SANTAVACTM: Summary of Research and Development




Abstract
:1. Introduction
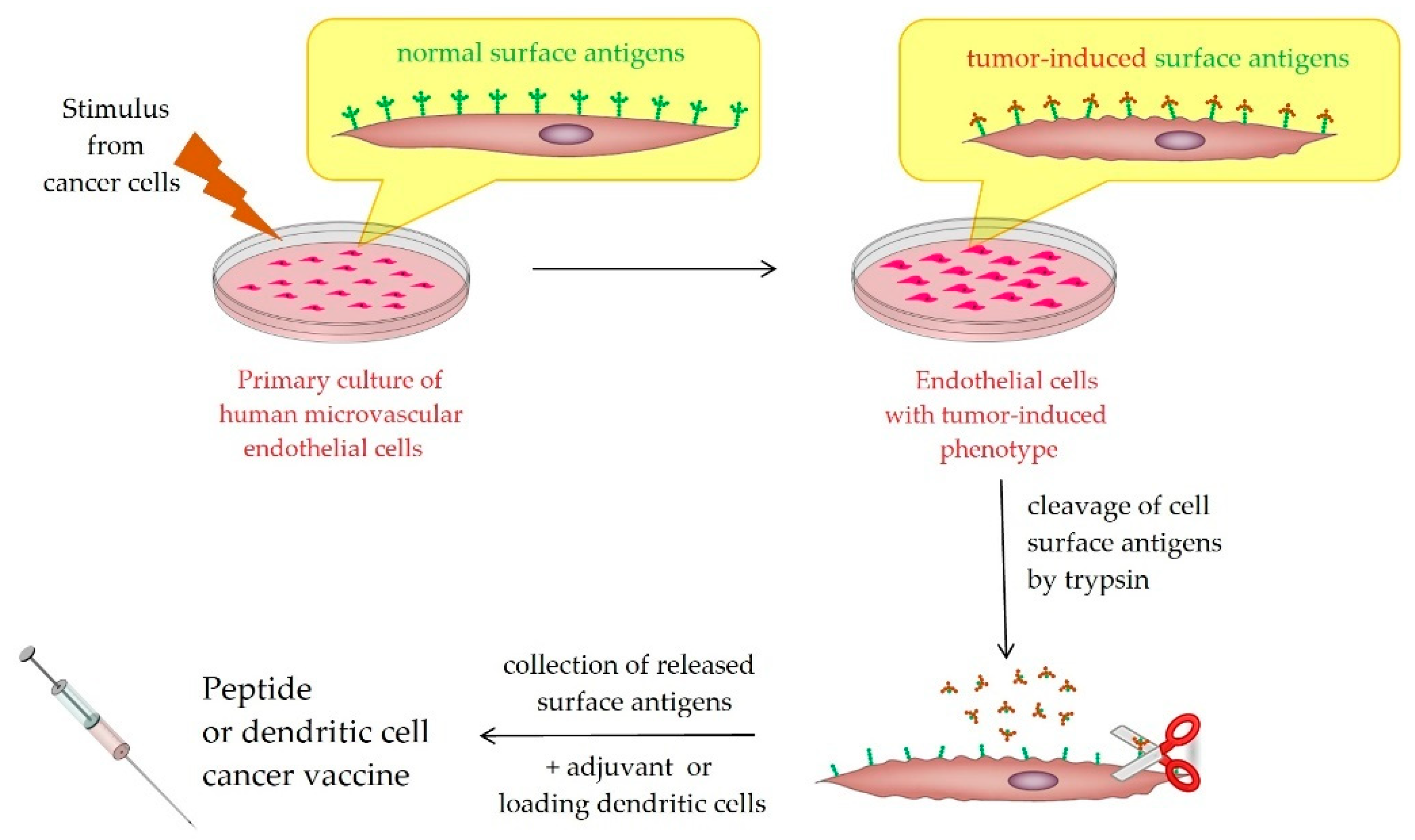
2. Innovation
3. SANTAVAC Equivalence to Cell Antigens
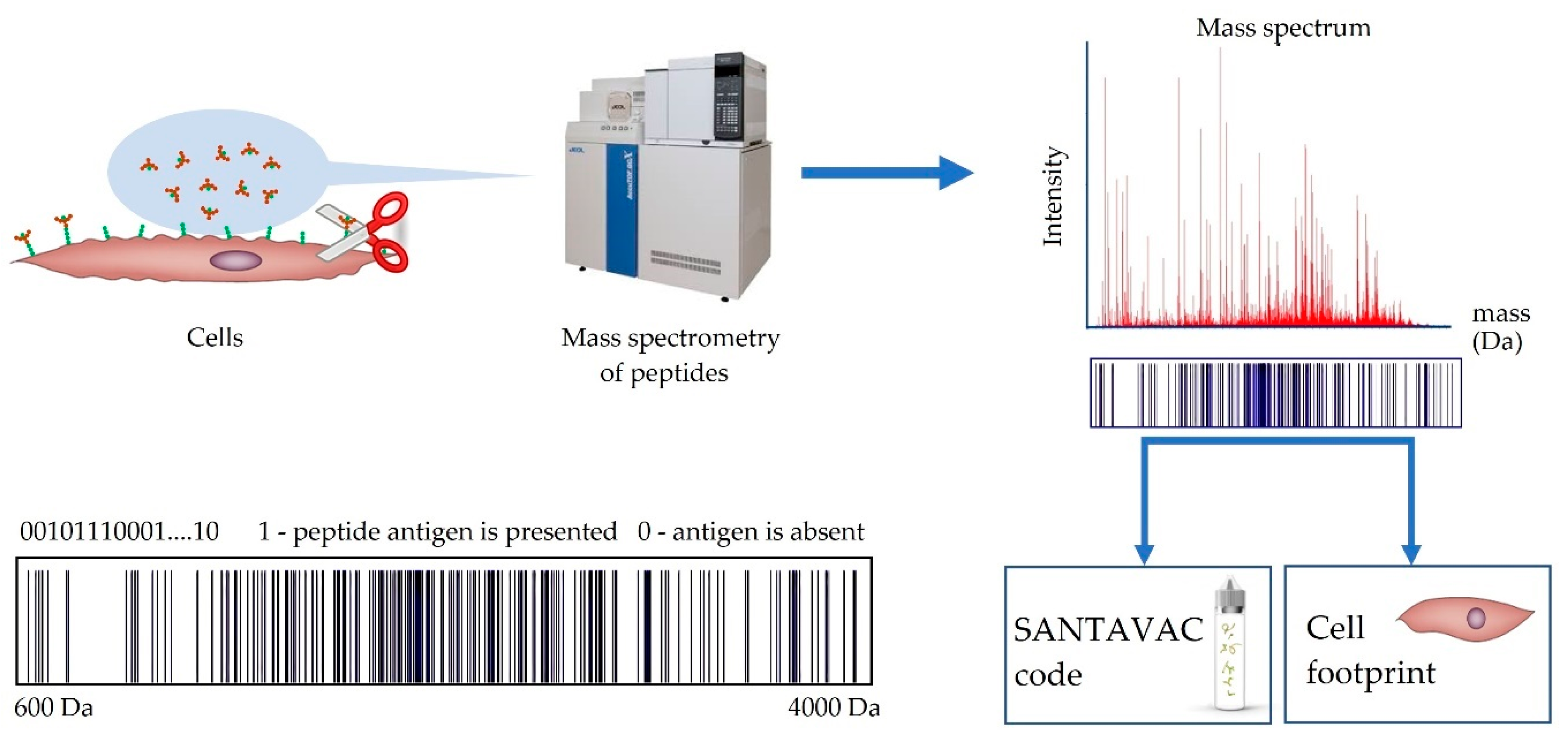
4. Method to Control SANTAVAC Composition
5. SANTAVAC Composition and Strength of Immune Response
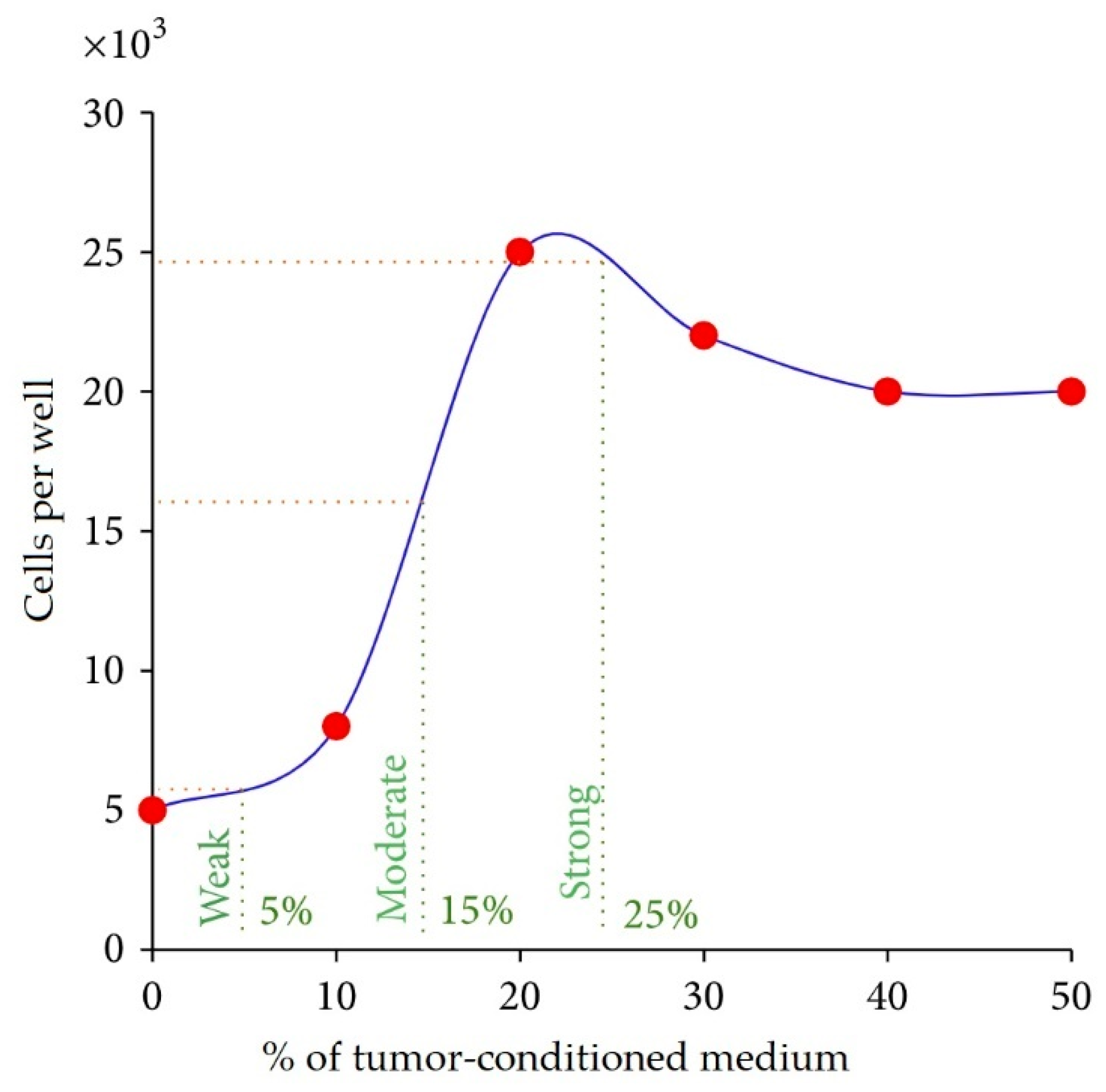
6. Antiangiogenic SANTAVAC Vaccines
6.1. Background, Rationale, and Significance
6.2. Development of Antiangiogenic SANTAVAC Vaccines
7. Antiangiogenic SANTAVAC Production and Physical Properties
8. Control of Antiangiogenic SANTAVAC Composition
9. Development Plans
- Therapeutic SANTAVAC15%-loaded allogeneic dendritic cell vaccine is an autologous cell therapy for which primary culture HMECs can be purchased to produce SANTAVAC. DCs should be obtained from patient blood.
- Preventive SANTAVAC25%-loaded allogeneic dendritic cell vaccine, similar to therapeutic vaccine, is an autologous cell therapy for which commercially available HMECs and DCs from patient blood can be used.
- Therapeutic SANTAVAC15%-loaded autologous dendritic cell vaccine is an autologous cell therapy prepared from the patient’s own biomaterial. HMECs for primary culture can be isolated from abdominal adipose by needle biopsy. DCs are obtained from the blood of the patient.
- Therapeutic SANTAVAC vaccine is allogeneic SANTAVAC15% mixed with adjuvant(s).
- Preventive SANTAVAC vaccine is allogeneic SANTAVAC25% mixed with adjuvant(s).
