The Interplay between Dengue Virus and the Human Innate Immune System: A Game of Hide and Seek


{kind=link}
Abstract
:1. Introduction
1.1. Disease Burden
1.2. Clinical Manifestation
1.3. Dengue Virus Life Cycle
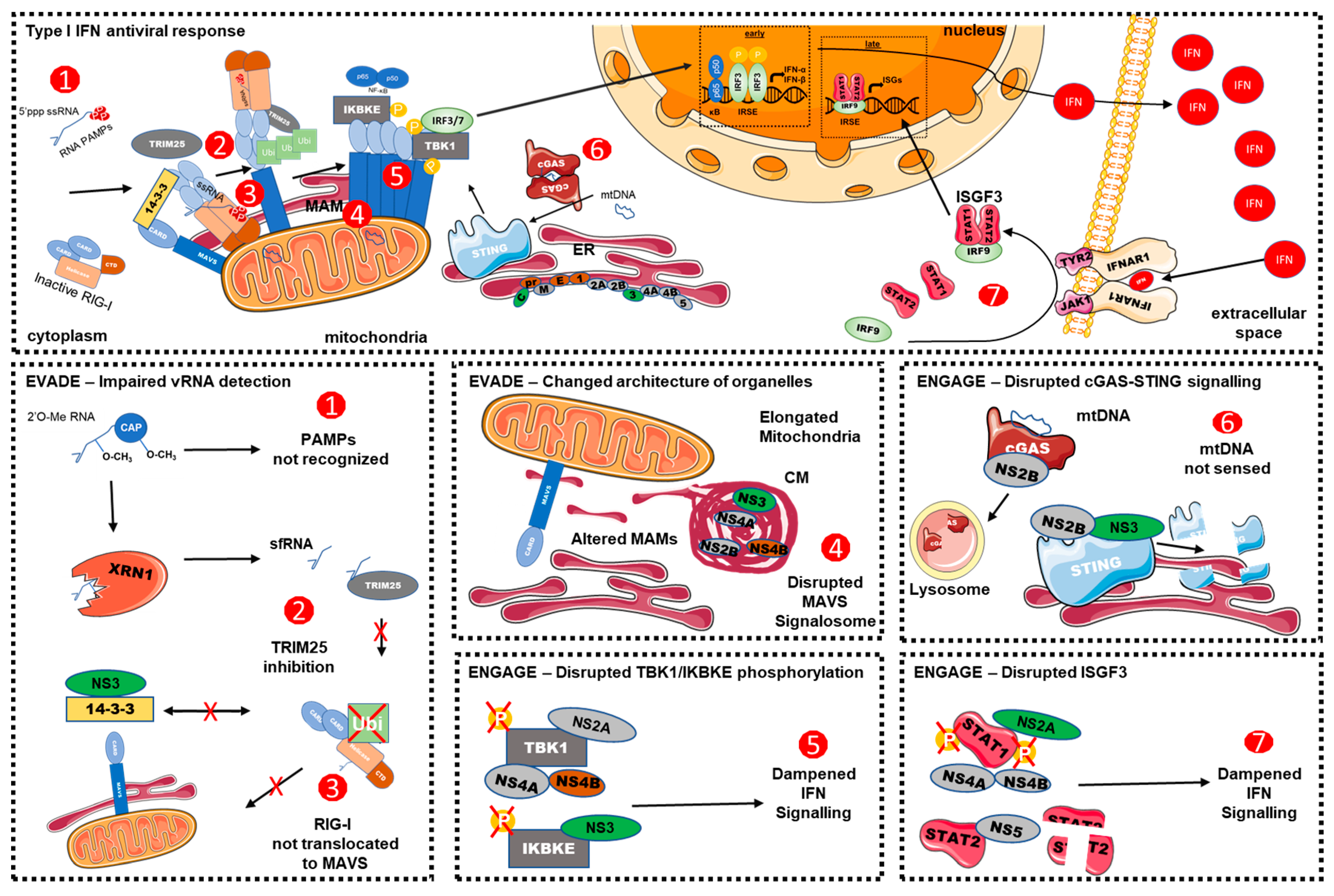
2. Recognition of DENV by Pathogen Recognition Receptors
2.1. DENV vRNA-Recognition
2.2. The DNA Sensing Pathway and DENV Infection
3. Viral Countermeasures
3.1. Interference with RLR-Dependent Signalling
3.2. Interference with the cGAS/STING Pathway
3.3. Interference with the IFN Signalling
3.4. Interference with Other Mechanisms
4. Importance for Vaccine Development
4.1. DENVΔ30 Recombinant Dengue Virus
4.2. 2′-O-methyltransferase-Deficient Dengue Vaccine
4.3. Immunocompetent Mouse Model for Denv Vaccine research
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Brady, O.J.; Gething, P.W.; Bhatt, S.; Messina, J.P.; Brownstein, J.S.; Hoen, A.G.; Moyes, C.L.; Farlow, A.W.; Scott, T.W.; Hay, S.I. Refining the Global Spatial Limits of Dengue Virus Transmission by Evidence-Based Consensus. PLoS Negl. Trop. Dis. 2012, 6, e1760. [Google Scholar] [CrossRef] [PubMed]
- Stanaway, J.D.; Shepard, D.S.; Undurraga, E.A.; Halasa, Y.A.; Coffeng, L.E.; Brady, O.J.; Hay, S.I.; Bedi, N.; Bensenor, I.M.; Castañeda-Orjuela, C.A.; et al. The global burden of dengue: An analysis from the Global Burden of Disease Study 2013. Lancet Infect. Dis. 2016, 16, 712–723. [Google Scholar] [PubMed]
- Ramos-Castañeda, J.; Barreto Dos Santos, F.; Martínez-Vega, R.; Galvão de Araujo, J.M.; Joint, G.; Sarti, E. Dengue in Latin America: Systematic Review of Molecular Epidemiological Trends. PLoS Negl. Trop. Dis. 2017, 11, e0005224. [Google Scholar] [CrossRef] [PubMed]
- San Martín, J.L.; Brathwaite, O.; Zambrano, B.; Solórzano, J.O.; Bouckenooghe, A.; Dayan, G.H.; Guzmán, M.G. The epidemiology of dengue in the americas over the last three decades: A worrisome reality. Am. J. Trop. Med. Hyg. 2010, 82, 128–135. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control; World Health Organization: Geneva, Switzerland, 2009; ISBN 9789241547871. [Google Scholar]
- Gubler, D.J.; Suharyono, W.; Tan, R.; Abidin, M.; Sie, A. Viraemia in patients with naturally acquired dengue infection. Bull. World Health Organ. 1981, 59, 623–630. [Google Scholar] [PubMed]
- Gómez-Ochoa, S.A. Viremia en plasma como factor asociado a gravedad en la infección por el virus del dengue: Revisión sistemática de la literatura. Revista Chilena de Infectología 2018, 35, 176–183. [Google Scholar] [PubMed]
- Vaughn, D.W.; Green, S.; Kalayanarooj, S.; Innis, B.L.; Nimmannitya, S.; Suntayakorn, S.; Endy, T.P.; Raengsakulrach, B.; Rothman, A.L.; Ennis, F.A.; et al. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J. Infect. Dis. 2000, 181, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Srikiatkhachorn, A.; Mathew, A.; Rothman, A.L. Immune-mediated cytokine storm and its role in severe dengue. Semin. Immunopathol. 2017, 39, 563–574. [Google Scholar] [CrossRef]
- Apte-Sengupta, S.; Sirohi, D.; Kuhn, R.J. Coupling of replication and assembly in flaviviruses. Curr. Opin. Virol. 2014, 9, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Mazeaud, C.; Freppel, W.; Chatel-Chaix, L. The Multiples Fates of the Flavivirus RNA Genome During Pathogenesis. Front. Genet. 2018, 9, 595. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Flaviviridae Replication Organelles: Oh, What a Tangled Web We Weave. Annu. Rev. Virol. 2015, 2, 289–310. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Bartenschlager, R. Dengue virus- and hepatitis C virus-induced replication and assembly compartments: The enemy inside–caught in the web. J. Virol. 2014, 88, 5907–5911. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.E.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Bühler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef]
- Wathelet, M.G.; Lin, C.H.; Parekh, B.S.; Ronco, L.V.; Howley, P.M.; Maniatis, T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1998, 1, 507–518. [Google Scholar] [CrossRef]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 1998, 18, 2986–2996. [Google Scholar] [CrossRef]
