BCG Provides Short-Term Protection from Experimental Cerebral Malaria in Mice


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Mice
2.2. BCG Vaccination
2.3. Parasites and Sporozoite Isolation
2.4. Parasitic Infection and Evaluation of Disease
2.5. Assessment of Blood Brain Barrier Integrity by Evans Blue
2.6. Cell Isolation and Purification from Brain, Spleen and Blood
2.7. Flow Cytometry
2.8. Multiplex Assay
2.9. RNA Isolation and RT-PCR
- Gapdh forward ATTGTCAGCAATGCATCCTG; Gapdh reverse ATGGACTGTGGTCATGAGCC;
- PbA 18S rRNA forward AAGCATTAAATAAAGCGAATACATCCTTAC; PbA 18S rRNA reverse GGAGATTGGTTTTGACGTTTATGTG;
- Ifng forward TCAAGTGGCATAGATGTGGAAGAA; Ifng reverse TGGCTCTGCAGGATTTTCATG:
- Tnf forward CCACCACGCTCTTCTGTCTAC; Tnf reverse AGGGTCTGGGCCATAGAACT;
- Lfa forward CTCCAGGAGGACAACTCAGC; Lfa reverse CTAGTGTGGGCATGTTGTGG;
- Il10 forward GAGGTGAGTGGCTGTCTGTG; Il10 reverse CAGAGAAGCATGGCCCAGAA
- Icam1 forward GGGACCACGGAGCCAATT; Icam1 reverse CTCGGAGACATTAGAGAACAATGC;
- Vcam1 forward ACAAGTCTACATCTCTCCCAGGA; Vcam1 reverse AAGGTGAGGGTGGCATTTCC;
- CD8a forward CAAATGTCCCAGGCCGCTA; CD8a reverse GAACTGTCCCCATCACACCC;
- Gzmb forward GGACAAAGGCAGGGGAGATC; Gzmb reverse TCACAGTGAGCAGCAGTCAG.
2.10. Statistical Analysis
3. Results
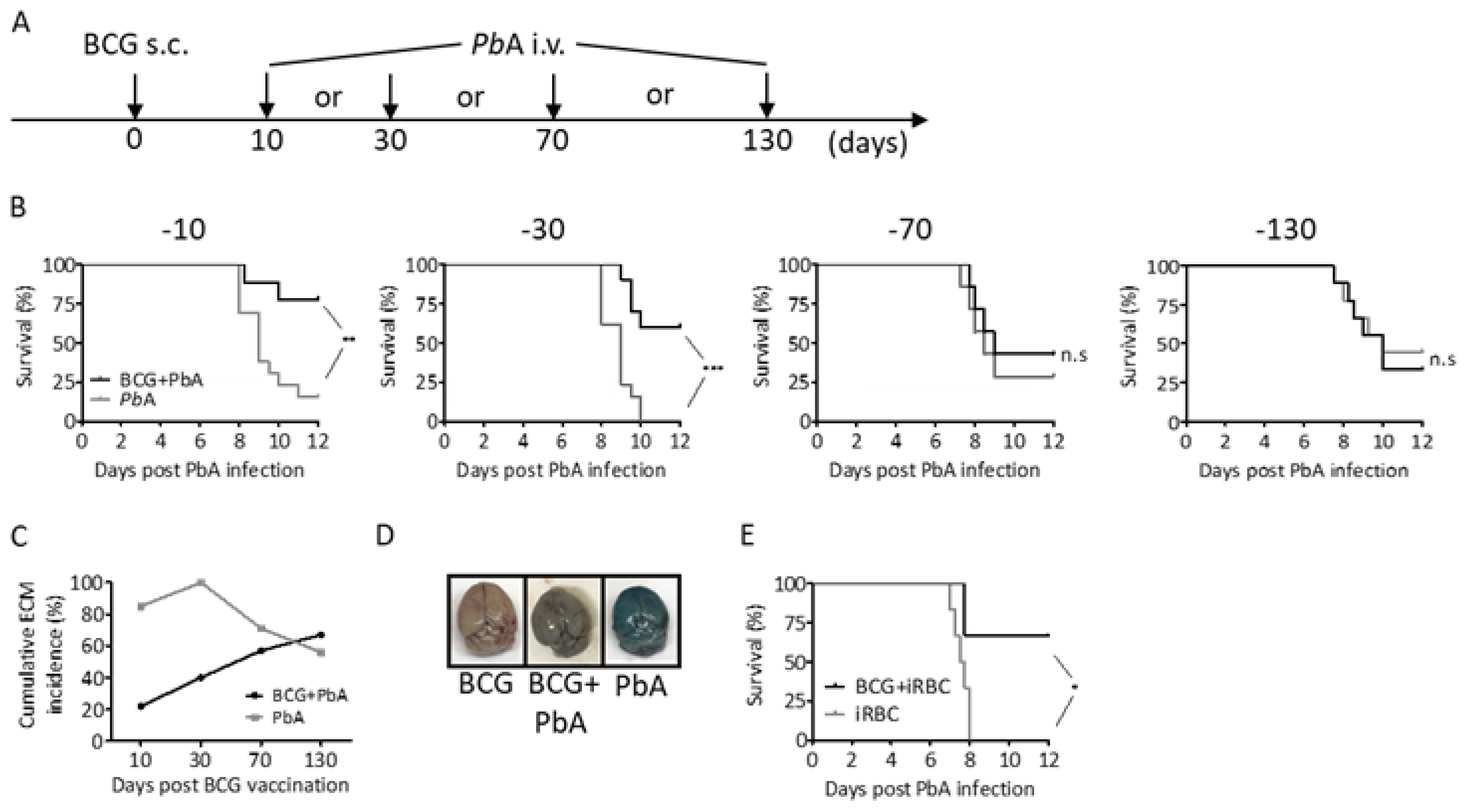
3.1. BCG Partly Protects C57BL/6 Mice from P. berghei ANKA Induced ECM
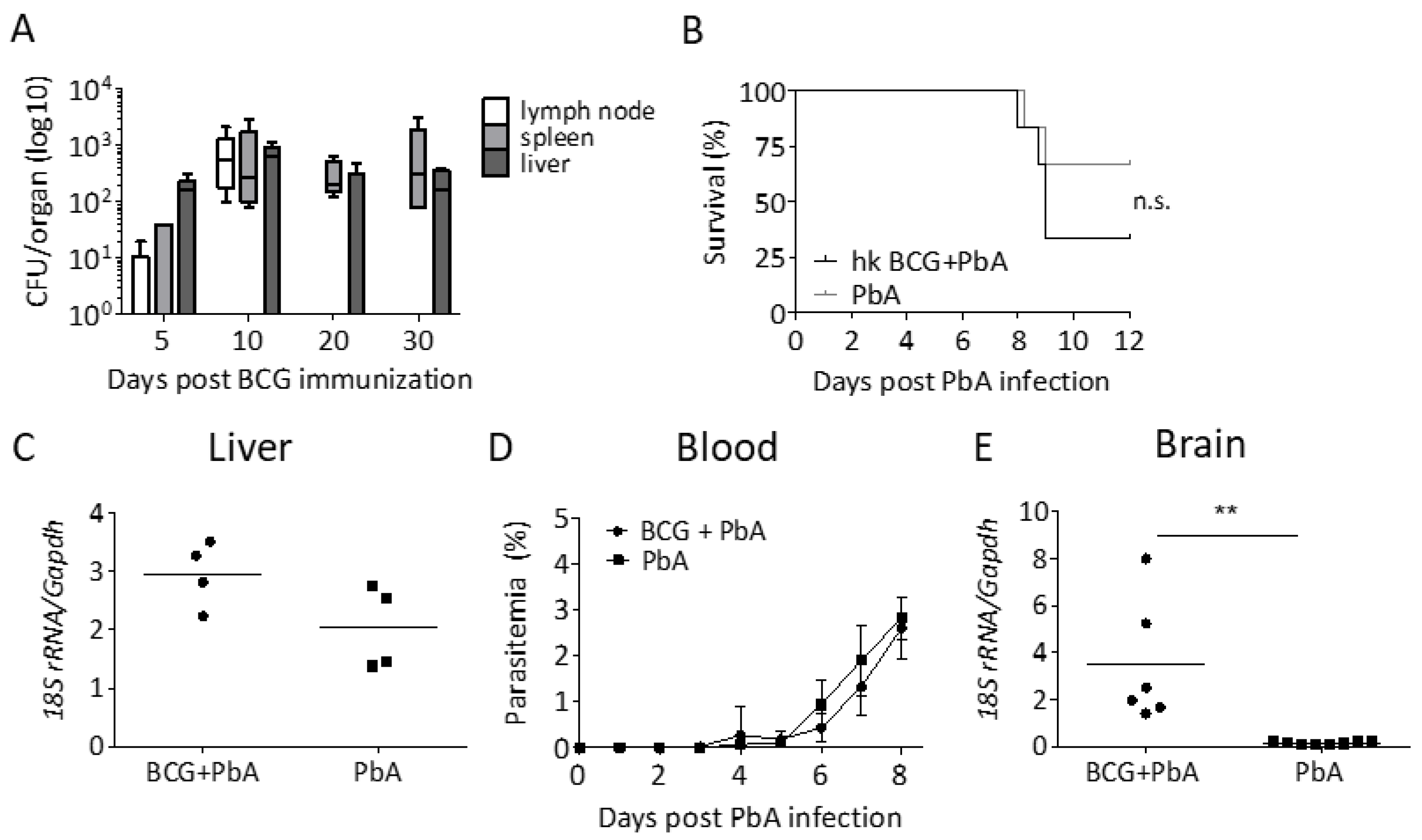
3.2. Protection from ECM Is Associated with the Recovery of Viable BCG
3.3. BCG Vaccination Does Not Lead to a Reduction in Parasite Load
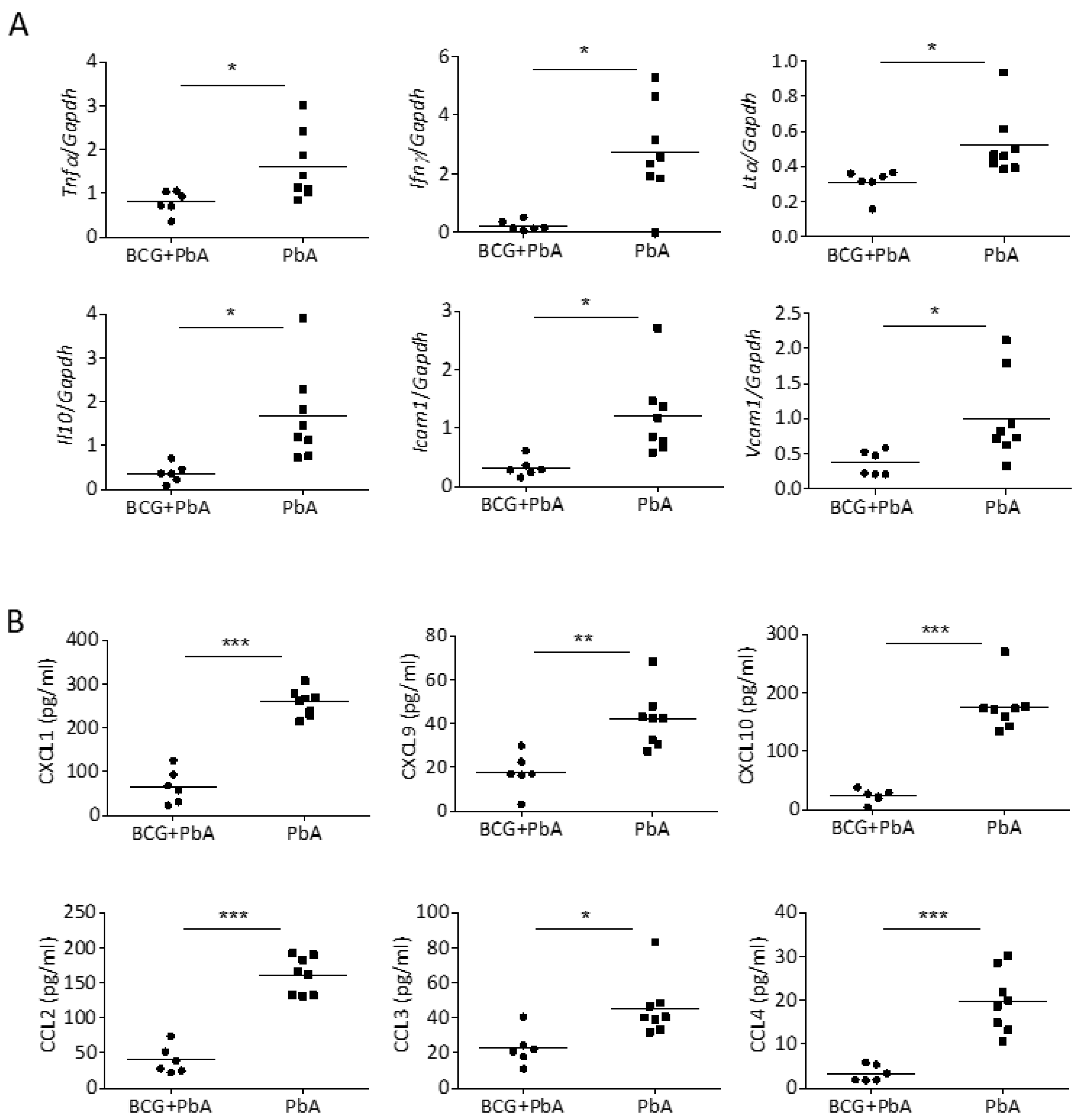
3.4. BCG-Mediated Protection from ECM Is Associated with Reduced Pro-Inflammatory Mediators in the Brain
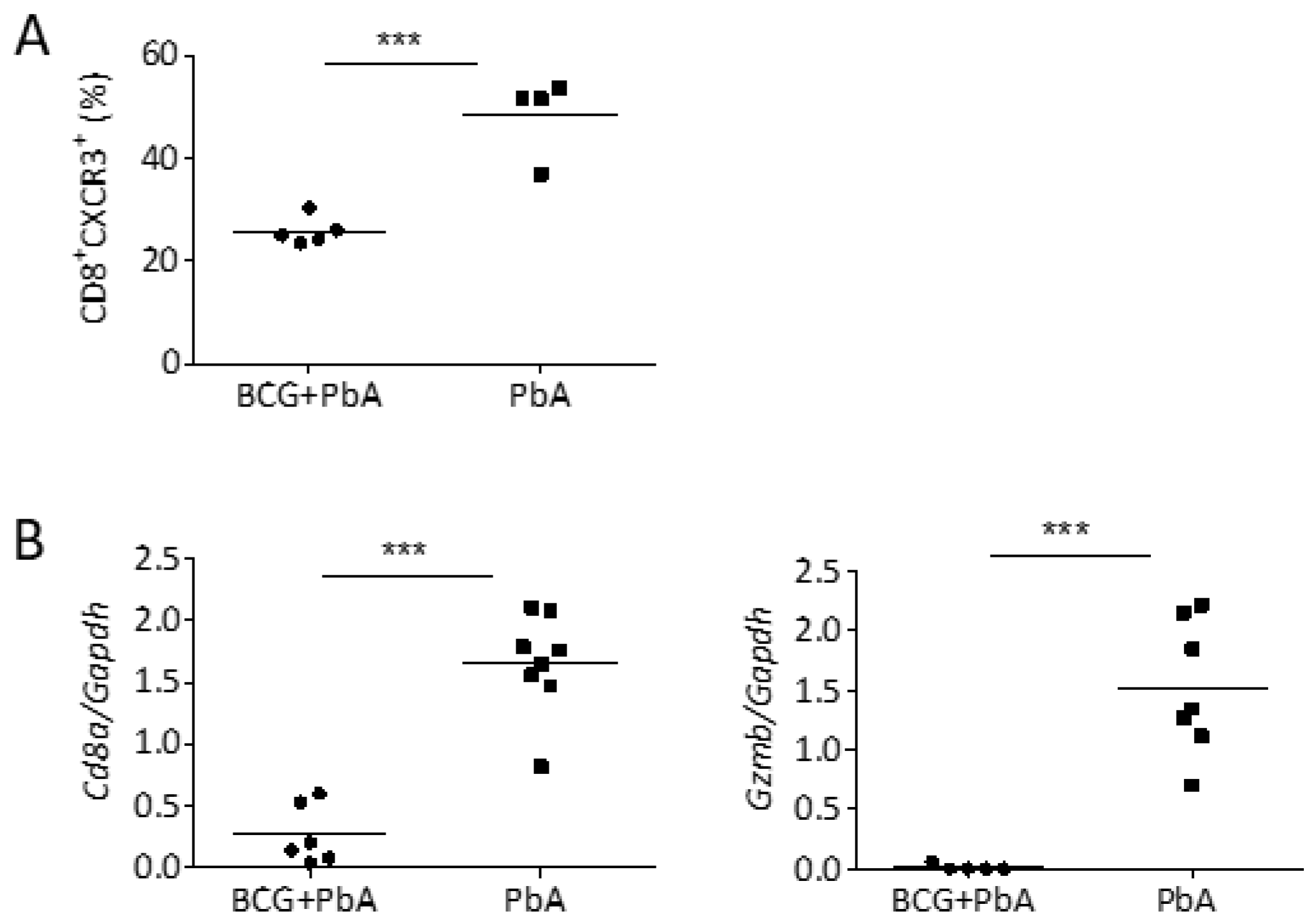
3.5. BCG Vaccination Reduced Cellular Influx into the Brains upon PbA-Infection
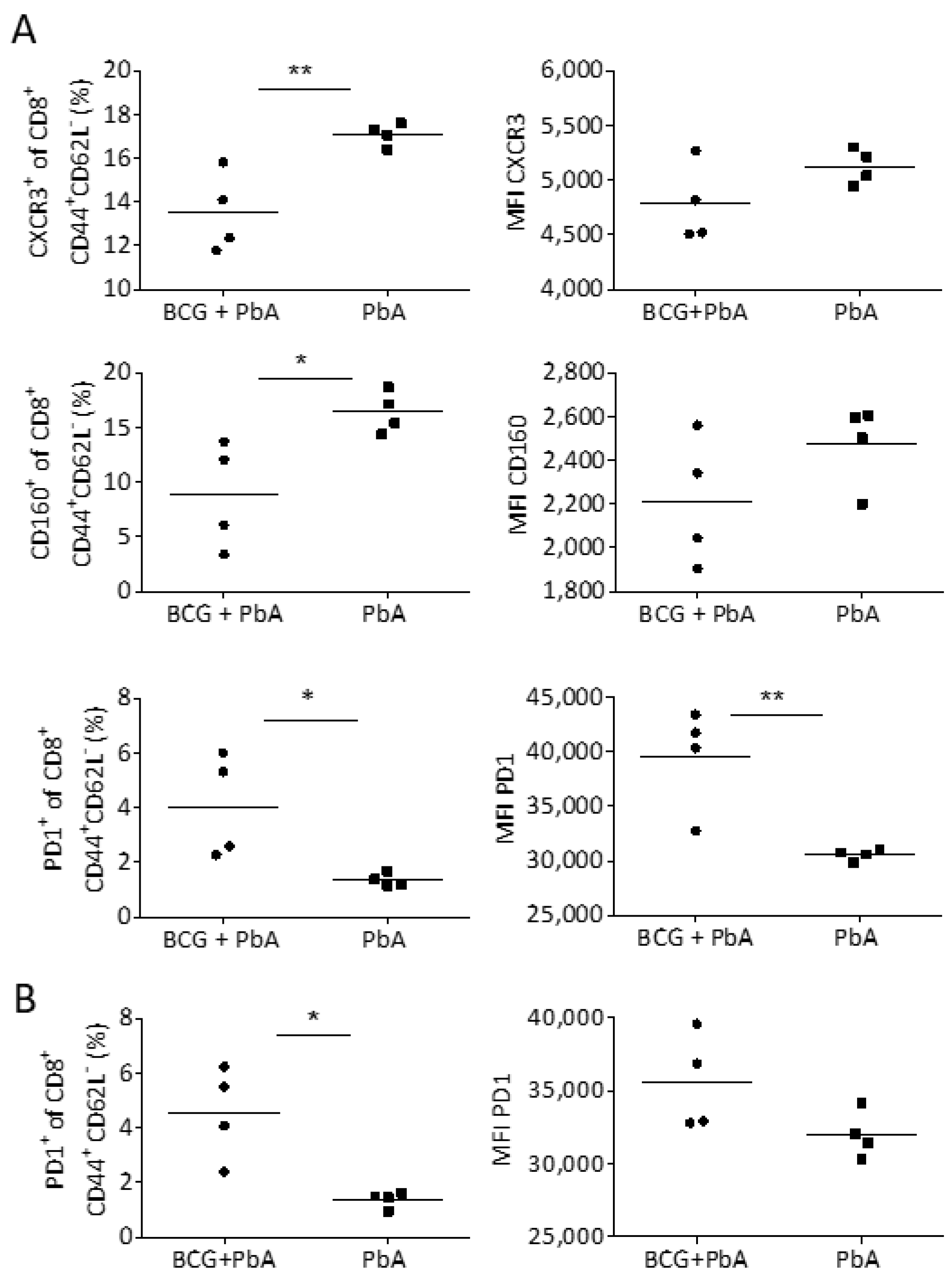
3.6. BCG Changes T Cell Phenotype in Blood and Spleen Prior to the Onset of ECM
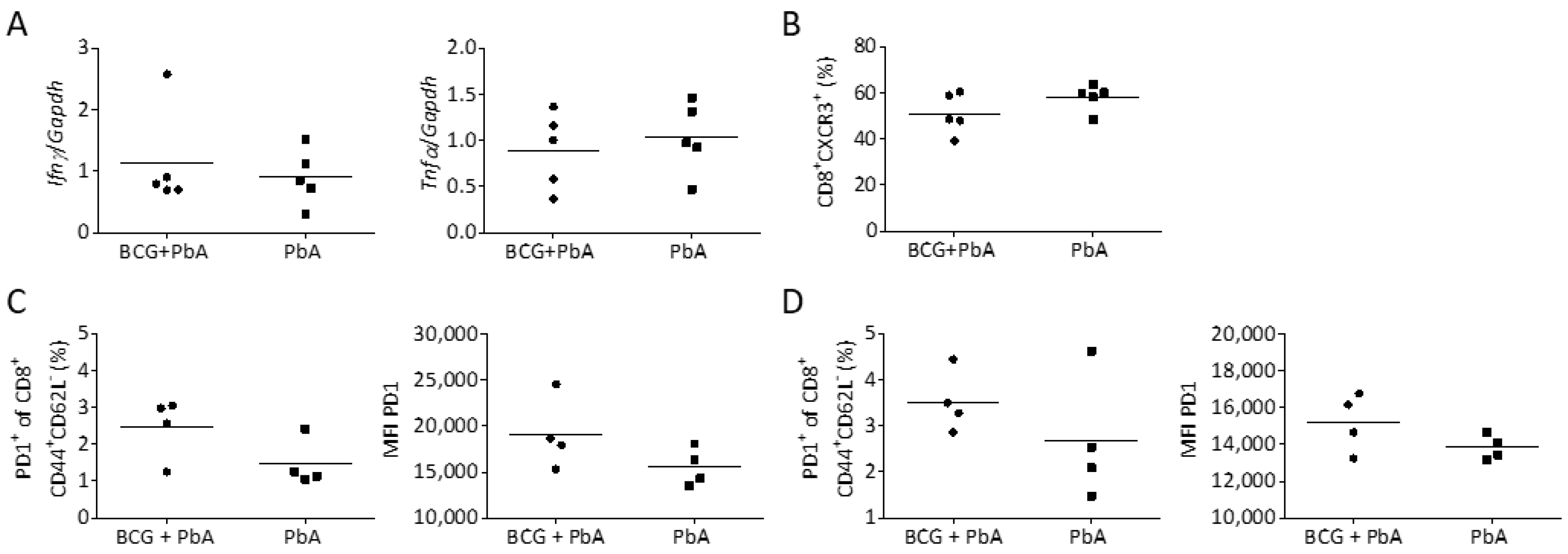
3.7. BCG-Induced Immunomodulation Is Lost When PbA Infection Was Performed after 130 Days