10. Conclusions
11. Patents
- Lokhov P.G. “Method for producing an antitumoral vaccine based on surface endothelial cell antigens”, 2007, Eurasian patent №009327.
- Lokhov P.G. Balashova E.E. “Method for testing cell culture quality”, 2007, Eurasian patent №009326.
- Lokhov P.G. Balashova E.E. “Method for producing an antitumoral vaccine”, 2009, Eurasian patent №011421.
- Lokhov P.G. “Method for producing an antitumoral vaccine based on surface endothelial cell antigens”, 2012, Japanese patent №5154641.
- Lokhov P.G. “Method for producing an antitumoral vaccine based on surface endothelial cell antigens”, 2013, Korean patent №10-1290641.
- Lokhov P.G. “Method for producing an antitumoral vaccine based on surface endothelial cell antigens”, 2015, European patent №2140873 (Protection in Switzerland/Liechtenstein, Germany, Spain, France, the United Kingdom, Ireland and Italy).
- Lokhov P.G. “Method for producing an antitumoral vaccine based on surface endothelial cell antigens”, 2017, USA patent №9844586.
Patents, Related to Same Technology Based on Cancer Cells
- 8.
- Lokhov P.G. “Antitumoral vaccine, method for producing an antitumoral vaccine and method for carry out antitumoral immunotherapy”, 2007, Eurasian patent №009326.
- 9.
- Lokhov PG. “Tumor vaccine, a method for producing a tumor vaccine and a method for carrying out antitumor immunotherapy”, 2013, Japanese patent №5172864.
- 10.