- Yang, H.; Lin, C.H.; Ma, G.; Baffi, M.O.; Wathelet, M.G. Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators. J. Biol. Chem. 2003, 278, 15495–15504. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.S.; Veals, S.A.; Fu, X.Y.; Levy, D.E. Interferon-alpha regulates nuclear translocation and DNA-binding affinity of ISGF3, a multimeric transcriptional activator. Genes Dev. 1990, 4, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.R.; Levy, O. 3—Innate immunity. In Clinical Immunology, 4th ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Content Repository Only: London, UK, 2013; pp. 35–46. ISBN 9780723436911. [Google Scholar]
- Chow, K.T.; Gale, M., Jr.; Loo, Y.-M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef] [PubMed]
- Said, E.A.; Tremblay, N.; Al-Balushi, M.S.; Al-Jabri, A.A.; Lamarre, D. Viruses Seen by Our Cells: The Role of Viral RNA Sensors. J. Immunol. Res. 2018, 2018, 9480497. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Xu, H.; He, X.; Zheng, H.; Huang, L.J.; Hou, F.; Yu, Z.; de la Cruz, M.J.; Borkowski, B.; Zhang, X.; Chen, Z.J.; et al. Correction: Structural basis for the prion-like MAVS filaments in antiviral innate immunity. Elife 2015, 4. [Google Scholar] [CrossRef]
- Xu, L.-G.; Wang, Y.-Y.; Han, K.-J.; Li, L.-Y.; Zhai, Z.; Shu, H.-B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef]
- Onoguchi, K.; Onomoto, K.; Takamatsu, S.; Jogi, M.; Takemura, A.; Morimoto, S.; Julkunen, I.; Namiki, H.; Yoneyama, M.; Fujita, T. Virus-Infection or 5′ppp-RNA Activates Antiviral Signal through Redistribution of IPS-1 Mediated by MFN1. PLoS Pathog. 2010, 6, e1001012. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.-H.; Liao, C.-L.; Lin, Y.-L. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 2006, 8, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Nasirudeen, A.M.A.; Wong, H.H.; Thien, P.; Xu, S.; Lam, K.-P.; Liu, D.X. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl. Trop. Dis. 2011, 5, e926. [Google Scholar] [CrossRef] [PubMed]
- da Conceição, T.M.; Rust, N.M.; Berbel, A.C.E.R.; Martins, N.B.; do Nascimento Santos, C.A.; Da Poian, A.T.; de Arruda, L.B. Essential role of RIG-I in the activation of endothelial cells by dengue virus. Virology 2013, 435, 281–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olagnier, D.; Scholte, F.E.M.; Chiang, C.; Albulescu, I.C.; Nichols, C.; He, Z.; Lin, R.; Snijder, E.J.; van Hemert, M.J.; Hiscott, J. Inhibition of dengue and chikungunya virus infections by RIG-I-mediated type I interferon-independent stimulation of the innate antiviral response. J. Virol. 2014, 88, 4180–4194. [Google Scholar] [CrossRef] [PubMed]
- Chazal, M.; Beauclair, G.; Gracias, S.; Najburg, V.; Simon-Lorière, E.; Tangy, F.; Komarova, A.V.; Jouvenet, N. RIG-I Recognizes the 5′ Region of Dengue and Zika Virus Genomes. Cell Rep. 2018, 24, 320–328. [Google Scholar] [CrossRef]
- Errett, J.S.; Suthar, M.S.; McMillan, A.; Diamond, M.S.; Gale, M. The Essential, Nonredundant Roles of RIG-I and MDA5 in Detecting and Controlling West Nile Virus Infection. J. Virol. 2013, 87, 11416–11425. [Google Scholar] [CrossRef] [Green Version]
- Leifer, C.A.; Medvedev, A.E. Molecular mechanisms of regulation of Toll-like receptor signaling. J. Leukoc. Biol. 2016, 100, 927–941. [Google Scholar] [CrossRef]
- Gao, D.; Li, W. Structures and recognition modes of toll-like receptors. Proteins 2017, 85, 3–9. [Google Scholar] [CrossRef]
- Tsai, Y.-T.; Chang, S.-Y.; Lee, C.-N.; Kao, C.-L. Human TLR3 recognizes dengue virus and modulates viral replication in vitro. Cell. Microbiol. 2009, 11, 604–615. [Google Scholar] [CrossRef]
- Liang, Z.; Wu, S.; Li, Y.; He, L.; Wu, M.; Jiang, L.; Feng, L.; Zhang, P.; Huang, X. Activation of Toll-like receptor 3 impairs the dengue virus serotype 2 replication through induction of IFN-β in cultured hepatoma cells. PLoS ONE 2011, 6, e23346. [Google Scholar] [CrossRef]