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zwerling, A.; Behr, M.A.; Verma, A.; Brewer, T.F.; Menzies, D.; Pai, M. The BCG World Atlas: A database of global BCG vaccination policies and practices. PLoS Med. 2011, 8, e1001012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwenhuizen, N.E.; Kaufmann, S.H.E. Next-Generation Vaccines Based on Bacille Calmette-Guerin. Front. Immunol. 2018, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Butkeviciute, E.; Jones, C.E.; Smith, S.G. Heterologous effects of infant BCG vaccination: Potential mechanisms of immunity. Future Microbiol. 2018, 13, 1193–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garly, M.L.; Martins, C.L.; Bale, C.; Balde, M.A.; Hedegaard, K.L.; Gustafson, P.; Lisse, I.M.; Whittle, H.C.; Aaby, P. BCG scar and positive tuberculin reaction associated with reduced child mortality in West Africa. A non-specific beneficial effect of BCG? Vaccine 2003, 21, 2782–2790. [Google Scholar] [CrossRef]
- Vaugelade, J.; Pinchinat, S.; Guiella, G.; Elguero, E.; Simondon, F. Non-specific effects of vaccination on child survival: Prospective cohort study in Burkina Faso. BMJ 2004, 329, 1309. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, I.; Aaby, P.; Jensen, H. Routine vaccinations and child survival: Follow up study in Guinea-Bissau, West Africa. BMJ 2000, 321, 1435–1438. [Google Scholar] [CrossRef] [Green Version]
- Roth, A.; Garly, M.L.; Jensen, H.; Nielsen, J.; Aaby, P. Bacillus Calmette-Guerin vaccination and infant mortality. Expert Rev. Vaccines 2006, 5, 277–293. [Google Scholar] [CrossRef]
- Aaby, P.; Roth, A.; Ravn, H.; Napirna, B.M.; Rodrigues, A.; Lisse, I.M.; Stensballe, L.; Diness, B.R.; Lausch, K.R.; Lund, N.; et al. Randomized trial of BCG vaccination at birth to low-birth-weight children: Beneficial nonspecific effects in the neonatal period? J. Infect. Dis. 2011, 204, 245–252. [Google Scholar] [CrossRef]
- Roth, A.; Gustafson, P.; Nhaga, A.; Djana, Q.; Poulsen, A.; Garly, M.L.; Jensen, H.; Sodemann, M.; Rodriques, A.; Aaby, P. BCG vaccination scar associated with better childhood survival in Guinea-Bissau. Int. J. Epidemiol. 2005, 34, 540–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, M.L.; van Gijzel, S.W.; Smits, J.; de Mast, Q.; Aaby, P.; Benn, C.S.; Netea, M.G.; van der Ven, A.J. BCG vaccination is associated with reduced malaria prevalence in children under the age of 5 years in sub-Saharan Africa. BMJ Glob. Health 2019, 4, e001862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.R. Host defenses in murine malaria: Nonspecific resistance to Plasmodium berghei generated in response to Mycobacterium bovis infection or Corynebacterium parvum stimulation. Infect. Immun. 