- Lokhov P.G. “Tumor vaccine, a method for producing a tumor vaccine and a method for carrying out antitumor immunotherapy”, 2012, Chinese patent №CN101636174 B.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
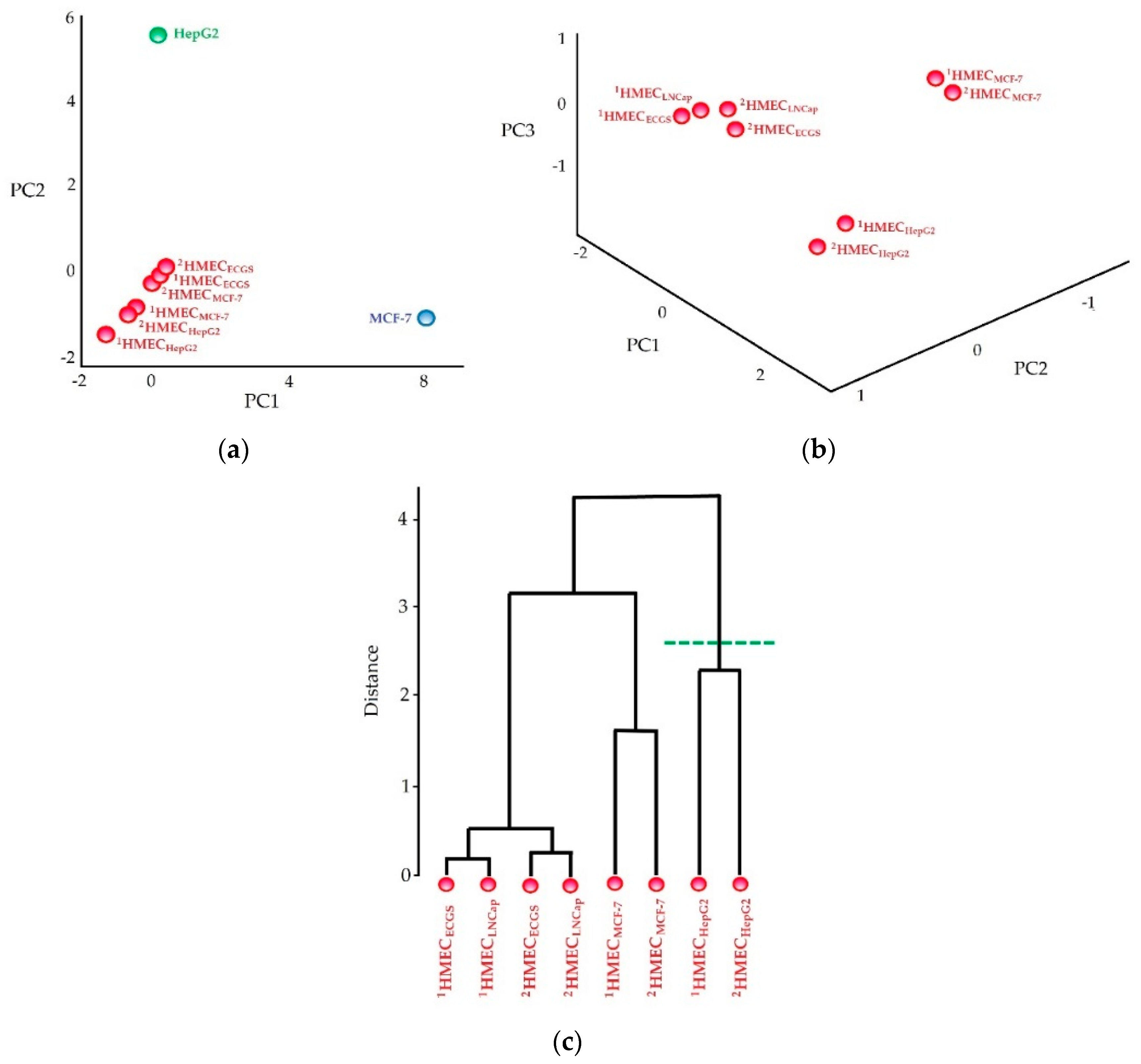
Appendix A.1. SANTAVAC Specificity to Cell Subtype
Appendix A.1.1. Experimental Procedure
- Cells were washed at least three times with 0.9% NaCl to remove traces of serum.
- Cells were quickly rinsed with cold trypsin solution (4–8 °C, 1 mg/mL, trypsin activity 5000 U/mg; Promega, USA) prepared in 0.9% NaCl followed by cell incubation at 37 °C and >95% humidity.
- Between 5 and 7 min of incubation, cells were rinsed with 0.9% NaCl (1 mL per 25 cm2 of flask surface) to wash off protein fragments released from cell surfaces. Cells at this stage must be attached to the bottom of the flask and have a round shape. If some cells are detached from the surface of the flask the samples should be quickly centrifuged to remove cells from the NaCl solution.
- The obtained NaCl solution with protein fragments was desalted using ZipTipC18 (Millipore Corp., USA) according to the manufacturer’s protocol. MALDI samples were prepared using a standard “dried droplet” method with 2,5-dihydroxybenzoic acid as the matrix. All mass spectra were acquired on a MicroFLEX MALDI-TOF mass spectrometer (Bruker Daltonik, Germany) in linear positive ion mode. Mass peak lists were formed manually. All peaks above the noise level were selected.
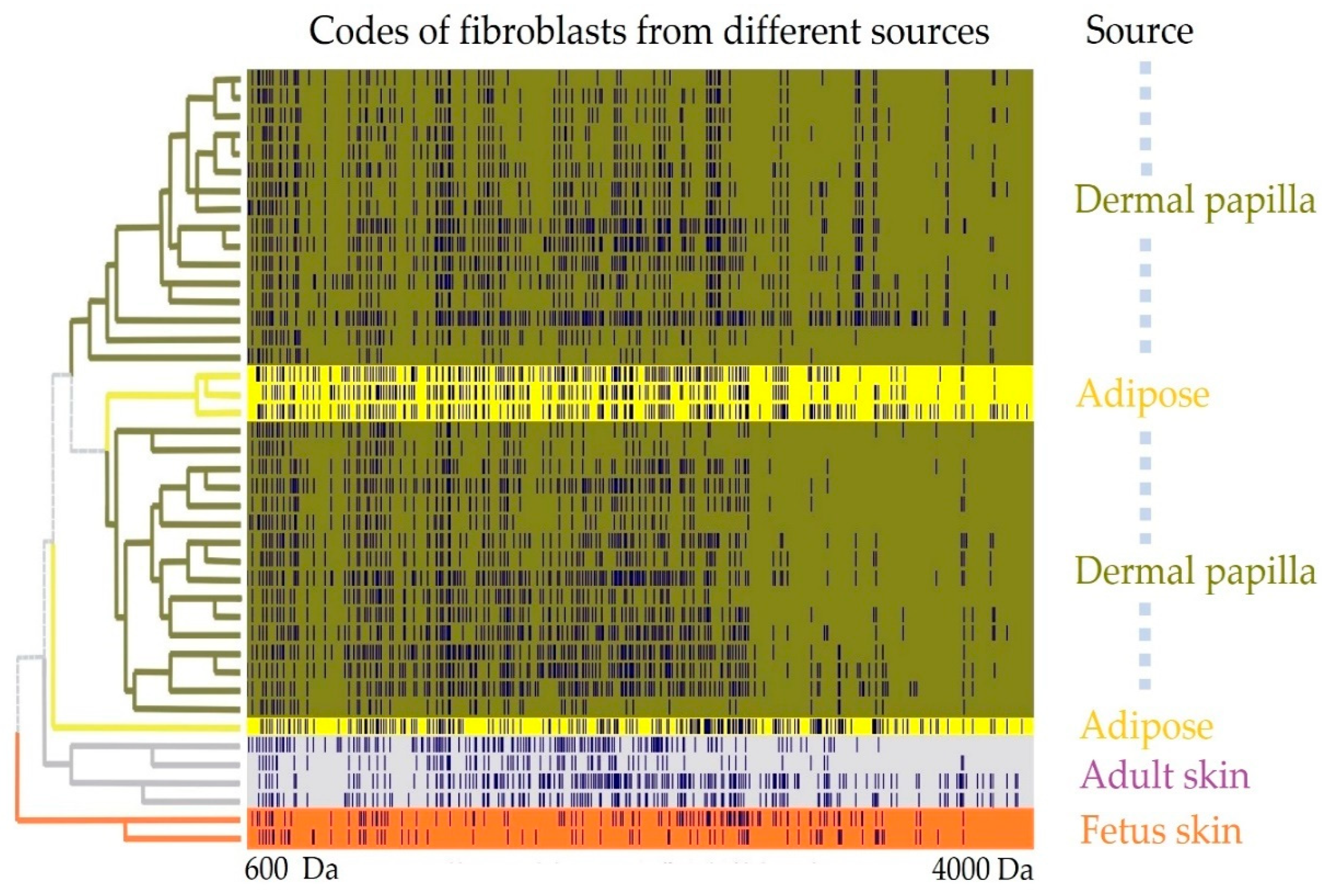
- The sets of the obtained mass spectra were binned in intervals of 0.2 Da and encoded into binary format, where 1 represents the presence of a measured peptide mass in an interval, and 0 the absence.