- Sariol, C.A.; Martínez, M.I.; Rivera, F.; Rodríguez, I.V.; Pantoja, P.; Abel, K.; Arana, T.; Giavedoni, L.; Hodara, V.; White, L.J.; et al. Decreased dengue replication and an increased anti-viral humoral response with the use of combined Toll-like receptor 3 and 7/8 agonists in macaques. PLoS ONE 2011, 6, e19323. [Google Scholar] [CrossRef]
- Dhanwani, R.; Takahashi, M.; Sharma, S. Cytosolic sensing of immuno-stimulatory DNA, the enemy within. Curr. Opin. Immunol. 2018, 50, 82–87. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Holm, C.K.; Rahbek, S.H.; Gad, H.H.; Bak, R.O.; Jakobsen, M.R.; Jiang, Z.; Hansen, A.L.; Jensen, S.K.; Sun, C.; Thomsen, M.K.; et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun. 2016, 7, 10680. [Google Scholar] [CrossRef]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef]
- Sun, L.; Xing, Y.; Chen, X.; Zheng, Y.; Yang, Y.; Nichols, D.B.; Clementz, M.A.; Banach, B.S.; Li, K.; Baker, S.C.; et al. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS ONE 2012, 7, e30802. [Google Scholar] [CrossRef]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.-J.; Kato, N.; Shu, H.-B.; Zhong, J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Chang, T.-H.; Liang, J.-J.; Chiang, R.-L.; Lee, Y.-L.; Liao, C.-L.; Lin, Y.-L. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017, 2, 17037. [Google Scholar] [CrossRef]
- Stabell, A.C.; Meyerson, N.R.; Gullberg, R.C.; Gilchrist, A.R.; Webb, K.J.; Old, W.M.; Perera, R.; Sawyer, S.L. Dengue viruses cleave STING in humans but not in nonhuman primates, their presumed natural reservoir. Elife 2018, 7, e31919. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6. [Google Scholar] [CrossRef]
- Wu, M.-F.; Chen, S.-T.; Yang, A.-H.; Lin, W.-W.; Lin, Y.-L.; Chen, N.-J.; Tsai, I.-S.; Li, L.; Hsieh, S.-L. CLEC5A is critical for dengue virus-induced inflammasome activation in human macrophages. Blood 2013, 121, 95–106. [Google Scholar] [CrossRef]
- Hottz, E.D.; Lopes, J.F.; Freitas, C.; Valls-de-Souza, R.; Oliveira, M.F.; Bozza, M.T.; Da Poian, A.T.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, F.A.; et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 2013, 122, 3405–3414. [Google Scholar] [CrossRef] [Green Version]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef]
- Nazmi, A.; Mukhopadhyay, R.; Dutta, K.; Basu, A. STING mediates neuronal innate immune response following Japanese encephalitis virus infection. Sci. Rep. 2012, 2, 347. [Google Scholar] [CrossRef]
- Thompson, M.R.; Sharma, S.; Atianand, M.; Jensen, S.B.; Carpenter, S.; Knipe, D.M.; Fitzgerald, K.A.; Kurt-Jones, E.A. Interferon γ-inducible Protein (IFI) 16 Transcriptionally Regulates Type I Interferons and Other Interferon-stimulated Genes and Controls the Interferon Response to both DNA and RNA Viruses. J. Biol. Chem. 2014, 289, 23568–23581. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Liu, Y.; Goulet, M.-L.; Sze, A.; Hadj, S.B.; Belgnaoui, S.M.; Lababidi, R.R.; Zheng, C.; Fritz, J.H.; Olagnier, D.; Lin, R. RIG-I-Mediated STING Upregulation Restricts Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 9406–9419. [Google Scholar] [CrossRef] [Green Version]
- Bradrick, S.S. Causes and Consequences of Flavivirus RNA Methylation. Front. Microbiol. 2017, 8, 2374. [Google Scholar] [CrossRef]
- Furuichi, Y.; Shatkin, A.J. Viral and cellular mRNA capping: Past and prospects. Adv. Virus Res. 2000, 55, 135–184. [Google Scholar]
- Dong, H.; Chang, D.C.; Hua, M.H.C.; Lim, S.P.; Chionh, Y.H.; Hia, F.; Lee, Y.H.; Kukkaro, P.; Lok, S.-M.; Dedon, P.C.; et al. 2′-O methylation of internal adenosine by flavivirus NS5 methyltransferase. PLoS Pathog. 2012, 8, e1002642. [Google Scholar] [CrossRef]
- Schmid, B.; Rinas, M.; Ruggieri, A.; Acosta, E.G.; Bartenschlager, M.; Reuter, A.; Fischl, W.; Harder, N.; Bergeest, J.-P.; Flossdorf, M.; et al. Live Cell Analysis and Mathematical Modeling Identify Determinants of Attenuation of Dengue Virus 2′-O-Methylation Mutant. PLoS Pathog. 2015, 11, e1005345. [Google Scholar] [CrossRef]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef]