1981, 33, 199–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, S.; Yukitake, H.; Kanbara, H.; Yamada, H.; Kitamura, A.; Yamada, T. Mycobacterium bovis bacillus calmette-guerin induces protective immunity against infection by Plasmodium yoelii at blood-stage depending on shifting immunity toward Th1 type and inducing protective IgG2a after the parasite infection. Vaccine 2000, 19, 779–787. [Google Scholar] [CrossRef]
- Leisewitz, A.L.; Rockett, K.; Kwiatkowski, D. BCG-malaria co-Infection has paradoxical effects on C57BL/6 and A/J mouse strains. Parasite Immunol. 2008, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Smrkovski, L.L. Effect of route of Mycobacterium bovis BCG administration on induction of suppression of sporozoite immunity in rodent malaria. Infect. Immun. 1981, 31, 408–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smrkovski, L.L.; Strickland, G.T. Rodent malaria: BCG-induced protection and immunosuppression. J. Immunol. 1978, 121, 1257–1261. [Google Scholar] [PubMed]
- Clark, I.A.; Allison, A.C.; Cox, F.E. Protection of mice against Babesia and Plasmodium with BCG. Nature 1976, 259, 309–311. [Google Scholar] [CrossRef]
- Parra, M.; Liu, X.; Derrick, S.C.; Yang, A.; Tian, J.; Kolibab, K.; Kumar, S.; Morris, S.L. Molecular analysis of non-specific protection against murine malaria induced by BCG vaccination. PLoS ONE 2013, 8, e66115. [Google Scholar] [CrossRef] [Green Version]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.; Ifrim, D.C.; Saeed, S.; Jacobs, C.; van Loenhout, J.; de Jong, D.; Stunnenberg, H.G.; et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 17537–17542. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Latz, E.; Mills, K.H.; O’Neill, L.A. Innate immune memory: A paradigm shift in understanding host defense. Nat. Immunol. 2015, 16, 675–679. [Google Scholar] [CrossRef]
