecules and the missing self. Scand. J. Immunol. 2002, 55, 221–228. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Blois, S.M.; Klapp, B.F.; Barrientos, G. Decidualization and angiogenesis in early pregnancy: Unravelling the functions of DC and NK cells. J. Reprod Immunol. 2011, 88, 86–92. [Google Scholar] [CrossRef]
- Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I.; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074. [Google Scholar] [CrossRef]
- Ferlazzo, G.; Morandi, B. Cross-Talks between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front. Immunol. 2014, 5, 159. [Google Scholar] [CrossRef] [Green Version]
- Schuster, I.S.; Coudert, J.D.; Andoniou, C.E.; Degli-Esposti, M.A. “Natural Regulators”: NK Cells as Modulators of T Cell Immunity. Front. Immunol. 2016, 7, 235. [Google Scholar] [CrossRef] [Green Version]
- Hasmim, M.; Messai, Y.; Ziani, L.; Thiery, J.; Bouhris, J.H.; Noman, M.Z.; Chouaib, S. Critical Role of Tumor Microenvironment in Shaping NK Cell Functions: Implication of Hypoxic Stress. Front. Immunol. 2015, 6, 482. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, N.H.; Manguso, R.T. Tumor-Derived PGE2 Gives NK Cells a Headache. Immunity 2020, 53, 1131–1132. [Google Scholar] [CrossRef]
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin. Cancer Res. 2014, 20, 4096–4106. [Google Scholar] [CrossRef] [Green Version]
- Slattery, K.; Gardiner, C.M. NK Cell Metabolism and TGFbeta-Implications for Immunotherapy. Front. Immunol. 2019, 10, 2915. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Wensveen, F.M.; Jelencic, V.; Polic, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef]
- Lanuza, P.M.; Pesini, C.; Arias, M.A.; Calvo, C.; Ramirez-Labrada, A.; Pardo, J. Recalling the Biological Significance of Immune Checkpoints on NK Cells: A Chance to Overcome LAG3, PD1, and CTLA4 Inhibitory Pathways by Adoptive NK Cell Transfer? Front. Immunol. 2019, 10, 3010. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Tian, Z. NK Cell Dysfunction and Checkpoint Immunotherapy. Front. Immunol. 2019, 10, 1999. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, X.; Jin, T.; Tian, Y.; Dai, C.; Widarma, C.; Song, R.; Xu, F. Immune checkpoint molecules in natural killer cells as potential targets for cancer immunotherapy. Signal. Transduct. Target. Ther. 2020, 5, 250. [Google Scholar] [CrossRef]
- Raulet, D.H.; Vance, R.E. Self-tolerance of natural killer cells. Nat. Rev. Immunol. 2006, 6, 520–531. [Google Scholar] [CrossRef]
- Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 2013, 15, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radomska-Lesniewska, D.M.; Bialoszewska, A.; Kaminski, P. Angiogenic Properties of NK Cells in Cancer and Other Angiogenesis-Dependent Diseases. Cells 2021, 10, 1621. [Google Scholar] [CrossRef] [PubMed]
- Gallazzi, M.; Baci, D.; Mortara, L.; Bosi, A.; Buono, G.; Naselli, A.; Guarneri, A.; Deho, F.; Capogrosso, P.; Albini, A.; et al. Prostate Cancer Peripheral Blood NK Cells Show Enhanced CD9, CD49a, CXCR4, CXCL8, MMP-9 Production and Secrete Monocyte-Recruiting and Polarizing Factors. Front. Immunol 2020, 11, 586126. [Google Scholar] [CrossRef]
- Bosi, A.; Zanellato, S.; Bassani, B.; Albini, A.; Musco, A.; Cattoni, M.; Desio, M.; Nardecchia, E.; D’Urso, D.G.; Imperatori, A.; et al. Natural Killer Cells from Malignant Pleural Effusion Are Endowed with a Decidual-Like Proangiogenic Polarization. J. Immunol. Res. 2018, 2018, 2438598. [Google Scholar] [CrossRef] [Green Version]
- Bruno, A.; Bassani, B.; D’Urso, D.G.; Pitaku, I.; Cassinotti, E.; Pelosi, G.; Boni, L.; Dominioni, L.; Noonan, D.M.; Mortara, L.; et al. Angiogenin and the MMP9-TIMP2 axis are up-regulated in proangiogenic, decidual NK-like cells from patients with colorectal cancer. FASEB J. 2018, 32, 5365–5377. [Google Scholar] [CrossRef] [Green Version]
- Bruno, A.; Ferlazzo, G.; Albini, A.; Noonan, D.M. A think tank of TINK/TANKs: Tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J. Natl. Cancer Inst. 2014, 106, dju200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal. Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef]