- Züst, R.; Dong, H.; Li, X.-F.; Chang, D.C.; Zhang, B.; Balakrishnan, T.; Toh, Y.-X.; Jiang, T.; Li, S.-H.; Deng, Y.-Q.; et al. Rational Design of a Live Attenuated Dengue Vaccine: 2′-O-Methyltransferase Mutants Are Highly Attenuated and Immunogenic in Mice and Macaques. PLoS Pathog. 2013, 9, e1003521. [Google Scholar] [CrossRef]
- Chang, D.C.; Hoang, L.T.; Naim, A.N.M.; Dong, H.; Schreiber, M.J.; Hibberd, M.L.; Tan, M.J.A.; Shi, P.-Y. Evasion of early innate immune response by 2′- O -methylation of dengue genomic RNA. Virology 2016, 499, 259–266. [Google Scholar] [CrossRef]
- Clarke, B.D.; Roby, J.A.; Slonchak, A.; Khromykh, A.A. Functional non-coding RNAs derived from the flavivirus 3′ untranslated region. Virus Res. 2015, 206, 53–61. [Google Scholar] [CrossRef]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.-Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Zou, J.; Zhang, B.; Yuan, Z. Dengue virus subgenomic RNA induces apoptosis through the Bcl-2-mediated PI3k/Akt signaling pathway. Virology 2014, 448, 15–25. [Google Scholar] [CrossRef]
- Moon, S.L.; Anderson, J.R.; Kumagai, Y.; Wilusz, C.J.; Akira, S.; Khromykh, A.A.; Wilusz, J. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 2012, 18, 2029–2040. [Google Scholar] [CrossRef] [Green Version]
- Schnettler, E.; Sterken, M.G.; Leung, J.Y.; Metz, S.W.; Geertsema, C.; Goldbach, R.W.; Vlak, J.M.; Kohl, A.; Khromykh, A.A.; Pijlman, G.P. Noncoding flavivirus RNA displays RNA interference suppressor activity in insect and Mammalian cells. J. Virol. 2012, 86, 13486–13500. [Google Scholar] [CrossRef]
- Fan, Y.-H.; Nadar, M.; Chen, C.-C.; Weng, C.-C.; Lin, Y.-T.; Chang, R.-Y. Small noncoding RNA modulates Japanese encephalitis virus replication and translation in trans. Virol. J. 2011, 8, 492. [Google Scholar] [CrossRef]
- Chang, R.-Y.; Hsu, T.-W.; Chen, Y.-L.; Liu, S.-F.; Tsai, Y.-J.; Lin, Y.-T.; Chen, Y.-S.; Fan, Y.-H. Japanese encephalitis virus non-coding RNA inhibits activation of interferon by blocking nuclear translocation of interferon regulatory factor 3. Vet. Microbiol. 2013, 166, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Schuessler, A.; Funk, A.; Lazear, H.M.; Cooper, D.A.; Torres, S.; Daffis, S.; Jha, B.K.; Kumagai, Y.; Takeuchi, O.; Hertzog, P.; et al. West Nile virus noncoding subgenomic RNA contributes to viral evasion of the type I interferon-mediated antiviral response. J. Virol. 2012, 86, 5708–5718. [Google Scholar] [CrossRef] [PubMed]
- Donald, C.L.; Brennan, B.; Cumberworth, S.L.; Rezelj, V.V.; Clark, J.J.; Cordeiro, M.T.; Freitas de Oliveira França, R.; Pena, L.J.; Wilkie, G.S.; Da Silva Filipe, A.; et al. Full Genome Sequence and sfRNA Interferon Antagonist Activity of Zika Virus from Recife, Brazil. PLoS Negl. Trop. Dis. 2016, 10, e0005048. [Google Scholar] [CrossRef] [PubMed]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.-S.; Huang, I.-C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Gack, M.U. A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nat. Immunol. 2016, 17, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Loo, Y.-M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The mitochondrial targeting chaperone 14-3-3ε regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhu, X.; Wen, W.; Yuan, J.; Hu, Y.; Chen, J.; An, S.; Dong, X.; Lin, C.; Yu, J.; et al. Dengue Virus Subverts Host Innate Immunity by Targeting Adaptor Protein MAVS. J. Virol. 2016, 90, 7219–7230. [Google Scholar] [CrossRef] [Green Version]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef]
- Lee, H.; Yoon, Y. Mitochondrial fission: Regulation and ER connection. Mol. Cells 2014, 37, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Y.; Liang, J.-J.; Li, J.-K.; Lee, Y.-L.; Chang, B.-L.; Su, C.-I.; Huang, W.-J.; Lai, M.M.C.; Lin, Y.-L. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PLoS Pathog. 2015, 11, e1005350. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Syed, G.H.; Khan, M.; Chiu, W.-W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, T.; Enquist, L.W. Alphaherpesvirus infection disrupts mitochondrial transport in neurons. Cell Host Microbe 2012, 11, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Hou, S.; Malik-Soni, N.; Xu, Z.; Kumar, A.; Rachubinski, R.A.; Frappier, L.; Hobman, T.C. Flavivirus Infection Impairs Peroxisome Biogenesis and Early Antiviral Signaling. J. Virol. 