
Differential Immune Response Patterns Induced by Anionic and Cationic Lipid Adjuvants in Intranasal Anti-Influenza Immunization

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Use and Experimental Groups
2.2. Vaccine and Adjuvant Formulation and Administration Procedure
2.3. Flow Cytometry
2.4. Serology Screening
2.5. Hemagglutination Inhibition Assay
2.6. Statistical Analyses
3. Results
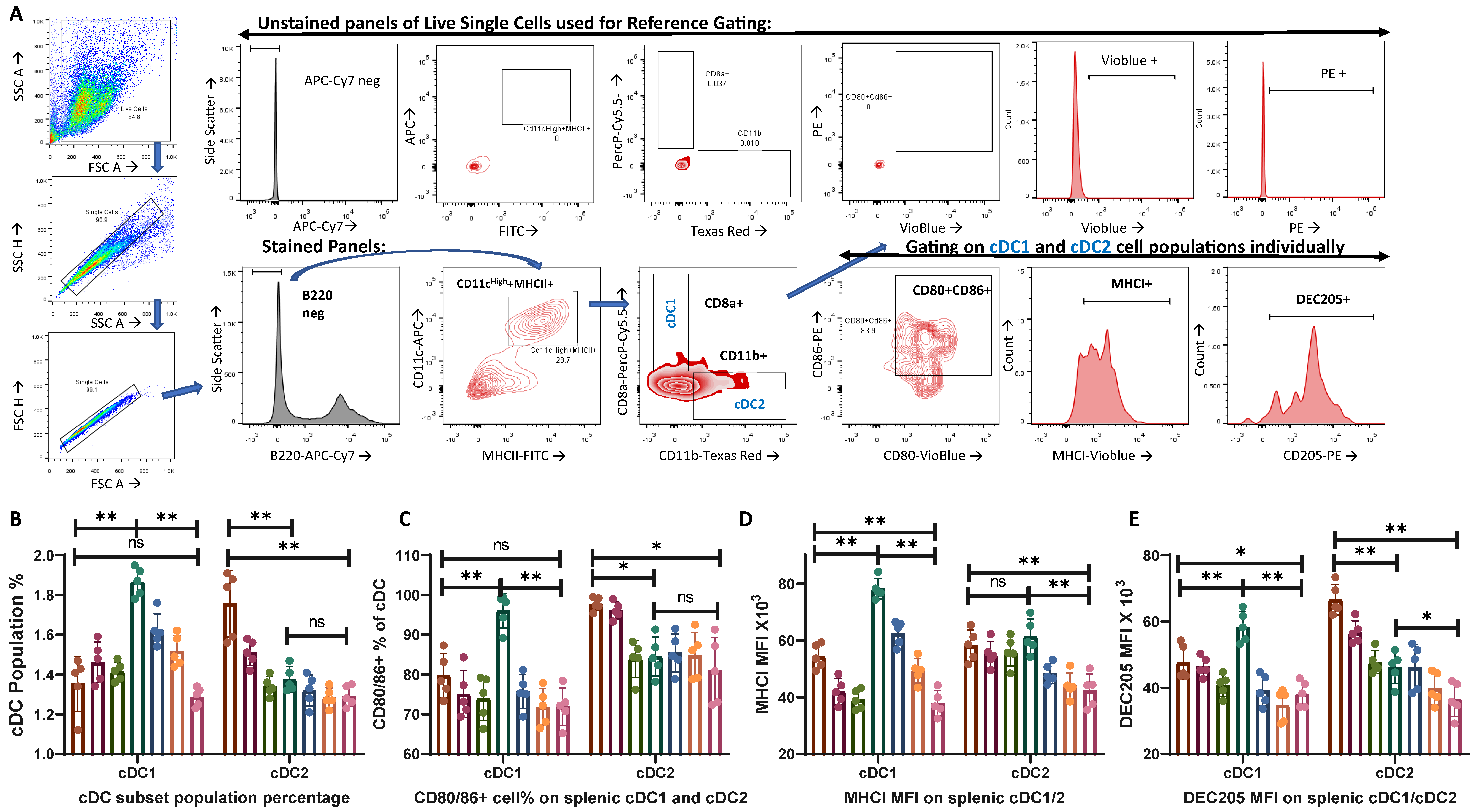
3.1. Anionic (L3) Adjuvant-Treated Group Showed Enhanced Activation of cDC2 While Cationic (N3) Adjuvant-Treated Group Facilitated cDC1 Population Activation
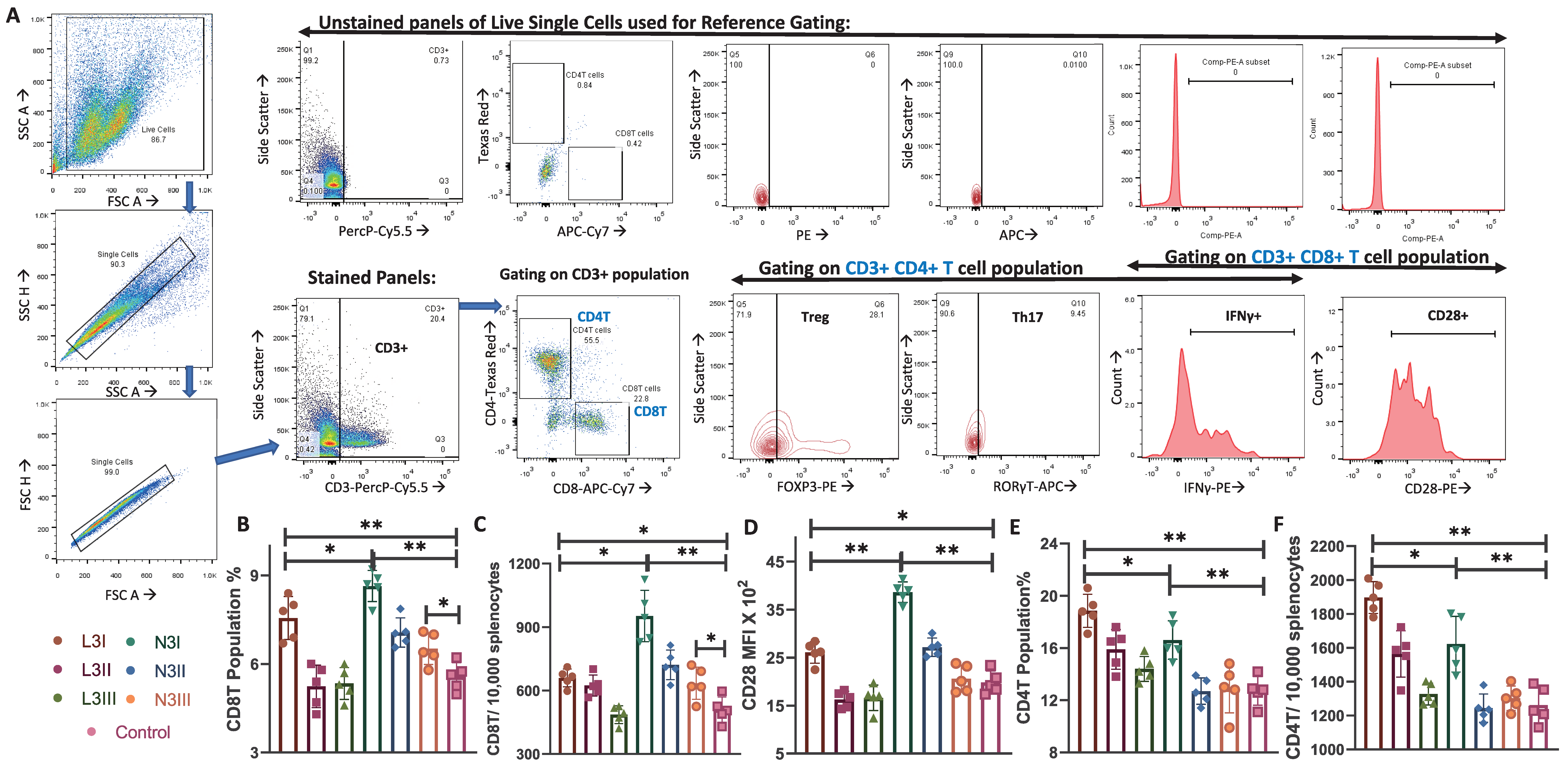
3.2. Increase in Cytotoxic CD8T Cells and Costimulatory Marker CD28 Expression in the N3 Treated Groups Observed
3.3. Higher Population Percentage of CD4T Cells in the Anionic Adjuvant Treatment with Altered Skew of the Treg:Th17 Ratio