- Binary encoded mass spectra were partitioned into different groups by hierarchical cluster analysis using the Ward method, and correlations between spectra were presented as a distance matrix.
Appendix A.1.2. Results

Appendix B
An Example of Angiogeneic SANTAVAC Production
Appendix C

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Study | Species | SANTAVAC Type | Summary of Significant Findings |
|---|---|---|---|
| Studies of Equivalence SANTAVAC to Cellular Antigens | |||
| In vitro | Human | Against cancer cells | SANTAVAC contains 0.7% of total cell protein, and in cytotoxicity assays stimulates anti-tumor response which kills 10–40% more cancer cells than samples stimulated with total cell lysate. From these results, it was concluded that SANTAVAC contains the essential antigens to induce an immune-mediated anti-tumor effect, and therefore, is a candidate for anti-tumor vaccine development [39]. |
| In vitro | Human | Antiangiogenic | HMEC lysate and SANTAVAC produced from the equivalent number of cells had total protein concentrations of 135 and 2 µg/mL, respectively. Despite this dramatic difference in concentration, SANTAVAC was able to stimulate immune cells in cytotoxicity assays better than the HMEC lysate. Moreover, SANTAVAC was able to stimulate an immune response toward tumor-activated endothelial cells. Based on these results, it was concluded that SANTAVAC provides a comprehensive set of surface antigens that are able to induce targeted, immune-mediated cytotoxic effects against tumor endothelial cells [41]. |
| Studies of Targeting Immune Response by SANTAVAC | |||
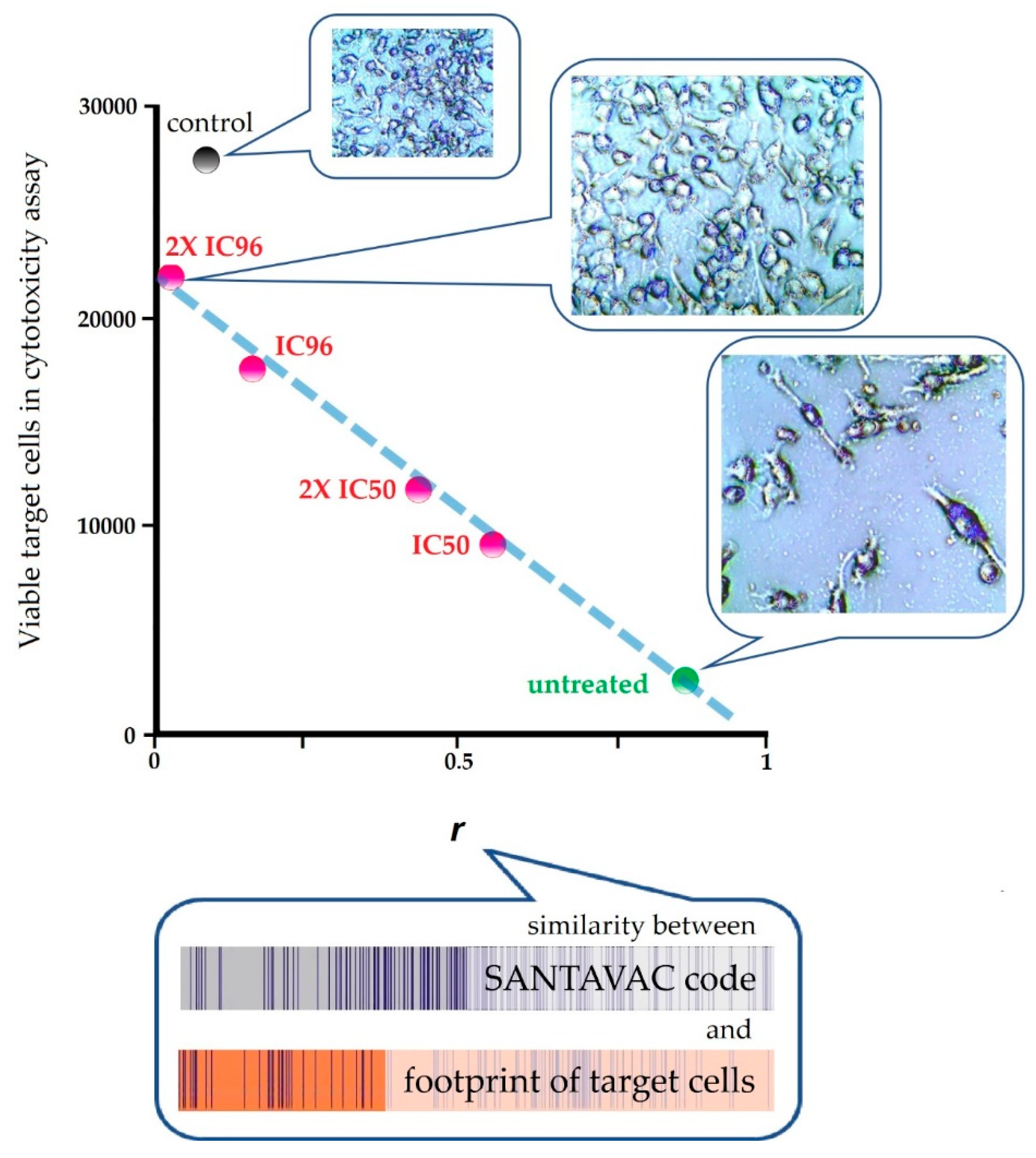
| In vitro | Human | Against cancer cells | To reveal the connection between SANTAVAC composition and immune response, a pairwise correlation analysis was performed of cell footprints of target cancer cells and SANTAVAC code. Correlation data were plotted against the results of cytotoxicity assays, wherein the surface profile of target cancer cells was gradually changed by drug selective pressure. Results clearly showed that the rate of cancer cell escape from immune response depends on the similarity between SANTAVAC composition and footprints of target cells (SANTAVAC/target similarity). This relationship between escape rate and SANTAVAC/target similarity was linearly approximated, with R2 almost equal to 1 [51]. |
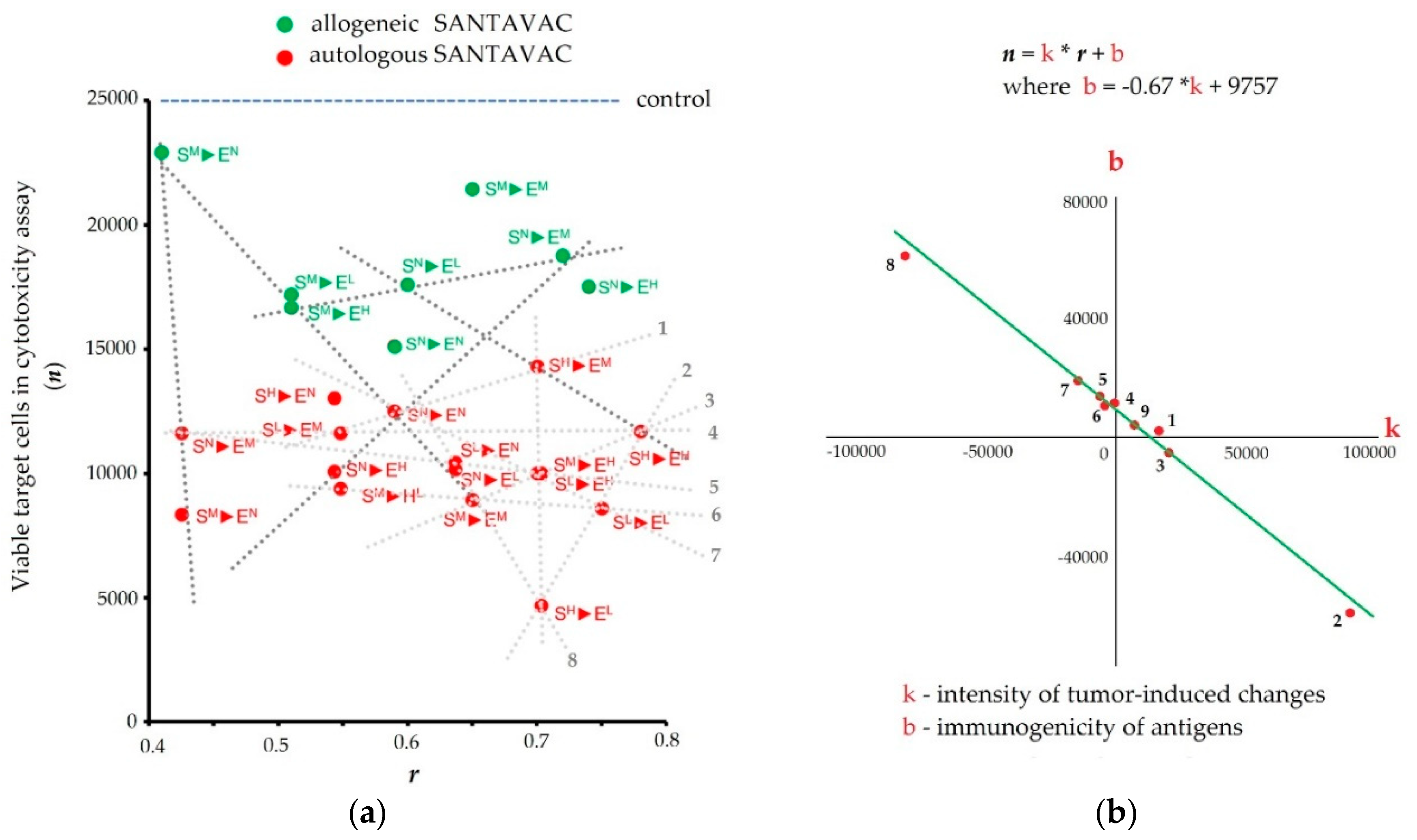
| In vitro | Human | Antiangiogenic, autologous, allogeneic | It was found that tumors induce pronounced, tumor type-dependent changes to HMEC surface that in an in vitro model of human antiangiogenic vaccination directly facilitated HMEC escape from SANTAVAC-mediated cell death (the mathematical model was proposed). Furthermore, it was found that tumors influence the HMEC phenotype unidirectionally and that HMEC immunogenicity was reciprocal to the intensity of tumor-induced changes to the HMEC surface. These findings provided data for the design of SANTAVAC with sufficient immunogenicity without posing a risk of autoimmunity [90,91,99]. |
| Efficacy Study of Final SANTAVAC Compositions | |||
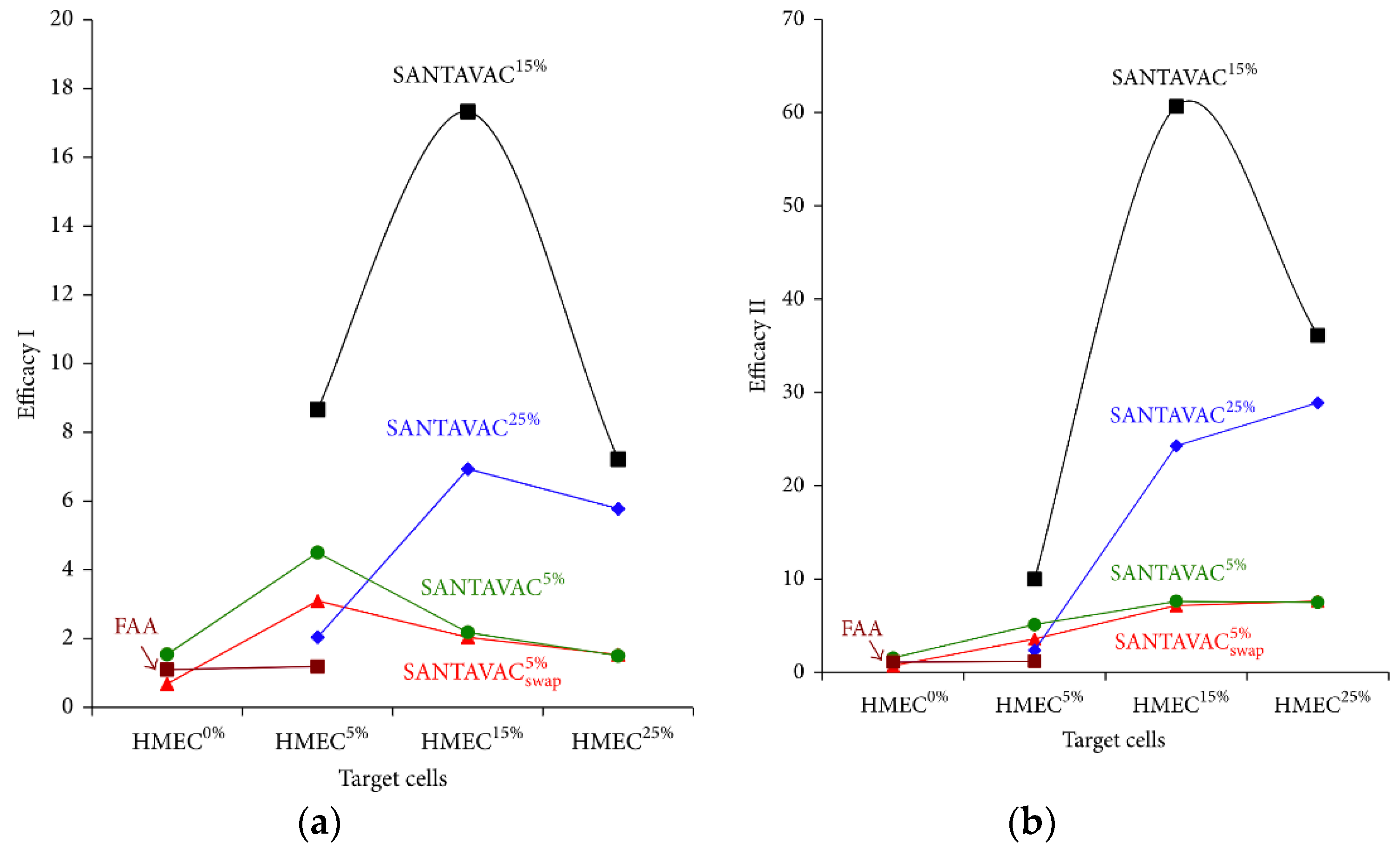
| In vitro | Human | Antiangiogenic, allogeneic | It was demonstrated that the allogeneic SANTAVAC is a perfect candidate for the development of a universal cancer vaccine (UCV) with outstanding efficacy and safety. The SANTAVAC formulation described achieved efficacy equal to 17 and 60 in relation to in vitro prediction of vaccine safety and capacity to arrest tumor growth, respectively. Criteria critical to the development of such efficient allogeneic SANTAVAC may be directly used for preparing UCV for clinical trials [92]. |
References
- Galmarini, D.; Galmarini, C.M.; Galmarini, F.C. Cancer chemotherapy: A critical analysis of its 60 years of history. Crit. Rev. Oncol. Hematol. 2012, 84, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Li, W.W.A.; Mooney, D.J.; Dranoff, G. Advances in Therapeutic Cancer Vaccines. Adv. Immunol. 2016, 130, 191–249. [Google Scholar] [PubMed]
- DeMaria, P.J.; Bilusic, M. Cancer Vaccines. Hematol. Oncol. Clin. N. Am. 2019, 33, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Palucka, K. Immunotherapy: Cancer vaccines on the move. Nat. Rev. Clin. Oncol. 2018, 15, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Xiang, R.; Luo, Y.; Niethammer, A.G.; Reisfeld, R.A. Oral DNA vaccines target the tumor vasculature and microenvironment and suppress tumor growth and metastasis. Immunol. Rev. 2008, 222, 117–128. [Google Scholar] [CrossRef]