- van der Meer, J.W.; Joosten, L.A.; Riksen, N.; Netea, M.G. Trained immunity: A smart way to enhance innate immune defence. Mol. Immunol. 2015, 68, 40–44. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; van der Meer, J.W. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, R.J.W.; Moorlag, S.; Novakovic, B.; Li, Y.; Wang, S.Y.; Oosting, M.; Kumar, V.; Xavier, R.J.; Wijmenga, C.; Joosten, L.A.B.; et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe 2018, 23, 89–100 e105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walk, J.; de Bree, L.C.J.; Graumans, W.; Stoter, R.; van Gemert, G.J.; van de Vegte-Bolmer, M.; Teelen, K.; Hermsen, C.C.; Arts, R.J.W.; Behet, M.C.; et al. Outcomes of controlled human malaria infection after BCG vaccination. Nat. Commun. 2019, 10, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renia, L.; Howland, S.W.; Claser, C.; Charlotte Gruner, A.; Suwanarusk, R.; Hui Teo, T.; Russell, B.; Ng, L.F. Cerebral malaria: Mysteries at the blood-brain barrier. Virulence 2012, 3, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazanfari, N.; Mueller, S.N.; Heath, W.R. Cerebral Malaria in Mouse and Man. Front. Immunol. 2018, 9, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idro, R.; Kakooza-Mwesige, A.; Balyejjussa, S.; Mirembe, G.; Mugasha, C.; Tugumisirize, J.; Byarugaba, J. Severe neurological sequelae and behaviour problems after cerebral malaria in Ugandan children. BMC Res. Notes 2010, 3, 104. [Google Scholar] [CrossRef] [Green Version]
- Idro, R.; Marsh, K.; John, C.C.; Newton, C.R. Cerebral malaria: Mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr. Res. 2010, 68, 267–274. [Google Scholar] [CrossRef]
- McQuillan, J.A.; Mitchell, A.J.; Ho, Y.F.; Combes, V.; Ball, H.J.; Golenser, J.; Grau, G.E.; Hunt, N.H. Coincident parasite and CD8 T cell sequestration is required for development of experimental cerebral malaria. Int. J. Parasitol. 2011, 41, 155–163. [Google Scholar] [CrossRef]
- Mitchell, A.J.; Hansen, A.M.; Hee, L.; Ball, H.J.; Potter, S.M.; Walker, J.C.; Hunt, N.H. Early cytokine production is associated with protection from murine cerebral malaria. Infect. Immun. 2005, 73, 5645–5653. [Google Scholar] [CrossRef] [Green Version]
- Baptista, F.G.; Pamplona, A.; Pena, A.C.; Mota, M.M.; Pied, S.; Vigario, A.M. Accumulation of Plasmodium berghei-infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect. Immun. 2010, 78, 4033–4039. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.; Best, S.E.; Unosson, K.; Amante, F.H.; de Labastida, F.; Anstey, N.M.; Karupiah, G.; Smyth, M.J.; Heath, W.R.; Engwerda, C.R. Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J. Immunol. 2011, 186, 6148–6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settles, E.W.; Moser, L.A.; Harris, T.H.; Knoll, L.J. Toxoplasma gondii upregulates interleukin-12 to prevent Plasmodium berghei-induced experimental cerebral malaria. Infect. Immun. 2014, 82, 1343–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rest, J.R. Cerebral malaria in inbred mice. I. A new model and its pathology. Trans. R. Soc. Trop. Med. Hyg. 1982, 76, 410–415. [Google Scholar] [CrossRef]
- Carroll, R.W.; Wainwright, M.S.; Kim, K.Y.; Kidambi, T.; Gomez, N.D.; Taylor, T.; Haldar, K. A rapid murine coma and behavior scale for quantitative assessment of murine cerebral malaria. PLoS ONE 2010, 5, e13124. [Google Scholar] [CrossRef] [Green Version]
- Blank, J.; Behrends, J.; Jacobs, T.; Schneider, B.E. Mycobacterium tuberculosis Coinfection Has No Impact on Plasmodium berghei ANKA-Induced Experimental Cerebral Malaria in C57BL/6 Mice. Infect. Immun. 2015, 84, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Heiss, K.; Maier, M.I.; Hoffmann, A.; Frank, R.; Bendszus, M.; Mueller, A.K.; Pfeil, J. Protection from experimental cerebral malaria with a single intravenous or subcutaneous whole-parasite immunization. Sci. Rep. 2018, 8, 3085. [Google Scholar] [CrossRef] [Green Version]
- Howland, S.W.; Poh, C.M.; Gun, S.Y.; Claser, C.; Malleret, B.; Shastri, N.; Ginhoux, F.; Grotenbreg, G.M.; Renia, L. Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol. Med. 2013, 5, 916–931. [Google Scholar] [CrossRef]
- Sierro, F.; Grau, G.E.R. The Ins and Outs of Cerebral Malaria Pathogenesis: Immunopathology, Extracellular Vesicles, Immunometabolism, and Trained Immunity. Front. Immunol. 2019, 10, 830. [Google Scholar] [CrossRef] [Green Version]
- Dunst, J.; Kamena, F.; Matuschewski, K. Cytokines and Chemokines in Cerebral Malaria Pathogenesis. Front. Cell Infect. Microbiol. 2017, 7, 324. [Google Scholar] [CrossRef]