3.4. N3 Treatment Induces Significantly More Interferon-Gamma (IFNγ)-Producing T Cell Subsets
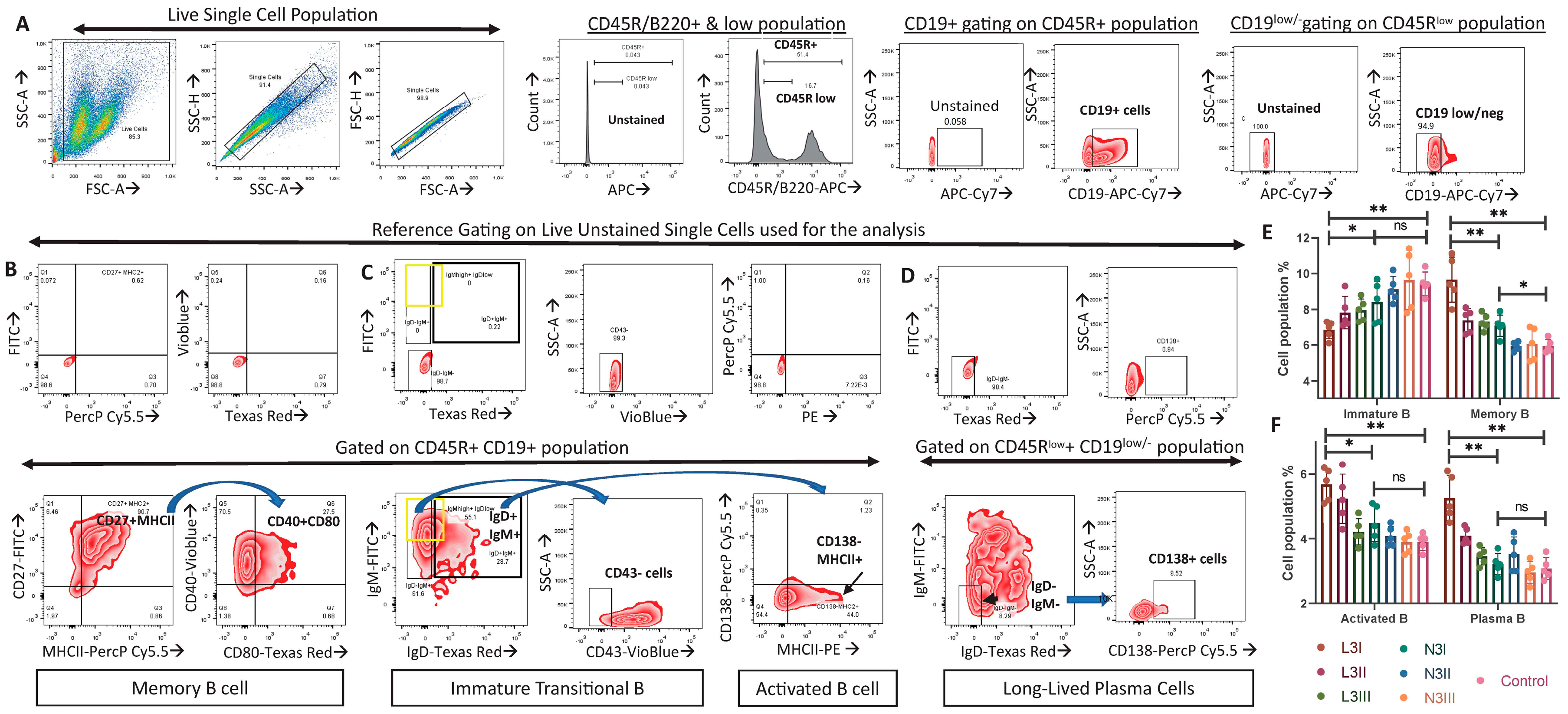
3.5. Significantly Fewer Immature B Cells and More Activated B and Plasma Cells in the L3-Treated Group Than in the N3 Groups Indicated an Elevated B Cell Response in Anionic-Adjuvant-Treated Mice
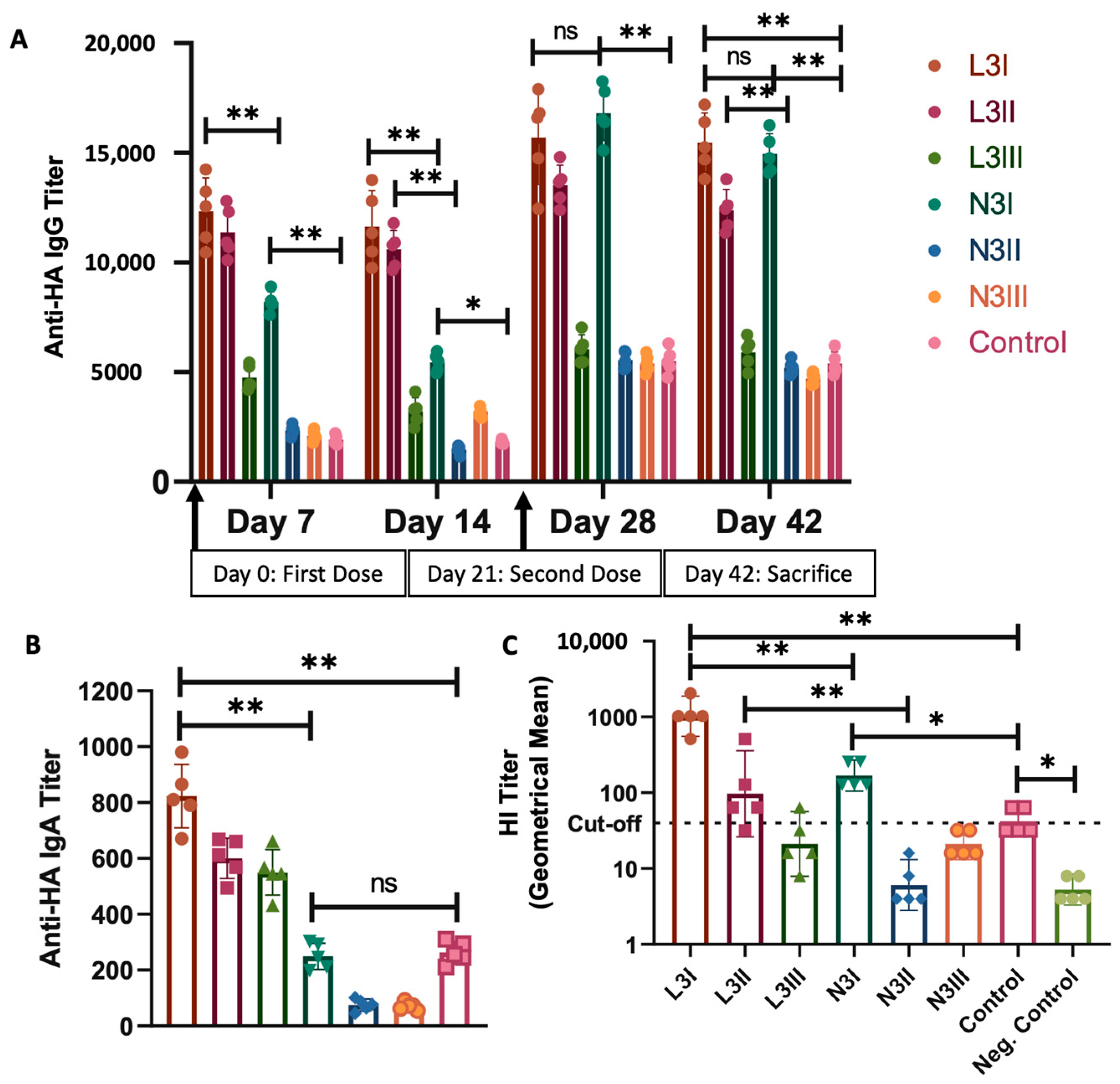
3.6. Serum IgG Levels against Influenza HA Were Significantly High in Both the L3I and N3I Groups, with Significantly Higher IgA Levels and Virus Neutralization Capacity in the L3 Group
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bok, K.; Sitar, S.; Graham, B.S.; Mascola, J.R. Accelerated COVID-19 vaccine development: Milestones, lessons, and prospects. Immunity 2021, 54, 1636–1651. [Google Scholar] [CrossRef]
- Pulendran, B.; Arunachalam, P.S.; O’Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef]