- Sparkes, J.; Guest, N.; Csizer, Z.; Barreto, L. Stability of BCG vaccine (intravesical) theracys®/BCG therapeutic immucyst® and its importance in clinical efficacy. Dev. Biol. Stand. 1992, 77, 217–222. [Google Scholar]
- Cheever, M.A.; Higano, C.S. PROVENGE (sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef]
- Copier, J.; Dalgleish, A. Overview of tumor cell-based vaccines. Int. Rev. Immunol. 2006, 25, 297–319. [Google Scholar] [CrossRef]
- De Gruijl, T.D.; Van Den Eertwegh, A.J.M.; Pinedo, H.M.; Scheper, R.J. Whole-cell cancer vaccination: From autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol. Immunother. 2008, 57, 1569–1577. [Google Scholar] [CrossRef]
- Chiang, C.L.L.; Benencia, F.; Coukos, G. Whole tumor antigen vaccines. Semin. Immunol. 2010, 22, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Keenan, B.P.; Jaffee, E.M. Whole cell vaccines—Past progress and future strategies. Semin. Oncol. 2012, 39, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Copier, J.; Dalgleish, A. Whole-cell vaccines: A failure or a success waiting to happen? Curr. Opin. Mol. Ther. 2010, 12, 14–20. [Google Scholar] [PubMed]
- Bodey, B.; Bodey, B.J.; Siegel, S.E.; Kaiser, H.E. Failure of cancer vaccines: The significant limitations of this approach to immunotherapy. Anticancer Res. 2000, 20, 2665–2676. [Google Scholar] [PubMed]
- Lokhov, P.G.; Balashova, E.E. Cellular cancer vaccines: An update on the development of vaccines generated from cell surface antigens. J. Cancer 2010, 1, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, M.; Ambrose, E.J. The Surface Properties of Cancer Cells: A Review. Cancer Res. 1962, 22, 525–548. [Google Scholar]
- Calman, F. Ultrastructural comparison of the cell coat in normal and chronic lymphocytic leukemic blood lymphocytes by concanavalin A labelling and cationic staining. Path. Eur. 1975, 10, 203–214. [Google Scholar]
- Gasic, G.J.; Loebel, F. Cytochemical identification of protein amino acids in the cell coat of mouse ascites tumor cells. Lab. Investig. 1966, 15, 1310–1319. [Google Scholar]
- Mallucci, L.; Poste, G.H.; Wells, V. Synthesis of cell coat in normal and transformed cells. Nat. New Biol. 1972, 235, 222–223. [Google Scholar] [CrossRef]
- Rittenhouse, H.G.; Rittenhouse, J.W.; Takemoto, L. Characterization of the cell coat of Ehrlich ascites tumor cells. Biochemistry 1978, 17, 829–837. [Google Scholar] [CrossRef]
- Ruiz-Cabello, F.; Cabrera, T.; Lopez-Nevot, M.A.; Garrido, F. Impaired surface antigen presentation in tumors: Implications for T cell-based immunotherapy. Semin. Cancer Biol. 2002, 12, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.T.; Restifo, N.P. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 2002, 3, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Bast, J.C. Principles of cancer biology: Tumor immunology. In Cancer Principles and Practice of Oncology; DeVita, V., Hellman, S.S.R., Eds.; J. B. Lippincott Co.: Philadelphia, PA, USA, 1985; pp. 125–150. [Google Scholar]
- Putnam, J.B.; Roth, J.A. Identification and characterization of a tumor-derived immunosuppressive glycoprotein from murine melanoma K-1735. Cancer Immunol. Immunother. 1985, 19, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.J.; Macchi, B.; Papazoglou, S.; Oldfield, E.H.; Kornblith, P.L.; Smith, B.H.; Gately, M.K. Lymphoid cell-glioma cell interaction enhances cell coat production by human gliomas: Novel suppressor mechanism. Science 1983, 220, 739–742. [Google Scholar] [CrossRef]
- Liu, F.T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- LeGrue, S.J. Noncytolytic extraction of cell surface antigens using butanol. Cancer Metastasis Rev. 1985, 4, 209–219. [Google Scholar] [CrossRef]
- Ben-Or, S.; Doljanski, F. Single-cell suspensions as tissue antigens. Exp. Cell Res. 1960, 20, 641–644. [Google Scholar] [CrossRef]
- Quash, G.; Delain, E.; Huppert, J. Effect of antipolyamine antibodies on mammalian cells in tissue culture. Exp. Cell Res. 1971, 66, 426–432. [Google Scholar] [CrossRef]
- Nairn, R.C.; Richmond, H.G.; Mc, E.M.; Fothergill, J.E. Immunological differences between normal and malignant cells. Br. Med. J. 1960, 2, 1335–1340. [Google Scholar] [CrossRef]
- Fakhri, O.; Tan, R.S. The effect of trypsin on cell surface antigens. Cell Immunol. 1975, 15, 452–456. [Google Scholar] [CrossRef]
- Molinari, J.A.; Platt, D. Modification of surface membrane antigens by trypsin. Proc. Soc. Exp. Biol. Med. 1975, 148, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Kohnert-Stavenhagen, E.; Zimmermann, B. Changes in the surface coat of mesenchymal cells of mouse limb buds after enzymatic cell separation. J. Embryol. Exp. Morphol. 1980, 59, 145–155. [Google Scholar] [PubMed]