- O’Sullivan, D.; Sanin, D.E.; Pearce, E.J.; Pearce, E.L. Metabolic interventions in the immune response to cancer. Nat. Rev. Immunol. 2019, 19, 324–335. [Google Scholar] [CrossRef]
- Yin, Z.; Bai, L.; Li, W.; Zeng, T.; Tian, H.; Cui, J. Targeting T cell metabolism in the tumor microenvironment: An anti-cancer therapeutic strategy. J. Exp. Clin. Cancer Res. 2019, 38, 403. [Google Scholar] [CrossRef]
- Molon, B.; Cali, B.; Viola, A. T Cells and Cancer: How Metabolism Shapes Immunity. Front. Immunol. 2016, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Makowski, L.; Chaib, M.; Rathmell, J.C. Immunometabolism: From basic mechanisms to translation. Immunol. Rev. 2020, 295, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehla, K.; Singh, P.K. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Zeng, H.; Horng, T. Metabolism as a guiding force for immunity. Nat. Cell Biol. 2019, 21, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, K.; Chawla, A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef] [Green Version]
- Caputa, G.; Castoldi, A.; Pearce, E.J. Metabolic adaptations of tissue-resident immune cells. Nat. Immunol. 2019, 20, 793–801. [Google Scholar] [CrossRef]
- Injarabian, L.; Devin, A.; Ransac, S.; Marteyn, B.S. Neutrophil Metabolic Shift during their Lifecycle: Impact on their Survival and Activation. Int. J. Mol. Sci. 2019, 21, 287. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Dikshit, M. Metabolic Insight of Neutrophils in Health and Disease. Front. Immunol. 2019, 10, 2099. [Google Scholar] [CrossRef]
- Sadiku, P.; Willson, J.A.; Ryan, E.M.; Sammut, D.; Coelho, P.; Watts, E.R.; Grecian, R.; Young, J.M.; Bewley, M.; Arienti, S.; et al. Neutrophils Fuel Effective Immune Responses through Gluconeogenesis and Glycogenesis. Cell Metab. 2021, 33, 411–423 e414. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Rice, C.M.; Davies, L.C.; Subleski, J.J.; Maio, N.; Gonzalez-Cotto, M.; Andrews, C.; Patel, N.L.; Palmieri, E.M.; Weiss, J.M.; Lee, J.M.; et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat. Commun. 2018, 9, 5099. [Google Scholar] [CrossRef] [Green Version]
- Hsu, B.E.; Tabaries, S.; Johnson, R.M.; Andrzejewski, S.; Senecal, J.; Lehuede, C.; Annis, M.G.; Ma, E.H.; Vols, S.; Ramsay, L.; et al. Immature Low-Density Neutrophils Exhibit Metabolic Flexibility that Facilitates Breast Cancer Liver Metastasis. Cell Rep. 2019, 27, 3902–3915.e3906. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, L.P.; Qin, W.T.; Yang, Q.; Chen, D.Y.; Li, Q.; Zhou, K.X.; Huang, P.Q.; Xu, C.J.; Li, J.; et al. Identification of a subset of immunosuppressive P2RX1-negative neutrophils in pancreatic cancer liver metastasis. Nat. Commun. 2021, 12, 174. [Google Scholar] [CrossRef]
- Terren, I.; Orrantia, A.; Vitalle, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10, 2278. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019, 19, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Terren, I.; Orrantia, A.; Vitalle, J.; Astarloa-Pando, G.; Zenarruzabeitia, O.; Borrego, F. Modulating NK cell metabolism for cancer immunotherapy. Semin. Hematol. 2020, 57, 213–224. [Google Scholar] [CrossRef]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Gnanaprakasam, J.N.R.; Sherman, J.W.; Wang, R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017, 35, 63–70. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, S.M.; Barra, N.G.; Ashkar, A.A.; Schertzer, J.D. Immunometabolism of T cells and NK cells: Metabolic control of effector and regulatory function. Inflamm. Res. 2018, 67, 813–828. [Google Scholar] [CrossRef]
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018, 28, 243–255.e5. [Google Scholar] [CrossRef] [Green Version]
- Gerbec, Z.J.; Hashemi, E.; Nanbakhsh, A.; Holzhauer, S.; Yang, C.; Mei, A.; Tsaih, S.W.; Lemke, A.; Flister, M.J.; Riese, M.J.; et al. Conditional Deletion of PGC-1alpha Results in Energetic and Functional Defects in NK Cells. iScience 2020, 23, 101454. [Google Scholar] [CrossRef]