- Mascola, J.R.; Fauci, A.S. Novel vaccine technologies for the 21st century. Nat. Rev. Immunol. 2020, 20, 87–88. [Google Scholar] [CrossRef]
- Weinberger, B. Adjuvant strategies to improve vaccination of the elderly population. Curr. Opin. Pharmacol. 2018, 41, 34–41. [Google Scholar] [CrossRef]
- Ten Threats to Global Health in 2019. Available online: https://www.who.int/news-room/spotlight/ten-threats-to-global-health-in-2019 (accessed on 1 February 2024).
- Lavelle, E.C.; Ward, R.W. Mucosal vaccines—Fortifying the frontiers. Nat. Rev. Immunol. 2021, 22, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Aoshi, T. Modes of Action for Mucosal Vaccine Adjuvants. Viral Immunol. 2017, 30, 463–470. [Google Scholar] [CrossRef]
- Mettelman, R.C.; Allen, E.K.; Thomas, P.G. Mucosal immune responses to infection and vaccination in the respiratory tract. Immunity 2022, 55, 749–780. [Google Scholar] [CrossRef] [PubMed]
- Calzas, C.; Chevalier, C. Innovative Mucosal Vaccine Formulations against Influenza A Virus Infections. Front. Immunol. 2019, 10, 448507. [Google Scholar] [CrossRef]
- Bernasconi, V.; Norling, K.; Bally, M.; Höök, F.; Lycke, N.Y. Mucosal Vaccine Development Based on Liposome Technology. J. Immunol. Res. 2016, 2016, 5482087. [Google Scholar] [CrossRef] [PubMed]
- Schroder, U. Immunstimulating Lipid Formulation. Patent CA2258017C, 24 May 2011. Available online: https://patents.google.com/patent/CA2258017C/en?q=(eurocine+adjuvant+influenza)&assignee=Eurocine+Vaccines+Ab (accessed on 1 February 2024).
- Arwidsson, H.; Schroder, U. Improved Vaccine Compositions. Patent AU2011310090B2, 18 June 2015. Available online: https://patents.google.com/patent/AU2011310090B2/en?q=(eurocine+adjuvant+influenza)&assignee=Eurocine+Vaccines+Ab (accessed on 1 February 2024).