- Nishanth, G.; Schluter, D. Blood-Brain Barrier in Cerebral Malaria: Pathogenesis and Therapeutic Intervention. Trends Parasitol. 2019, 35, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Villegas-Mendez, A.; Greig, R.; Shaw, T.N.; de Souza, J.B.; Gwyer Findlay, E.; Stumhofer, J.S.; Hafalla, J.C.; Blount, D.G.; Hunter, C.A.; Riley, E.M.; et al. IFN-gamma-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J. Immunol. 2012, 189, 968–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belnoue, E.; Kayibanda, M.; Vigario, A.M.; Deschemin, J.C.; van Rooijen, N.; Viguier, M.; Snounou, G.; Renia, L. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J. Immunol. 2002, 169, 6369–6375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscate, F.; Stetter, N.; Schramm, C.; Schulze Zur Wiesch, J.; Bosurgi, L.; Jacobs, T. HVEM and CD160: Regulators of Immunopathology During Malaria Blood-Stage. Front. Immunol. 2018, 9, 2611. [Google Scholar] [CrossRef] [PubMed]
- Uthayakumar, D.; Paris, S.; Chapat, L.; Freyburger, L.; Poulet, H.; De Luca, K. Non-specific Effects of Vaccines Illustrated Through the BCG Example: From Observations to Demonstrations. Front. Immunol. 2018, 9, 2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, R.I.; Spadaro-Antonelli, M.A.; Lawrence, V.J.; Quagliata, F. Immunosuppressive activity of BCG. effects of adjuvant disease, lymphocyte subpopulations, and homing of thoracic duct cells in rats. Infect. Immun. 1979, 25, 310–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.A.; Rao, V.S.; Mitchell, M.S. Systemic bacillus Calmette-Guerin (BCG) activates natural suppressor cells. Proc. Natl. Acad. Sci. USA 1978, 75, 5142–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalewicz-Kulbat, M.; Locht, C. BCG and protection against inflammatory and auto-immune diseases. Expert Rev. Vaccines 2017, 16, 1–10. [Google Scholar] [CrossRef]
- Ristori, G.; Buzzi, M.G.; Sabatini, U.; Giugni, E.; Bastianello, S.; Viselli, F.; Buttinelli, C.; Ruggieri, S.; Colonnese, C.; Pozzilli, C.; et al. Use of Bacille Calmette-Guerin (BCG) in multiple sclerosis. Neurology 1999, 53, 1588–1589. [Google Scholar] [CrossRef]
- Ristori, G.; Romano, S.; Cannoni, S.; Visconti, A.; Tinelli, E.; Mendozzi, L.; Cecconi, P.; Lanzillo, R.; Quarantelli, M.; Buttinelli, C.; et al. Effects of Bacille Calmette-Guerin after the first demyelinating event in the CNS. Neurology 2014, 82, 41–48. [Google Scholar] [CrossRef]
- Lippens, C.; Garnier, L.; Guyonvarc’h, P.M.; Santiago-Raber, M.L.; Hugues, S. Extended Freeze-Dried BCG Instructed pDCs Induce Suppressive Tregs and Dampen EAE. Front. Immunol. 2018, 9, 2777. [Google Scholar] [CrossRef]