- Zheng, X.; Qian, Y.; Fu, B.; Jiao, D.; Jiang, Y.; Chen, P.; Shen, Y.; Zhang, H.; Sun, R.; Tian, Z.; et al. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat. Immunol. 2019, 20, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Zecca, A.; Barili, V.; Canetti, D.; Regina, V.; Olivani, A.; Carone, C.; Capizzuto, V.; Zerbato, B.; Trenti, T.; Dalla Valle, R.; et al. Energy metabolism and cell motility defect in NK-cells from patients with hepatocellular carcinoma. Cancer Immunol. Immunother 2020, 69, 1589–1603. [Google Scholar] [CrossRef]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, H.O.; Roy, E.; Comerci, A.J.; van der Windt, D.J.; Zhang, H.; Huang, H.; Loughran, P.; Shiva, S.; Geller, D.A.; Bartlett, D.L.; et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res. 2019, 79, 5626–5639. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Lu, M.; Shi, J.; Gong, Z.; Hua, L.; Li, Q.; Lim, B.; Zhang, X.H.; Chen, X.; Li, S.; et al. Lung mesenchymal cells elicit lipid storage in neutrophils that fuel breast cancer lung metastasis. Nat. Immunol. 2020, 21, 1444–1455. [Google Scholar] [CrossRef]
- Charni, S.; de Bettignies, G.; Rathore, M.G.; Aguilo, J.I.; van den Elsen, P.J.; Haouzi, D.; Hipskind, R.A.; Enriquez, J.A.; Sanchez-Beato, M.; Pardo, J.; et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. J. Immunol. 2010, 185, 3498–3503. [Google Scholar] [CrossRef] [Green Version]
- Catalan, E.; Charni, S.; Jaime, P.; Aguilo, J.I.; Enriquez, J.A.; Naval, J.; Pardo, J.; Villalba, M.; Anel, A. MHC-I modulation due to changes in tumor cell metabolism regulates tumor sensitivity to CTL and NK cells. Oncoimmunology 2015, 4, e985924. [Google Scholar] [CrossRef] [Green Version]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Tai, L.H.; Alkayyal, A.A.; Leslie, A.L.; Sahi, S.; Bennett, S.; Tanese de Souza, C.; Baxter, K.; Angka, L.; Xu, R.; Kennedy, M.A.; et al. Phosphodiesterase-5 inhibition reduces postoperative metastatic disease by targeting surgery-induced myeloid derived suppressor cell-dependent inhibition of Natural Killer cell cytotoxicity. Oncoimmunology 2018, 7, e1431082. [Google Scholar] [CrossRef] [PubMed]
- Niavarani, S.R.; Lawson, C.; Bakos, O.; Boudaud, M.; Batenchuk, C.; Rouleau, S.; Tai, L.H. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer 2019, 19, 823. [Google Scholar] [CrossRef]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chen, S.; Liu, W.; Ma, Y.; Li, J.; Fisher, P.B.; Fang, X.; Wang, X.Y. Immunometabolism: A new target for improving cancer immunotherapy. Adv. Cancer Res. 2019, 143, 195–253. [Google Scholar] [CrossRef] [PubMed]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschlager, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef]
- Zaiatz-Bittencourt, V.; Finlay, D.K.; Gardiner, C.M. Canonical TGF-beta Signaling Pathway Represses Human NK Cell Metabolism. J. Immunol. 2018, 200, 3934–3941. [Google Scholar] [CrossRef]
- Viel, S.; Marcais, A.; Guimaraes, F.S.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Nunez Cortes, A.K.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.L.; Malmberg, K.J.; et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem. Cell 2020, 27, 224–237.e6. [Google Scholar] [CrossRef]
- Bahr, I.; Spielmann, J.; Quandt, D.; Kielstein, H. Obesity-Associated Alterations of Natural Killer Cells and Immunosurveillance of Cancer. Front. Immunol. 2020, 11, 245. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E.; Wolski, K. Rosiglitazone revisited: An updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch. Intern. Med. 2010, 170, 1191–1201. [Google Scholar] [CrossRef]
- Kobayashi, T.; Lam, P.Y.; Jiang, H.; Bednarska, K.; Gloury, R.; Murigneux, V.; Tay, J.; Jacquelot, N.; Li, R.; Tuong, Z.K.; et al. Increased lipid metabolism impairs NK cell function and mediates adaptation to the lymphoma environment. Blood 2020, 136, 3004–3017. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Li, B.; Chen, A.; Zheng, M.; Xu, T.; Zhang, H.; Dong, J.; Wu, J.; Yu, D.; Wei, J. Targeting aerobic glycolysis by dichloroacetate improves Newcastle disease virus-mediated viro-immunotherapy in hepatocellular carcinoma. Br. J. Cancer 2020, 122, 111–120. [Google Scholar] [CrossRef]