- Lundemosem, A.; Ökvist, A.; Ove, A.; Hovden, M.; Grønvold, S. Methods of Immunization with a Vaccine Inducing a Humoral Immune Response and with a Vaccine Inducing a Cellular Immune Response. EU Patent WO2015110659A1, 30 July 2015. Available online: https://patents.google.com/patent/WO2015110659A1/en?q=(eurocine+adjuvant+influenza)&oq=eurocine+adjuvant+influenza (accessed on 1 February 2024).
- Hans, A.; Maltais, A.-K. Vaccine Composition for Naive Subjects. Patent JP2019112448A, 11 July 2019. Available online: https://patents.google.com/patent/JP2019112448A/en?q=(eurocine+adjuvant+influenza)&assignee=Eurocine+Vaccines+Ab (accessed on 1 February 2024).
- Study to Assess the Safety, Tolerability and Immune Response Following Vaccination with ImmunoseTM FLU in Older Adults. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03437304 (accessed on 1 February 2024).
- Study to Assess the Safety, Tolerability and Immune Response Following Vaccination with ImmunoseTM FLU. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02998996 (accessed on 1 February 2024).
- Falkeborn, T. Nasal Vaccination Using Novel Mucosal Adjuvants—With Main Focus on Influenza A Virus. Ph.D. Thesis, Linköping University, Linköping, Sweden, 2015. [Google Scholar]
- Falkeborn, T.; Bråve, A.; Larsson, M.; Åkerlind, B.; Schröder, U.; Hinkula, J. EndocineTM, N3OA and N3OASq; three mucosal adjuvants that enhance the immune response to nasal influenza vaccination. PLoS ONE 2013, 8, e70527. [Google Scholar] [CrossRef] [PubMed]
- Hinkula, J.; Nyström, S.; Devito, C.; Bråve, A.; Applequist, S.E. Long-Lasting Mucosal and Systemic Immunity against Influenza A Virus Is Significantly Prolonged and Protective by Nasal Whole Influenza Immunization with Mucosal Adjuvant N3 and DNA-Plasmid Expressing Flagellin in Aging In- and Outbred Mice. Vaccines 2019, 7, 64. [Google Scholar] [CrossRef]
- Maltais, A.-K.; Stittelaar, K.J.; Kroeze, E.J.V.; van Amerongen, G.; Dijkshoorn, M.L.; Krestin, G.P.; Hinkula, J.; Arwidsson, H.; Lindberg, A.; Osterhaus, A.D. Intranasally administered Endocine™ formulated 2009 pandemic influenza H1N1 vaccine induces broad specific antibody responses and confers protection in ferrets. Vaccine 2014, 32, 3307–3315. [Google Scholar] [CrossRef]
- Petersson, P.; Hedenskog, M.; Alves, D.; Brytting, M.; Schröder, U.; Linde, A.; Lundkvist, Å. The Eurocine® L3 adjuvants with subunit influenza antigens induce protective immunity in mice after intranasal vaccination. Vaccine 2010, 28, 6491–6497. [Google Scholar] [CrossRef] [PubMed]
- Comparison of the Mucosal Adjuvant EndocineTM with Two Well-Known Adjuvants: Cholera Toxin and Alum. Available online: https://liu.diva-portal.org/smash/record.jsf?pid=diva2%3A812606&dswid=4406 (accessed on 1 February 2024).
- Schröder, U.; Schröder, U.; Svenson, S.B. Nasal and parenteral immunizations with diphtheria toxoid using monoglyceride/fatty acid lipid suspensions as adjuvants. Vaccine 1999, 17, 2096–2103. [Google Scholar] [CrossRef] [PubMed]
- Haile, M.; Schröder, U.; Hamasur, B.; Pawlowski, A.; Jaxmar, T.; Källenius, G.; Svenson, S.B. Immunization with heat-killed Mycobacterium bovis bacille Calmette–Guerin (BCG) in EurocineTM L3 adjuvant protects against tuberculosis. Vaccine 2004, 22, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Hinkula, J.; Devito, C.; Zuber, B.; Benthin, R.; Ferreira, D.; Wahren, B.; Schröder, U. A novel DNA adjuvant, N3, enhances mucosal and systemic immune responses induced by HIV-1 DNA and peptide immunizations. Vaccine 2006, 24, 4494–4497. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Azharuddin, M.; Cardona, M.E.; Devito, C.; von Castelmur, E.; Wehlin, A.; Al-Otaibi, N. Intranasal Coronavirus SARS-CoV-2 Immunization with Lipid Adjuvants Provides Systemic and Mucosal Immune Response against SARS-CoV-2 S1 Spike and Nucleocapsid Protein. Vaccines 2022, 10, 504. [Google Scholar]