- Lacan, G.; Dang, H.; Middleton, B.; Horwitz, M.A.; Tian, J.; Melega, W.P.; Kaufman, D.L. Bacillus Calmette-Guerin vaccine-mediated neuroprotection is associated with regulatory T-cell induction in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J. Neurosci. Res. 2013, 91, 1292–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, J.; Lacan, G.; Dang, H.; Hsieh, T.; Middleton, B.; Wasserfall, C.; Tian, J.; Melega, W.P.; Kaufman, D.L. BCG vaccine-induced neuroprotection in a mouse model of Parkinson’s disease. PLoS ONE 2011, 6, e16610. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Qi, F.; Yang, J.; Wang, X.; Wu, Y.; Wen, Y.; Yuan, Q.; Zou, J.; Guo, K.; Yao, Z.B. Immunization with Bacillus Calmette-Guerin (BCG) alleviates neuroinflammation and cognitive deficits in APP/PS1 mice via the recruitment of inflammation-resolving monocytes to the brain. Neurobiol. Dis. 2017, 101, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Koeken, V.A.; de Bree, L.C.J.; Mourits, V.P.; Moorlag, S.J.; Walk, J.; Cirovic, B.; Arts, R.J.; Jaeger, M.; Dijkstra, H.; Lemmers, H.; et al. BCG vaccination in humans inhibits systemic inflammation in a sex-dependent manner. J. Clin. Investig. 2020, 130, 5591–5602. [Google Scholar] [CrossRef] [PubMed]
- Engwerda, C.R.; Mynott, T.L.; Sawhney, S.; De Souza, J.B.; Bickle, Q.D.; Kaye, P.M. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J. Exp. Med. 2002, 195, 1371–1377. [Google Scholar] [CrossRef]
- Amani, V.; Vigario, A.M.; Belnoue, E.; Marussig, M.; Fonseca, L.; Mazier, D.; Renia, L. Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur. J. Immunol. 2000, 30, 1646–1655. [Google Scholar] [CrossRef]
- Grau, G.E.; Heremans, H.; Piguet, P.F.; Pointaire, P.; Lambert, P.H.; Billiau, A.; Vassalli, P. Monoclonal antibody against interferon gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1989, 86, 5572–5574. [Google Scholar] [CrossRef] [Green Version]
- Bauer, P.R.; Van Der Heyde, H.C.; Sun, G.; Specian, R.D.; Granger, D.N. Regulation of endothelial cell adhesion molecule expression in an experimental model of cerebral malaria. Microcirculation 2002, 9, 463–470. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Del Portillo, H.A.; Ferrer, M.; Brugat, T.; Martin-Jaular, L.; Langhorne, J.; Lacerda, M.V. The role of the spleen in malaria. Cell Microbiol. 