- Gasic, G.; Gasic, T. Removal and regeneration of the cell coating in tumour cells. Nature 1962, 196, 170. [Google Scholar] [CrossRef] [PubMed]
- Uhlenbruck, G. Action of proteolytic enzymes on the human erythrocyte surface. Nature 1961, 190, 181. [Google Scholar] [CrossRef] [PubMed]
- Anglhileri, L.J. Dermietzel Cell coat in tumor cells—Effects of trypsin and EDTA: A biochemical and morphological study. Oncology 1976, 33, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, N.; Economou, G.C.; Boone, C.W. Accelerated regeneration of trypsin-treated surface antigens of simian virus 40-transformed BALB/3T3 cells induced by X-irradiation. Cancer Res. 1976, 36, 1258–1262. [Google Scholar]
- Baumann, H.; Doyle, D. Effect of trypsin on the cell surface proteins of hepatoma tissue culture cells. Characterization of a carbohydrate-rich glycopeptide released from a calcium binding membrane glycoprotein. J. Biol. Chem. 1979, 254, 3935–3946. [Google Scholar]
- Balashova, E.E.; Lokhov, P.G. Proteolytically-cleaved fragments of cell-surface proteins from live tumor cells stimulate anti-tumor immune response in vitro. J. Carcinog. Mutagen. 2010, 1, 1–3. [Google Scholar] [CrossRef]
- Balashova, E.; Lokhov, P. SANTAVAC TM: A Novel Universal Antigen Composition for Developing Cancer Vaccines. Recent Pat. Biotechnol. 2017, 11, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Balashova, E.E.; Lokhov, P.G. Proteolytically-cleaved fragments of cell surface proteins stimulate a cytotoxic immune response against tumor-activated endothelial cells in vitro. J. Cancer Sci. Ther. 2010, 2, 126–131. [Google Scholar] [CrossRef]
- Stacey, G.N.; Masters, J.R.W.; Hay, R.J.; Drexler, H.G.; MacLeod, R.A.F.; Freshney, R.I. Cell contamination leads to inaccurate data: We must take action now. Nature 2000, 403, 356. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, R.A.F.; Dirks, W.G.; Matsuo, Y.; Kaufmann, M.; Milch, H.; Drexler, H.G. Widespread intraspecies cross-contamination of human tumor cell lines arising at source. Int. J. Cancer 1999, 83, 555–563. [Google Scholar] [CrossRef]
- Identity crisis. Nature 2009, 457, 935–936. Available online: https://www.nature.com/articles/457935b (accessed on 17 November 2019). [CrossRef] [PubMed] [Green Version]
- Cabrera, C.M.; Cobo, F.; Nieto, A.; Cortés, J.L.; Montes, R.M.; Catalina, P.; Concha, A. Identity tests: Determination of cell line cross-contamination. Cytotechnology 2006, 51, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Wright, W.E.; Shay, J.W. Historical claims and current interpretations of replicative aging. Nat. Biotechnol. 2002, 20, 682–688. [Google Scholar] [CrossRef]
- Huggins, J.W.; Chestnut, R.W.; Durham, N.N.; Carraway, K.L. Molecular changes in cell surface membranes resulting from trypsinization of sarcoma 180 tumor cells. BBA Biomembr. 1976, 426, 630–637. [Google Scholar] [CrossRef]
- Angus, R.; Collins, C.M.P.; Symes, M.O. Expression of major histocompatibility complex (MHC) antigens and their loss on culture in renal carcinoma. Eur. J. Cancer 1993, 29, 2158–2160. [Google Scholar] [CrossRef]
- Lokhov, P.; Balashova, E.; Dashtiev, M. Cell proteomic footprint. Rapid Commun. Mass Spectrom. 2009, 23, 680–682. [Google Scholar] [CrossRef] [Green Version]
- Balashova, E.E.; Dashtiev, M.I.; Lokhov, P.G. Proteomic Footprinting of Drug-Treated Cancer Cells as a Measure of Cellular Vaccine Efficacy for the Prevention of Cancer Recurrence. Mol. Cell. Proteom. 2012, 11, M111.014480. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Wentink, M.Q.; Huijbers, E.J.M.; de Gruijl, T.D.; Verheul, H.M.W.; Olsson, A.K.; Griffioen, A.W. Vaccination approach to anti-angiogenic treatment of cancer. Biochim. Biophys. Acta Rev. Cancer 2015, 1855, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Allegra, C.J.; Yothers, G.; O’Connell, M.J.; Sharif, S.; Petrelli, N.J.; Lopa, S.H.; Wolmark, N. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J. Clin. Oncol. 2013, 31, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Moserle, L.; Jiménez-Valerio, G.; Casanovas, O. Antiangiogenic therapies: Going beyond their limits. Cancer Discov. 2014, 4, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W. Therapeutic Approaches of Angiogenesis Inhibition: Are We Tackling the Problem at the Right Level? Trends Cardiovasc. Med. 2007, 17, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Bridges, E.M.; Harris, A.L. The angiogenic process as a therapeutic target in cancer. Biochem. Pharmacol. 2011, 81, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Seidi, K.; Jahanban-Esfahlan, R.; Zarghami, N. Tumor rim cells: From resistance to vascular targeting agents to complete tumor ablation. Tumor Biol. 2017, 39, 1010428317691001. [Google Scholar] [CrossRef] [Green Version]