- Wasan, E.K.; Syeda, J.; Strom, S.; Cawthray, J.; Hancock, R.E.; Wasan, K.M.; Gerdts, V. A lipidic delivery system of a triple vaccine adjuvant enhances mucosal immunity following nasal administration in mice. Vaccine 2019, 37, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Corthésy, B.; Bioley, G. Lipid-Based Particles: Versatile Delivery Systems for Mucosal Vaccination against Infection. Front. Immunol. 2018, 9, 431. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.B.; Zhou, H.; Ponce, A.; Chen, W. Mucosal and systemic immune responses by intranasal immunization using archaeal lipid-adjuvanted vaccines. Vaccine 2007, 25, 8622–8636. [Google Scholar] [CrossRef]
- Bailoo, J.D.; Murphy, E.; Boada-Saña, M.; Varholick, J.A.; Hintze, S.; Baussière, C.; Hahn, K.C.; Göpfert, C.; Palme, R.; Voelkl, B.; et al. Effects of Cage Enrichment on Behavior, Welfare and Outcome Variability in Female Mice. Front. Behav. Neurosci. 2018, 12, 232. [Google Scholar] [CrossRef] [PubMed]
- Olsson, I.A.S.; Dahlborn, K. Improving housing conditions for laboratory mice: A review of ‘environmental enrichment’. Lab. Anim. 2002, 36, 243–270. [Google Scholar] [CrossRef]
- Franco, N.H.; Correia-Neves, M.; Olsson, I.A.S. How “Humane” Is Your Endpoint?—Refining the Science-Driven Approach for Termination of Animal Studies of Chronic Infection. PLoS Pathog. 2012, 8, e1002399. [Google Scholar] [CrossRef] [PubMed]
- Devito, C.; Zuber, B.; Schröder, U.; Benthin, R.; Okuda, K.; Broliden, K.; Wahren, B.; Hinkula, J. Intranasal HIV-1-gp160-DNA/gp41 Peptide Prime-Boost Immunization Regimen in Mice Results in Long-Term HIV-1 Neutralizing Humoral Mucosal and Systemic Immunity. J. Immunol. 2004, 173, 7078–7089. [Google Scholar] [CrossRef] [PubMed]
- Boonyo, W.; Junginger, H.E.; Waranuch, N.; Polnok, A.; Pitaksuteepong, T. Chitosan and trimethyl chitosan chloride (TMC) as adjuvants for inducing immune responses to ovalbumin in mice following nasal administration. J. Control. Release 2007, 121, 168–175. [Google Scholar] [CrossRef]
- Phua, K.K.L.; Staats, H.F.; Leong, K.W.; Nair, S.K. Intranasal mRNA nanoparticle vaccination induces prophylactic and therapeutic anti-tumor immunity. Sci. Rep. 2014, 4, 5128. [Google Scholar] [CrossRef]
- Sengupta, A.; Mukherjee, S.; Ghosh, S.; Keswani, T.; Sarkar, S.; Majumdar, G.; Bhattacharyya, A. Partial impairment of late-stage autophagic flux in murine splenocytes leads to sqstm1/p62 mediated nrf2-keap1 antioxidant pathway activation and induced proteasome-mediated degradation in malaria. Microb. Pathog. 2020, 147, 104289. [Google Scholar] [CrossRef]
- Sengupta, A.; Keswani, T.; Sarkar, S.; Ghosh, S.; Mukherjee, S.; Bhattacharyya, A. Autophagic induction modulates splenic plasmacytoid dendritic cell mediated immune response in cerebral malarial infection model. Microbes Infect. 2019, 21, 475–484. [Google Scholar] [CrossRef]
- Available online: https://www.miltenyibiotec.com/SE-en/resources/macs-handbook/mouse-cells-and-organs/mouse-cell-types/dendritic-cells-mouse.html (accessed on 1 February 2024).
- Available online: https://www.biolegend.com/en-us/dendritic-cells (accessed on 1 February 2024).
- Abcam. Effector T Cell Markers Poster. Available online: https://www.abcam.com/primary-antibodies/effector-t-cell-markers (accessed on 1 February 2024).
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef]
- Available online: https://www.bdbiosciences.com/content/dam/bdb/marketing-documents/Bcell_Brochure.pdf (accessed on 1 February 2024).
- Available online: https://resources.rndsystems.com/images/site/rnd-systems-bcells-br.pdf (accessed on 1 February 2024).