2012, 14, 343–355. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Shen, Y.; Liang, J.; Li, Y.; Huang, Y.; Liu, X.; Jiang, D.; Yang, S.; Zhao, Y.; et al. PDL1 Fusion Protein Protects Against Experimental Cerebral Malaria via Repressing Over-Reactive CD8(+) T Cell Responses. Front. Immunol. 2018, 9, 3157. [Google Scholar] [CrossRef] [PubMed]
- Civil, R.H.; Warren, K.S.; Mahmoud, A.A. Conditions for bacille Calmette-Guerin-induced resistance to infection with Schistosoma mansoni in mice. J. Infect. Dis. 1978, 137, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Blanden, R.V.; Lefford, M.J.; Mackaness, G.B. The host response to Calmette-Guerin bacillus infection in mice. J. Exp. Med. 1969, 129, 1079–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaveh, D.A.; Garcia-Pelayo, M.C.; Hogarth, P.J. Persistent BCG bacilli perpetuate CD4 T effector memory and optimal protection against tuberculosis. Vaccine 2014, 32, 6911–6918. [Google Scholar] [CrossRef] [Green Version]
- Olsen, A.W.; Brandt, L.; Agger, E.M.; van Pinxteren, L.A.; Andersen, P. The influence of remaining live BCG organisms in vaccinated mice on the maintenance of immunity to tuberculosis. Scand. J. Immunol. 2004, 60, 273–277. [Google Scholar] [CrossRef]
- Converse, P.J.; Almeida, D.V.; Nuermberger, E.L.; Grosset, J.H. BCG-mediated protection against Mycobacterium ulcerans infection in the mouse. PLoS Negl. Trop. Dis. 2011, 5, e985. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witschkowski, J.; Behrends, J.; Frank, R.; Eggers, L.; von Borstel, L.; Hertz, D.; Mueller, A.-K.; Schneider, B.E. BCG Provides Short-Term Protection from Experimental Cerebral Malaria in Mice. Vaccines 2020, 8, 745. https://doi.org/10.3390/vaccines8040745
Witschkowski J, Behrends J, Frank R, Eggers L, von Borstel L, Hertz D, Mueller A-K, Schneider BE. BCG Provides Short-Term Protection from Experimental Cerebral Malaria in Mice. Vaccines. 2020; 8(4):745. https://doi.org/10.3390/vaccines8040745
Chicago/Turabian StyleWitschkowski, Julia, Jochen Behrends, Roland Frank, Lars Eggers, Linda von Borstel, David Hertz, Ann-Kristin Mueller, and Bianca E. Schneider. 2020. "BCG Provides Short-Term Protection from Experimental Cerebral Malaria in Mice" Vaccines 8, no. 4: 745. https://doi.org/10.3390/vaccines8040745