- Van Beijnum, J.R.; Dings, R.P.; Van Der Linden, E.; Zwaans, B.M.M.; Ramaekers, F.C.S.; Mayo, K.H.; Griffioen, A.W. Gene expression of tumor angiogenesis dissected: Specific targeting of colon cancer angiogenic vasculature. Blood 2006, 108, 2339–2348. [Google Scholar] [CrossRef] [Green Version]
- Seaman, S.; Stevens, J.; Yang, M.Y.; Logsdon, D.; Graff-Cherry, C.; St Croix, B. Genes that Distinguish Physiological and Pathological Angiogenesis. Cancer Cell 2007, 11, 539–554. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.Q.; Wang, Q.R.; Zhao, X.; Yang, L.; Tian, L.; Lu, Y.; Kang, B.; Lu, C.J.; Huang, M.J.; Lou, Y.Y.; et al. Immunotherapy of tumors with xenogeneic endothelial cells as a vaccine. Nat. Med. 2000, 6, 1160–1166. [Google Scholar] [CrossRef]
- Okaji, Y.; Tsuno, N.H.; Kitayama, J.; Saito, S.; Takahashi, T.; Kawai, K.; Yazawa, K.; Asakage, M.; Hori, N.; Watanabe, T.; et al. Vaccination with autologous endothelium inhibits angiogenesis and metastasis of colon cancer through autoimmunity. Cancer Sci. 2004, 95, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Okaji, Y.; Tsuno, N.H.; Tanaka, M.; Yoneyama, S.; Matsuhashi, M.; Kitayama, J.; Saito, S.; Nagura, Y.; Tsuchiya, T.; Yamada, J.; et al. Pilot study of anti-angiogenic vaccine using fixed whole endothelium in patients with progressive malignancy after failure of conventional therapy. Eur. J. Cancer 2008, 44, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Uldry, E.; Faes, S.; Demartines, N.; Dormond, O. Fine-tuning tumor endothelial cells to selectively kill cancer. Int. J. Mol. Sci. 2017, 18, 1401. [Google Scholar] [CrossRef] [Green Version]
- Algire, G.H.; Chalkley, H.W.; Legallais, F.Y.; Park, H.D. Vasculae reactions of normal and malignant tissues in vivo. i. vascular reactions of mice to wounds and to normal and neoplastic transplants. J. Natl. Cancer Inst. 1945, 6, 73–85. [Google Scholar] [CrossRef]
- Boehm, T.; Folkman, J.; Browder, T.; O’Reilly, M.S. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390, 404–407. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Folkman, J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 1990, 82, 4–6. [Google Scholar] [CrossRef]
- Pluda, J.M. Tumor-associated angiogenesis: Mechanisms, clinical implications, and therapeutic strategies. Semin. Oncol. 1997, 24, 203–218. [Google Scholar]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodarev, N.N.; Yu, J.; Labay, E.; Darga, T.; Brown, C.K.; Mauceri, H.J.; Yassari, R.; Gupta, N.; Weichselbaum, R.R. Tumour-endothelium interactions in co-culture: Coordinated changes of gene expression profiles and phenotypic properties of endothelial cells. J. Cell Sci. 2003, 116, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhati, R.; Patterson, C.; Livasy, C.A.; Fan, C.; Ketelsen, D.; Hu, Z.; Reynolds, E.; Tanner, C.; Moore, D.T.; Gabrielli, F.; et al. Molecular characterization of human breast tumor vascular cells. Am. J. Pathol. 2008, 172, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- St Croix, B.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montgomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes expressed in human tumor endothelium. Science 2000, 289, 1197–1202. [Google Scholar] [CrossRef]
- Kumar, S.; West, D.C.; Ager, A. Heterogeneity in endothelial cells from large vessels and microvessels. Differentiation 1987, 36, 57–70. [Google Scholar] [CrossRef]
- Lang, I.; Pabst, M.A.; Hiden, U.; Blaschitz, A.; Dohr, G.; Hahn, T.; Desoye, G. Heterogeneity of microvascular endothelial cells isolated from human term placenta and macrovascular umbilical vein endothelial cells. Eur. J. Cell Biol. 2003, 82, 163–173. [Google Scholar] [CrossRef]
- Swerlick, R.A.; Lee, K.H.; Wick, T.M.; Lawley, T.J. Human dermal microvascular endothelial but not human umbilical vein endothelial cells express CD36 in vivo and in vitro. J. Immunol. 1992, 148, 78–83. [Google Scholar]
- Swerlick, R.A.; Lee, K.H.; Li, L.J.; Sepp, N.T.; Caughman, S.W.; Lawley, T.J. Regulation of vascular cell adhesion molecule 1 on human dermal microvascular endothelial cells. J. Immunol. 1992, 149, 698–705. [Google Scholar]
- Lee, K.H.; Lawley, T.J.; Xu, Y.L.; Swerlick, R.A. VCAM-1-, ELAM-1-, and ICAM-1-independent adhesion of melanoma cells to cultured human dermal microvascular endothelial cells. J. Investig. Dermatol. 1992, 98, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pötzsch, B.; Grulich-Henn, J.; Rössing, R.; Wille, D.; Müller-Berghaus, G. Identification of endothelial and mesothelial cells in human omental tissue and in omentum-derived cultured cells by specific cell markers. Lab. Investig. 1990, 63, 841–852. [Google Scholar] [PubMed]