- Sengupta, A.; Al-Otaibi, N.; Hinkula, J. Sex-Specific Immune Responses to Seasonal Influenza Vaccination in Diabetic Individuals: Implications for Vaccine Efficacy. J. Immunol. Res. 2023, 2023, 3111351. [Google Scholar] [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef]
- Durai, V.; Murphy, K.M. Functions of Murine Dendritic Cells. Immunity 2016, 45, 719–736. [Google Scholar] [CrossRef] [PubMed]
- Keswani, T.; Sarkar, S.; Sengupta, A.; Bhattacharyya, A. Role of TGF-β and IL-6 in dendritic cells, Treg and Th17 mediated immune response during experimental cerebral malaria. Cytokine 2016, 88, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, J.D.; Ito, J.T.; de Martins, M.A.; de Tibério, I.F.L.C.; dos Lopes, F.D.T.Q.S. Th17/Treg Imbalance in Chronic Obstructive Pulmonary Disease: Clinical and Experimental Evidence. Front. Immunol. 2021, 12, 804919. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Jin, H.; Xu, Y. T Cell Metabolism: A New Perspective on Th17/Treg Cell Imbalance in Systemic Lupus Erythematosus. Front. Immunol. 2020, 11, 542018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Swiggard, W.J.; Heufler, C.; Peng, M.; Mirza, A.; Steinman, R.M.; Nussenzweig, M.C. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 1995, 375, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, K.; Guo, M.; Lee, S.; Sepulveda, H.; Swain, S.L.; Nussenzweig, M.; Steinman, R.M. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 2000, 151, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Lagergård, T.; Yang, Y.; Lindblad, M.; Fredriksson, M.; Wallerström, G.; Holmgren, J. Effect of pre-existing immunity for systemic and mucosal immune responses to intranasal immunization with group B Streptococcus type III capsular polysaccharide-cholera toxin B subunit conjugate. Vaccine 2001, 19, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Lagergård, T.; Yang, Y.; Lindblad, M.; Fredriksson, M.; Holmgren, J. Systemic and Mucosal Immune Responses in Mice after Mucosal Immunization with Group B Streptococcus Type III Capsular Polysaccharide-Cholera Toxin B Subunit Conjugate Vaccine. Infect. Immun. 2000, 68, 5749–5755. [Google Scholar] [CrossRef]
- Macpherson, A.J.; McCoy, K.D.; Johansen, F.-E.; Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal Immunol. 2008, 1, 11–22. [Google Scholar] [CrossRef]
- Serum Cross-Reactive Antibody Response to a Novel Influenza A (H1N1) Virus after Vaccination with Seasonal Influenza Vaccine. Available online: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm5819a1.htm (accessed on 1 February 2024).
- Green, M.D.; Hussain Al-Humadi, N. Preclinical Toxicology of Vaccines. A Comprehensive Guide to Toxicology in Preclinical Drug Development; Academic Press: Cambridge, MA, USA, 2013; p. 619. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A. The physiology of the lymphatic system. Adv. Drug Deliv. Rev. 2001, 50, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhu, L.; Li, Y.; Zhang, X.; Xu, S.; Yang, G.; Delair, T. Chitosan-based Colloidal Polyelectrolyte Complexes for Drug Delivery: A Review. Carbohydr. Polym. 2020, 238, 116126. [Google Scholar] [CrossRef] [PubMed]
- Laera, D.; HogenEsch, H.; O’Hagan, D.T. Aluminum Adjuvants—‘Back to the Future’. Pharmaceutics 2023, 15, 1884. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Pérez-Betancourt, Y. Cationic Nanostructures for Vaccines Design. Biomimetics 2020, 5, 32. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Name | Antigen (HA) Dose | Adjuvant 2% | Sample Size |
|---|---|---|---|
| L3I | 1 μg | L3 Anionic | 5 |
| L3II | 0.1 μg | L3 Anionic | 5 |
| L3III | 0.01 μg | L3 Anionic | 5 |
| N3I | 1 μg | N3 Cationic | 5 |
| N3II | 0.1 μg | N3 Cationic | 5 |
| N3III | 0.01 μg | N3 Cationic | 5 |
| Control | 1 μg | No Adjuvant/ Buffer | 5 |
| Flow Cytometry Antibody Combinations | Phenotypes/Activation Marker | Antibodies Used (Catalogue No.) * | Refs. |
|---|---|---|---|
| Dendritic Cells | B220-APC-Vio770 (130-102-267) MHCII-FITC (130-112-386) CD11c-APC (130-119-762) CD11b-PE-Vio615 (130-113-807) CD8a-PerCP-Vio700 (130-128-616) CD80-VioBlue/PB (Biolegend: 104723) CD86-PE (130-123-724) MHCI-Vioblue (130-125-228) DEC205-PE (130-112-272) | [38,39] | |
| B220-MHCII+CD11c+CD11b−CD8a+ | Conventional dendritic cells 1 (cDC1) | ||