2015, 89, 12349–12361. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.P.; Xu, Z.; Hou, S.; Limonta, D.; Kumar, A.; Power, C.; Hobman, T.C. Interplay between Zika Virus and Peroxisomes during Infection. Cells 2019, 8, 725. [Google Scholar] [CrossRef] [PubMed]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.Y.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Reuter, A.; Eberle, F.; Einhorn, E.; Binder, M.; Bartenschlager, R. Activation of Type I and III Interferon Response by Mitochondrial and Peroxisomal MAVS and Inhibition by Hepatitis C Virus. PLoS Pathog. 2015, 11, e1005264. [Google Scholar] [CrossRef] [PubMed]
- Götte, K.; Girzalsky, W.; Linkert, M.; Baumgart, E.; Kammerer, S.; Kunau, W.H.; Erdmann, R. Pex19p, a farnesylated protein essential for peroxisome biogenesis. Mol. Cell. Biol. 1998, 18, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Angleró-Rodríguez, Y.I.; Pantoja, P.; Sariol, C.A. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IκB kinase epsilon interaction. Clin. Vaccine Immunol. 2014, 21, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, N.A.; Cimica, V.; Mackow, E.R. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. mBio 2015, 6. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Fischl, W.; Scaturro, P.; Cortese, M.; Kallis, S.; Bartenschlager, M.; Fischer, B.; Bartenschlager, R. A Combined Genetic-Proteomic Approach Identifies Residues within Dengue Virus NS4B Critical for Interaction with NS3 and Viral Replication. J. Virol. 2015, 89, 7170–7186. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Lee, L.T.; Wang, Q.Y.; Xie, X.; Lu, S.; Yau, Y.H.; Yuan, Z.; Geifman Shochat, S.; Kang, C.; Lescar, J.; et al. Mapping the Interactions between the NS4B and NS3 proteins of dengue virus. J. Virol. 2015, 89, 3471–3483. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Xie, X.; Wang, Q.-Y.; Dong, H.; Lee, M.Y.; Kang, C.; Yuan, Z.; Shi, P.-Y. Characterization of dengue virus NS4A and NS4B protein interaction. J. Virol. 2015, 89, 3455–3470. [Google Scholar] [CrossRef] [PubMed]
- Umareddy, I.; Chao, A.; Sampath, A.; Gu, F.; Vasudevan, S.G. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J. Gen. Virol. 2006, 87, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Sundström, K.B.; Chew, J.J.; Bist, P.; Gan, E.S.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7, 3594. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Y.; Chang, T.-H.; Liang, J.-J.; Chiang, R.-L.; Lee, Y.-L.; Liao, C.-L.; Lin, Y.-L. P122 Dengue virus targets the adaptor protein MITA to subvert host innate immunity. Cytokine 2012, 59, 558. [Google Scholar] [CrossRef]
- Bao, D.; Zhao, J.; Zhou, X.; Yang, Q.; Chen, Y.; Zhu, J.; Yuan, P.; Yang, J.; Qin, T.; Wan, S.; et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene 2019, 38, 5007–5020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Jordan, J.L.; Sánchez-Burgos, G.G.; Laurent-Rolle, M.; García-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Jordán, J.L.; Laurent-Rolle, M.; Ashour, J.; Martínez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; García-Sastre, A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.F.; Fernandez-Sesma, A.; García-Sastre, A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.-Y.; García-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef]
- Jones, M.; Davidson, A.; Hibbert, L.; Gruenwald, P.; Schlaak, J.; Ball, S.; Foster, G.R.; Jacobs, M. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J. Virol. 2005, 79, 5414–5420. [Google Scholar] [CrossRef]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.S.; Link, N.; Jang, G.M.; Sharp, P.P.; Zhu, T.; Swaney, D.L.; Johnson, J.R.; Von Dollen, J.; Ramage, H.R.; Satkamp, L.; et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175, 1931–1945.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maio, F.A.; Risso, G.; Iglesias, N.G.; Shah, P.; Pozzi, B.; Gebhard, L.G.; Mammi, P.; Mancini, E.; Yanovsky, M.J.; Andino, R.; et al. The Dengue Virus NS5 Protein Intrudes in the Cellular Spliceosome and Modulates Splicing. PLoS Pathog. 2016, 12, e1005841. [Google Scholar] [CrossRef] [PubMed]
