
Next-Generation TB Vaccines: Progress, Challenges, and Prospects



Abstract
:1. Introduction
2. Infection and Immunity to MTB
2.1. Innate Immune Responses Induced by MTB
2.2. Adaptive Immune Responses Induced by MTB
2.2.1. CD4+ T Cells and Their Differentiation and Balance
2.2.2. CD8+ T Cells and B Cells
3. The Clinical Pipeline of TB Vaccines
3.1. Inactivated TB Vaccines
3.1.1. MIP
3.1.2. RUTI Vaccine
3.1.3. Mycobacterium Vaccae
3.1.4. DAR-901
3.2. Attenuated TB Vaccines
3.2.1. MTBVAC
3.2.2. BCG Revaccination (Gates MRI-TBV01-201)
3.3. Recombinant BCG Vaccines
3.4. Subunit TB Vaccines
3.4.1. M72/AS01E
3.4.2. GamTBvac
3.4.3. H56:IC31
3.4.4. H4:IC31 (AERAS-404)
3.4.5. ID93+GLA-SE (QTP101)
3.4.6. AEC/BC02
3.5. Viral Vector-Based TB Vaccines
3.5.1. MVA85A
3.5.2. ChAdOx1.85A
3.5.3. TB/FLU-01L and TB/FLU-04L
3.5.4. AdHu5Ag85A (formerly Ad5Ag85A)
3.6. TB DNA Vaccines
4. Challenges and Prospects in the Research of Novel TB Vaccines
4.1. Unsustainability of TB Vaccine Clinical Trials
4.2. The Selection of Suitable Immunogenic Antigenic Epitopes Is the Focus and Challenge of TB Vaccine Research
4.3. Clinical Trials on TB Vaccines for Pregnant Women Is Lacking
4.4. Controversies in the Evaluation Endpoints of TB Vaccine Clinical Trials
4.5. The Choice of Vaccine Adjuvants or Delivery Systems Is Crucial for the Immunogenicity and Protective Efficacy of TB Vaccines
4.5.1. Liposomes and Emulsions
4.5.2. TLR-9 agonist CpG-ODN1a and IC31 Adjuvants
4.5.3. Possible Future Application for Adjuvant or Delivery Systems for TB Subunit Vaccines
4.6. The Choice of Animal Models for TB Vaccine Research
4.7. Deep Learning Empowers TB Vaccine Research
4.7.1. Inclusion Criteria for Clinical Trials in TB Diagnosis
4.7.2. Prediction of MTB Protein Structures
4.7.3. Prediction and Screening of MTB Epitopes
4.7.4. Prediction and Optimization of Vaccine Administration Timing
- Pathogen infection and immune status monitoring: Following pathogen infection, the human body initiates an immune response against the pathogen, with the timing and intensity of these responses often influenced by various factors such as the mode and dosage of infection. The timing and dosage selection for vaccine administration largely depend on the patient’s immune status. Therefore, monitoring a patient’s immune status is crucial for determining the timing and dosage of vaccine administration. Deep learning techniques can be applied to monitor a patient’s immune status and pathogen infection, providing accurate predictions and recommendations for optimizing the timing of vaccine administration. For example, medical researchers can utilize deep learning technology to perform comprehensive analysis of various diagnostic data, including pathogen detection and immunological assessment, to predict the timing and intensity of immune system responses, thereby determining the optimal timing for vaccine administration.
- T cell epitope immunogenicity prediction: Deep learning techniques can be applied to accurately analyze the complexity of T cell immune responses and predict the intensity and timing of future immune responses. Taking TB vaccine as an example, TB is characterized by chronic infection. After MTB infection, it resides within the host for an extended period and triggers immune responses at appropriate times. Therefore, predicting the immunogenicity of potential antigenic epitopes of MTB is a key aspect of constructing an ideal vaccine. Deep learning techniques can perform T cell epitope immunogenicity prediction from various perspectives, including models based on deep neural networks such as DeepImmuno-CNN, DeepImmuno-GAN, DeepNetBim, and DeepHLApan. These models can predict the immunogenicity of future T cell responses based on potential immune factors, thereby proposing more rational vaccine administration timing and strategies [297,298].