| B220-MHCII+CD11c+CD11b+ CD8a- | Conventional dendritic cells 2 (cDC2) | ||
| cDC1/cDC2+CD80+CD86 | Co-stimulatory expression | ||
| cDC1/cDC2+MHCI | MHC/co-stimulation marker | ||
| cDC1/cDC2+DEC205 | Activation/co-stimulation marker | ||
| T cells: | CD3-PerCP-Vio700 (130-120-826) CD8-APC-Vio770 (130-123-280) CD4-PE-Vio615 (130-118-455) FOXP3-PE (130-111-678) RORgT-APC (130-124-035) CD28-PE (130-102-601) INFγ-PE (130-123-700) | [40,41] | |
| CD3+CD8+ | Cytotoxic T cells | ||
| CD3+CD8+CD28 | Co-stimulatory expression | ||
| CD3+CD4+ | Helper T cells | ||
| CD3+CD4+FOXP3+ | Treg subset of CD4T cells | ||
| CD3+CD4+RORγT+ | Th17 subset of CD4T cells | ||
| B cells: | B220-APC (130-110-847) CD19-APC-Vio770 (130-123-572) MHCII-PerCP-Vio700 (130-112-391) MHCII-PE (130-112-387) IgM-FITC (130-095-906) IgD-BV605/TexasRed (Biolegend: 405727) CD138-PerCP-Cy5.5 (Biolegend: 142509) CD40-Vioblue/PB (Biolegend: 124625) CD80-PE-Vio615 (130-116-467) CD43-Vioblue (130-112-702) CD27-FITC (130-114-165) | [42,43] | |
| B220lowCD19 low/neg IgM-IgD-CD138+ | Long-lived plasma cells | ||
| B220/CD45R+CD19+IgM+IgD+MHCII+CD138- | Activated B cell | ||
| B220/CD45R+CD19+IgMhigh+IgDlow/neg+CD43- | Immature/Transitional B | ||
| CD45R/B220+CD19+ CD27+MHC2+CD80+CD40+ | Memory B cells | ||
| Parameters | L3 Anionic | N3 Cationic | Fig/Ref. |
|---|---|---|---|
| Dendritic cells | Increased cDC2% | Increase in cDC1% | Figure 1B |
| Higher CD80/86+ cDC2 | Higher CD80/86+ cDC1 | Figure 1C | |
| Increased in MHCI+ cDC1 than control | Significantly higher MHCI+ cDC1 than cDC2 | Figure 1D | |
| Increased DEC205 in cDC2 | Increased DEC205 in cDC1 | Figure 1E | |
| T lymphocytes | Significantly lower CD8T population and CD28+CD8T than N3 | Significantly high CD8T population and CD28+CD8T than L3 | Figure 2B–D |
| Significantly higher CD4T than N3 and control | Significantly lower than L3, higher than control | Figure 2E,F | |
| Treg: Significantly higher than N3 and control | Treg: No change compared to the control | Figure 3A | |
| Th17: No change than control | Th17: Significantly higher than L3 and control | Figure 3B | |
| Low IFNγ within CD8T, high within CD4T | High IFNγ within CD4T and CD8T | Figure 3D | |
| B lymphocytes | Few immature B and high memory B cells | More immature B and low memory B cells | Figure 4E |
| Significantly more activated B cells | No significant change compared to control | Figure 4F | |
| Significantly more plasma cells | No significant change compared to control | Figure 4F | |
| Antibody titer | Higher anti-HA IgG compared to N3 and control. | Significantly higher anti-HA IgG than control | Figure 5A |
| Significantly higher anti-HA IgA than N3. | Significantly lower anti-HA IgA than L3 and control | Figure 5B | |
| HIA titer | Significantly higher than N3 | Significantly lower than L3, higher than control | Figure 5C |
| Previous Report | 24-fold increase in HIA titer compared to non-adjuvanted formulation | 4-fold increase in HIA titer compared to non-adjuvanted formulation | Ref. [12] |
| ELISPOT: 7-fold increase in T cell response as pg/mL of INF-γ over non-adjuvanted formulation | 60-fold increase in T cell response as pg/mL of INF-γ over non-adjuvanted formulation | ||
| Conclusion | Generated a stronger humoral immune response. Recommended formulation for:
| Generated stronger cellular Th1/Th17 immunity. Recommended formulation for:
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sengupta, A.; Al-Otaibi, N.; Devito, C.; Lottersberger, F.; Hinkula, J. Differential Immune Response Patterns Induced by Anionic and Cationic Lipid Adjuvants in Intranasal Anti-Influenza Immunization. Vaccines 2024, 12, 320. https://doi.org/10.3390/vaccines12030320
Sengupta A, Al-Otaibi N, Devito C, Lottersberger F, Hinkula J. Differential Immune Response Patterns Induced by Anionic and Cationic Lipid Adjuvants in Intranasal Anti-Influenza Immunization. Vaccines. 2024; 12(3):320. https://doi.org/10.3390/vaccines12030320
Chicago/Turabian StyleSengupta, Anirban, Noha Al-Otaibi, Claudia Devito, Francisca Lottersberger, and Jorma Hinkula. 2024. "Differential Immune Response Patterns Induced by Anionic and Cationic Lipid Adjuvants in Intranasal Anti-Influenza Immunization" Vaccines 12, no. 3: 320. https://doi.org/10.3390/vaccines12030320