- Bidet, K.; Dadlani, D.; Garcia-Blanco, M.A. G3BP1, G3BP2 and CAPRIN1 Are Required for Translation of Interferon Stimulated mRNAs and Are Targeted by a Dengue Virus Non-coding RNA. PLoS Pathog. 2014, 10, e1004242. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.-Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452. [Google Scholar] [CrossRef]
- Habjan, M.; Hubel, P.; Lacerda, L.; Benda, C.; Holze, C.; Eberl, C.H.; Mann, A.; Kindler, E.; Gil-Cruz, C.; Ziebuhr, J.; et al. Sequestration by IFIT1 impairs translation of 2′ O-unmethylated capped RNA. PLoS Pathog. 2013, 9, e1003663. [Google Scholar] [CrossRef]
- Kimura, T.; Katoh, H.; Kayama, H.; Saiga, H.; Okuyama, M.; Okamoto, T.; Umemoto, E.; Matsuura, Y.; Yamamoto, M.; Takeda, K. Ifit1 Inhibits Japanese Encephalitis Virus Replication through Binding to 5′ Capped 2′-O Unmethylated RNA. J. Virol. 2013, 87, 9997–10003. [Google Scholar] [CrossRef]
- Kumar, P.; Sweeney, T.R.; Skabkin, M.A.; Skabkina, O.V.; Hellen, C.U.T.; Pestova, T.V. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res. 2014, 42, 3228–3245. [Google Scholar] [CrossRef]
- Pompon, J.; Manuel, M.; Ng, G.K.; Wong, B.; Shan, C.; Manokaran, G.; Soto-Acosta, R.; Bradrick, S.S.; Ooi, E.E.; Missé, D.; et al. Dengue subgenomic flaviviral RNA disrupts immunity in mosquito salivary glands to increase virus transmission. PLoS Pathog. 2017, 13, e1006535. [Google Scholar] [CrossRef]
- Moon, S.L.; Dodd, B.J.T.; Brackney, D.E.; Wilusz, C.J.; Ebel, G.D.; Wilusz, J. Flavivirus sfRNA suppresses antiviral RNA interference in cultured cells and mosquitoes and directly interacts with the RNAi machinery. Virology 2015, 485, 322–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göertz, G.P.; Fros, J.J.; Miesen, P.; Vogels, C.B.F.; van der Bent, M.L.; Geertsema, C.; Koenraadt, C.J.M.; van Rij, R.P.; van Oers, M.M.; Pijlman, G.P. Noncoding Subgenomic Flavivirus RNA Is Processed by the Mosquito RNA Interference Machinery and Determines West Nile Virus Transmission by Culex pipiens Mosquitoes. J. Virol. 2016, 90, 10145–10159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammon, D.B.; Mello, C.C. RNA interference-mediated antiviral defense in insects. Curr Opin Insect Sci 2015, 8, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Siomi, H.; Siomi, M.C. On the road to reading the RNA-interference code. Nature 2009, 457, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-M.; Chen, H.-W.; Hsiao, Y.-J.; Wu, S.-H.; Chung, H.-H.; Hsieh, C.-H.; Chong, P.; Leng, C.-H.; Pan, C.-H. The successful induction of T-cell and antibody responses by a recombinant measles virus-vectored tetravalent dengue vaccine provides partial protection against dengue-2 infection. Hum. Vaccin. Immunother. 2016, 12, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Recombinant Protein-Based Dengue Vaccines. Front. Immunol. 2018, 9, 1919. [Google Scholar] [CrossRef]
- McArthur, M.A.; Sztein, M.B.; Edelman, R. Dengue vaccines: Recent developments, ongoing challenges and current candidates. Expert Rev. Vaccines 2013, 12, 933–953. [Google Scholar] [CrossRef]
- Shukla, R.; Ramasamy, V.; Rajpoot, R.K.; Arora, U.; Poddar, A.; Ahuja, R.; Beesetti, H.; Swaminathan, S.; Khanna, N. Next generation designer virus-like particle vaccines for dengue. Expert Rev. Vaccines 2019, 18, 105–117. [Google Scholar] [CrossRef]
- Fahimi, H.; Mohammadipour, M.; Haddad Kashani, H.; Parvini, F.; Sadeghizadeh, M. Dengue viruses and promising envelope protein domain III-based vaccines. Appl. Microbiol. Biotechnol. 2018, 102, 2977–2996. [Google Scholar] [CrossRef]
- Lazo, L.; Valdes, I.; Guillén, G.; Hermida, L.; Gil, L. Aiming at the heart: The capsid protein of dengue virus as a vaccine candidate. Expert Rev. Vaccines 2019, 18, 161–173. [Google Scholar] [CrossRef]