- Hewett, P.W.; Murray, J.C.; Price, E.A.; Watts, M.E.; Woodcock, M. Isolation and characterization of microvessel endothelial cells from human mammary adipose tissue. Vitr. Cell. Dev. Biol. Anim. J. Soc. Vitr. Biol. 1993, 29A, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Hull, M.; Hewett, P.; Brough, J.; Hawkey, C. Isolation and culture of human gastric endothelial cells. Gastroenterology 1996, 111, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Hewett, P.; Murray, J. Immunomagnetic purification of human microvessel endothelial cells using Dynabeads coated with monoclonal antibodies to PECAM-1. Eur. J. Cell Biol. 1993, 62, 451–454. [Google Scholar]
- Hewett, P.W.; Murray, J.C. Human omental mesothelial cells: A simple method for isolation and discrimination from endothelial cells. Vitr. Cell. Dev. Biol. Anim. 1994, 30A, 145–147. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, Y.; Antonyak, M.; Peng, X. Isolation and characterization of vascular endothelial cells from murine heart and lung. Methods Mol. Biol. 2012, 843, 147–154. [Google Scholar]
- Jankowski, R. A Comparison of Commercially-Available Human Skeletal Muscle Cells and Media for Research Applications. Nat. Methods 2011. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Balashova, E.E. Tumor-induced endothelial cell surface heterogeneity directly affects endothelial cell escape from a cell-mediated immune response in vitro. Hum. Vaccin. Immunother. 2013, 9, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Lokhov, P.G.; Balashova, E.E. Design of universal cancer vaccines using natural tumor vessel-specific antigens (SANTAVAC). Hum. Vaccin. Immunother. 2015, 11, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Lokhov, P.G.; Balashova, E.E. Allogeneic antigen composition for preparing universal cancer vaccines. J. Immunol. Res. 2016, 5031529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nör, J.E.; Peters, M.C.; Christensen, J.B.; Sutorik, M.M.; Linn, S.; Khan, M.K.; Addison, C.L.; Mooney, D.J.; Polverini, P.J. Engineering and characterization of functional human microvessels in immunodeficient mice. Lab. Investig. 2001, 81, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Imai, A.; Krishnamurthy, S.; Zhang, Z.; Zeitlin, B.D.; Nör, J.E. Xenograft tumors vascularized with murine blood vessels may overestimate the effect of anti-tumor drugs: A pilot study. PLoS ONE 2013, 8, e84236. [Google Scholar] [CrossRef] [PubMed]
- Lü, Z.F.; Cai, S.Q.; Wu, J.J.; Zheng, M. Biological characterization of cultured dermal papilla cells and hair follicle regeneration in vitro and in vivo. Chin. Med. J. 2006, 119, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–288. [Google Scholar] [CrossRef] [Green Version]
- Kolokol’tseva, T.D.; Iurchenko, N.D.; Kolosov, N.G.; Nechaeva, E.A.; Shumakova, O.V.; Khristo, S.A. Prospects of use human fetal fibroblasts in the treatment of various etiology wounds. Vestn. Ross. Akad. Med. Nauk 1998, 3, 32–35. [Google Scholar]
- Morimoto, N.; Saso, Y.; Tomihata, K.; Taira, T.; Takahashi, Y.; Ohta, M.; Suzuki, S. Viability and function of autologous and allogeneic fibroblasts seeded in dermal substitutes after implantation. J. Surg. Res. 2005, 125, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Lokhov, P.G.; Balashova, E.E. Universal cancer vaccine: An update on the design of cancer vaccines generated from endothelial cells. Hum. Vaccin. Immunother. 2013, 9, 1549–1552. [Google Scholar] [CrossRef] [Green Version]








| Property | Value |
|---|---|
| Appearance (turbidity) | Clear |
| Appearance (color) | Clear |
| Appearance (form) | Solution |
| pH | 7.2–7.6 |
| Osmolality | 260–300 mOs/kg |
| Salt composition | Hank’s balanced salt solution |
| Endotoxin level | <1.0 EU/mL |
| Cytotoxicity overlay | Non-toxic |
| Polypeptides concentration | 2.5–3 mg/L |
| Trypsin autolysis products | traces |
| Activity | 0.8–0.9 U/mL 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lokhov, P.G.; Mkrtichyan, M.; Mamikonyan, G.; Balashova, E.E. SANTAVACTM: Summary of Research and Development. Vaccines 2019, 7, 186. https://doi.org/10.3390/vaccines7040186
Lokhov PG, Mkrtichyan M, Mamikonyan G, Balashova EE. SANTAVACTM: Summary of Research and Development. Vaccines. 2019; 7(4):186. https://doi.org/10.3390/vaccines7040186
Chicago/Turabian StyleLokhov, Petr G., Mikayel Mkrtichyan, Grigor Mamikonyan, and Elena E. Balashova. 2019. "SANTAVACTM: Summary of Research and Development" Vaccines 7, no. 4: 186. https://doi.org/10.3390/vaccines7040186