- Jian, S.L.; Chen, W.W.; Su, Y.C.; Su, Y.W.; Chuang, T.H.; Hsu, S.C.; Huang, L.R. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017, 8, e2779. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Eikawa, S.; Nishida, M.; Kunisada, Y.; Yoshida, A.; Fujiwara, T.; Kunisada, T.; Ozaki, T.; Udono, H. Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: Implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int. Immunol. 2019, 31, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- Ugolini, A.; Tyurin, V.A.; Tyurina, Y.Y.; Tcyganov, E.N.; Donthireddy, L.; Kagan, V.E.; Gabrilovich, D.I.; Veglia, F. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight 2020, 5, e138581. [Google Scholar] [CrossRef]
- Zuo, H.; Hou, Y.; Yu, Y.; Li, Z.; Liu, H.; Liu, C.; He, J.; Miao, L. Circumventing Myeloid-Derived Suppressor Cell-Mediated Immunosuppression Using an Oxygen-Generated and -Economized Nanoplatform. ACS Appl. Mater. Interfaces 2020, 12, 55723–55736. [Google Scholar] [CrossRef]
- Chiu, D.K.; Tse, A.P.; Xu, I.M.; Di Cui, J.; Lai, R.K.; Li, L.L.; Koh, H.Y.; Tsang, F.H.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Bai, L.; Li, W.; Cui, J. The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front. Oncol. 2020, 10, 605154. [Google Scholar] [CrossRef]
- Paudel, B.B.; Quaranta, V. Metabolic plasticity meets gene regulation. Proc. Natl. Acad. Sci. USA 2019, 116, 3370–3372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Liang, P.; Guo, G.; Huang, Z.; Niu, Y.; Dong, L.; Wang, C.; Zhang, J. Re-polarizing Myeloid-derived Suppressor Cells (MDSCs) with Cationic Polymers for Cancer Immunotherapy. Sci. Rep. 2016, 6, 24506. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Pino, M.D.; Dean, M.J.; Ochoa, A.C. Myeloid-derived suppressor cells (MDSC): When good intentions go awry. Cell Immunol. 2021, 362, 104302. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamoorthy, M.; Gerhardt, L.; Maleki Vareki, S. Immunosuppressive Effects of Myeloid-Derived Suppressor Cells in Cancer and Immunotherapy. Cells 2021, 10, 1170. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, N.; Tan, H.Y.; Chueng, F.; Zhang, Z.J.; Yuen, M.F.; Feng, Y. Modulation of gut microbiota mediates berberine-induced expansion of immuno-suppressive cells to against alcoholic liver disease. Clin. Transl. Med. 2020, 10, e112. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Butler, L.M.; Perone, Y.; Dehairs, J.; Lupien, L.E.; de Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv. Rev. 2020, 159, 245–293. [Google Scholar] [CrossRef] [PubMed]
- Laubichler, M.D. Tinkering: A conceptual and historical evaluation. Novartis Found. Symp. 2007, 284, 20–29, discussion 29–34, 110–115. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Lerma Barbaro, A.; Palano, M.T.; Cucchiara, M.; Gallazzi, M.; Mortara, L.; Bruno, A. Metabolic Rewiring in the Tumor Microenvironment to Support Immunotherapy: A Focus on Neutrophils, Polymorphonuclear Myeloid-Derived Suppressor Cells and Natural Killer Cells. Vaccines 2021, 9, 1178. https://doi.org/10.3390/vaccines9101178
De Lerma Barbaro A, Palano MT, Cucchiara M, Gallazzi M, Mortara L, Bruno A. Metabolic Rewiring in the Tumor Microenvironment to Support Immunotherapy: A Focus on Neutrophils, Polymorphonuclear Myeloid-Derived Suppressor Cells and Natural Killer Cells. Vaccines. 2021; 9(10):1178. https://doi.org/10.3390/vaccines9101178
Chicago/Turabian StyleDe Lerma Barbaro, Andrea, Maria Teresa Palano, Martina Cucchiara, Matteo Gallazzi, Lorenzo Mortara, and Antonino Bruno. 2021. "Metabolic Rewiring in the Tumor Microenvironment to Support Immunotherapy: A Focus on Neutrophils, Polymorphonuclear Myeloid-Derived Suppressor Cells and Natural Killer Cells" Vaccines 9, no. 10: 1178. https://doi.org/10.3390/vaccines9101178