- Hadinegoro, S.R.; Arredondo-García, J.L.; Capeding, M.R.; Deseda, C.; Chotpitayasunondh, T.; Dietze, R.; Muhammad Ismail, H.I.H.; Reynales, H.; Limkittikul, K.; Rivera-Medina, D.M.; et al. Efficacy and Long-Term Safety of a Dengue Vaccine in Regions of Endemic Disease. N. Engl. J. Med. 2015, 373, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Malisheni, M.; Khaiboullina, S.F.; Rizvanov, A.A.; Takah, N.; Murewanhema, G.; Bates, M. Clinical Efficacy, Safety, and Immunogenicity of a Live Attenuated Tetravalent Dengue Vaccine (CYD-TDV) in Children: A Systematic Review with Meta-analysis. Front. Immunol. 2017, 8, 863. [Google Scholar] [CrossRef] [PubMed]
- Diseases, T.L.I. The Lancet Infectious Diseases The dengue vaccine dilemma. Lancet Infect. Dis. 2018, 18, 123. [Google Scholar] [CrossRef]
- Dengue vaccine: WHO position paper, September 2018—Recommendations. Vaccine 2019, 37, 4848–4849. [CrossRef] [PubMed]
- Velumani, S.; Toh, Y.X.; Balasingam, S.; Archuleta, S.; Leo, Y.S.; Gan, V.C.; Thein, T.L.; Wilder-Smith, A.; Fink, K. Low antibody titers 5 years after vaccination with the CYD-TDV dengue vaccine in both pre-immune and naïve vaccinees. Hum. Vaccines Immunother. 2016, 12, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Sabin, A.B. Research on dengue during World War II. Am. J. Trop. Med. Hyg. 1952, 1, 30–50. [Google Scholar] [CrossRef]
- Snow, G.E.; Haaland, B.; Ooi, E.E.; Gubler, D.J. Review article: Research on dengue during World War II revisited. Am. J. Trop. Med. Hyg. 2014, 91, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Kuczera, D.; Assolini, J.P.; Tomiotto-Pellissier, F.; Pavanelli, W.R.; Silveira, G.F. Highlights for Dengue Immunopathogenesis: Antibody-Dependent Enhancement, Cytokine Storm, and Beyond. J. Interferon Cytokine Res. 2018, 38, 69–80. [Google Scholar] [CrossRef]
- St John, A.L.; Rathore, A.P.S. Adaptive immune responses to primary and secondary dengue virus infections. Nat. Rev. Immunol. 2019, 19, 218–230. [Google Scholar] [CrossRef]
- Blaney, J.E., Jr.; Durbin, A.P.; Murphy, B.R.; Whitehead, S.S. Development of a live attenuated dengue virus vaccine using reverse genetics. Viral Immunol. 2006, 19, 10–32. [Google Scholar] [CrossRef]
- Durbin, A.P.; Karron, R.A.; Sun, W.; Vaughn, D.W.; Reynolds, M.J.; Perreault, J.R.; Thumar, B.; Men, R.; Lai, C.J.; Elkins, W.R.; et al. Attenuation and immunogenicity in humans of a live dengue virus type-4 vaccine candidate with a 30 nucleotide deletion in its 3′-untranslated region. Am. J. Trop. Med. Hyg. 2001, 65, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Blaney, J.E.; Sathe, N.S.; Goddard, L.; Hanson, C.T.; Romero, T.A.; Hanley, K.A.; Murphy, B.R.; Whitehead, S.S. Dengue virus type 3 vaccine candidates generated by introduction of deletions in the 3′ untranslated region (3′-UTR) or by exchange of the DENV-3 3′-UTR with that of DENV-4. Vaccine 2008, 26, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.P.; Whitehead, S.S.; Shaffer, D.; Elwood, D.; Wanionek, K.; Thumar, B.; Blaney, J.E.; Murphy, B.R.; Schmidt, A.C. A single dose of the DENV-1 candidate vaccine rDEN1Δ30 is strongly immunogenic and induces resistance to a second dose in a randomized trial. PLoS Negl. Trop. Dis. 2011, 5, e1267. [Google Scholar] [CrossRef] [PubMed]
- Blaney, J.E., Jr.; Hanson, C.T.; Firestone, C.-Y.; Hanley, K.A.; Murphy, B.R.; Whitehead, S.S. Genetically modified, live attenuated dengue virus type 3 vaccine candidates. Am. J. Trop. Med. Hyg. 2004, 71, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.P.; McArthur, J.; Marron, J.A.; Blaney, J.E., Jr.; Thumar, B.; Wanionek, K.; Murphy, B.R.; Whitehead, S.S. The Live Attenuated Dengue Serotype 1 Vaccine rDEN1Δ30 is Safe and Highly Immunogenic in Healthy Adult Volunteers. Hum. Vaccin. 2006, 2, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.P.; McArthur, J.H.; Marron, J.A.; Blaney, J.E.; Thumar, B.; Wanionek, K.; Murphy, B.R. rDEN2/4Δ30 (ME), a live attenuated chimeric dengue serotype 2 vaccine, is safe and highly immunogenic in healthy dengue-naïve adults. Hum. Vaccin. 2006, 2, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.P.; Whitehead, S.S.; McArthur, J.; Perreault, J.R.; Blaney, J.E., Jr.; Thumar, B.; Murphy, B.R.; Karron, R.A. rDEN4delta30, a live attenuated dengue virus type 4 vaccine candidate, is safe, immunogenic, and highly infectious in healthy adult volunteers. J. Infect. Dis. 2005, 191, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Blaney, J.E.; Matro, J.M.; Murphy, B.R.; Whitehead, S.S. Recombinant, Live-Attenuated Tetravalent Dengue Virus Vaccine Formulations Induce a Balanced, Broad, and Protective Neutralizing Antibody Response against Each of the Four Serotypes in Rhesus Monkeys. J. Virol. 2005, 79, 5516–5528. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, B.D.; Whitehead, S.S.; Pierce, K.K.; Tibery, C.M.; Grier, P.L.; Hynes, N.A.; Larsson, C.J.; Sabundayo, B.P.; Talaat, K.R.; Janiak, A.; et al. The live attenuated dengue vaccine TV003 elicits complete protection against dengue in a human challenge model. Sci. Transl. Med. 2016, 8, 330ra36. [Google Scholar] [CrossRef]
- Troyer, J.M.; Durbin, A.P.; Strickman, D.; Hanley, K.A.; Karron, R.A.; Murphy, B.R.; Whitehead, S.S. A live attenuated recombinant dengue-4 virus vaccine candidate with restricted capacity for dissemination in mosquitoes and lack of transmission from vaccinees to mosquitoes. Am. J. Trop. Med. Hyg. 2001, 65, 414–419. [Google Scholar] [CrossRef]
- Bustos-Arriaga, J.; Gromowski, G.D.; Tsetsarkin, K.A.; Firestone, C.-Y.; Castro-Jiménez, T.; Pletnev, A.G.; Cedillo-Barrón, L.; Whitehead, S.S. Decreased accumulation of subgenomic RNA in human cells infected with vaccine candidate DEN4Δ30 increases viral susceptibility to type I interferon. Vaccine 2018, 36, 3460–3467. [Google Scholar] [CrossRef]
- Li, S.-H.; Dong, H.; Li, X.-F.; Xie, X.; Zhao, H.; Deng, Y.-Q.; Wang, X.-Y.; Ye, Q.; Zhu, S.-Y.; Wang, H.-J.; et al. Rational design of a flavivirus vaccine by abolishing viral RNA 2′-O methylation. J. Virol. 2013, 87, 5812–5819. [Google Scholar] [CrossRef] [PubMed]
- Gerdts, V.; Wilson, H.L.; Meurens, F.; van Drunen Littel-van den Hurk, S.; Wilson, D.; Walker, S.; Wheler, C.; Townsend, H.; Potter, A.A. Large animal models for vaccine development and testing. ILAR J. 2015, 56, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.J.; Roehrig, J.T. New mouse model for dengue virus vaccine testing. J. Virol. 1999, 73, 783–786. [Google Scholar]
- Sarathy, V.V.; White, M.; Li, L.; Gorder, S.R.; Pyles, R.B.; Campbell, G.A.; Milligan, G.N.; Bourne, N.; Barrett, A.D.T. A lethal murine infection model for dengue virus 3 in AG129 mice deficient in type I and II interferon receptors leads to systemic disease. J. Virol. 2015, 89, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Shresta, S.; Kyle, J.L.; Snider, H.M.; Basavapatna, M.; Beatty, P.R.; Harris, E. Interferon-Dependent Immunity Is Essential for Resistance to Primary Dengue Virus Infection in Mice, Whereas T- and B-Cell-Dependent Immunity Are Less Critical. J. Virol. 2004, 78, 2701–2710. [Google Scholar] [CrossRef] [Green Version]
- Gorman, M.J.; Caine, E.A.; Zaitsev, K.; Begley, M.C.; Weger-Lucarelli, J.; Uccellini, M.B.; Tripathi, S.; Morrison, J.; Yount, B.L.; Dinnon, K.H., 3rd; et al. An Immunocompetent Mouse Model of Zika Virus Infection. Cell Host Microbe 2018, 23, 672–685.e6. [Google Scholar] [CrossRef] [Green Version]
- Bowen, J.R.; Quicke, K.M.; Maddur, M.S.; O’Neal, J.T.; McDonald, C.E.; Fedorova, N.B.; Puri, V.; Shabman, R.S.; Pulendran, B.; Suthar, M.S. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog. 2017, 13, e1006164. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tremblay, N.; Freppel, W.; Sow, A.A.; Chatel-Chaix, L. The Interplay between Dengue Virus and the Human Innate Immune System: A Game of Hide and Seek. Vaccines 2019, 7, 145. https://doi.org/10.3390/vaccines7040145
Tremblay N, Freppel W, Sow AA, Chatel-Chaix L. The Interplay between Dengue Virus and the Human Innate Immune System: A Game of Hide and Seek. Vaccines. 2019; 7(4):145. https://doi.org/10.3390/vaccines7040145
Chicago/Turabian StyleTremblay, Nicolas, Wesley Freppel, Aïssatou Aïcha Sow, and Laurent Chatel-Chaix. 2019. "The Interplay between Dengue Virus and the Human Innate Immune System: A Game of Hide and Seek" Vaccines 7, no. 4: 145. https://doi.org/10.3390/vaccines7040145