- Vaccine dosage selection: Analyzing and predicting a patient’s immune status using deep learning techniques can assist physicians in making better decisions regarding vaccine dosage and administration. Deep learning can consider factors such as patient weight, age, and disease condition to predict the optimal vaccine dosage, thus determining the best vaccine administration timing and strategy.
4.7.5. Immune Repertoire Analysis
4.8. TB mRNA Vaccines
4.9. Virus-like Particle (VLP)-Based TB Vaccines
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zumla, A.; Raviglione, M.; Hafner, R.; von Reyn, C.F. Tuberculosis. N. Engl. J. Med. 2013, 368, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A. The white plague returns to London—With a vengeance. Lancet 2011, 377, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Barberis, I.; Bragazzi, N.L.; Galluzzo, L.; Martini, M. The history of tuberculosis: From the first historical records to the isolation of Koch’s bacillus. J. Prev. Med. Hyg. 2017, 58, E9–E12. [Google Scholar] [PubMed]
- WHO. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022; pp. 1–49. [Google Scholar]
- Bagcchi, S. WHO’s Global Tuberculosis Report 2022. Lancet Microbe 2023, 4, e20. [Google Scholar] [CrossRef]
- Glaziou, P.; Floyd, K.; Raviglione, M.C. Global Epidemiology of Tuberculosis. Semin. Respir. Crit. Care Med. 2018, 39, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.E. Vaccine Development Against Tuberculosis Over the Last 140 Years: Failure as Part of Success. Front. Microbiol. 2021, 12, 750124. [Google Scholar] [CrossRef]
- Adesanya, O.A.; Uche-Orji, C.I.; Adedeji, Y.A.; Joshua, J.I.; Adesola, A.A.; Chukwudike, C.J. Bacillus Calmette-Guerin (BCG): The adroit vaccine. AIMS Microbiol. 2021, 7, 96–113. [Google Scholar] [CrossRef]
- Colditz, G.A.; Brewer, T.F.; Berkey, C.S.; Wilson, M.E.; Burdick, E.; Fineberg, H.V.; Mosteller, F. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA 1994, 271, 698–702. [Google Scholar] [CrossRef]
- Fine, P.E. Variation in protection by BCG: Implications of and for heterologous immunity. Lancet 1995, 346, 1339–1345. [Google Scholar] [CrossRef]
- Singh, A.K.; Netea, M.G.; Bishai, W.R. BCG turns 100: Its nontraditional uses against viruses, cancer, and immunologic diseases. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Gong, W.; Wu, X. Differential Diagnosis of Latent Tuberculosis Infection and Active Tuberculosis: A Key to a Successful Tuberculosis Control Strategy. Front. Microbiol. 2021, 12, 745592. [Google Scholar] [CrossRef] [PubMed]
- Collins, H.L.; Kaufmann, S.H. Prospects for better tuberculosis vaccines. Lancet Infect. Dis. 2001, 1, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Fogel, N. Tuberculosis: A disease without boundaries. Tuberculosis 2015, 95, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Zhao, Y. Global Control of Tuberculosis: Current Status and Future Prospects. Zoonoses 2021, 2, 9. [Google Scholar] [CrossRef]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell. Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Deng, G.; Li, M.; Liu, X.; Wang, Y. Roles of Mucosal Immunity against Mycobacterium tuberculosis Infection. Tuberc. Res. Treat. 2012, 2012, 791728. [Google Scholar] [CrossRef] [Green Version]
- Chai, Q.; Lu, Z.; Liu, C.H. Host defense mechanisms against Mycobacterium tuberculosis. Cell. Mol. Life Sci. 2020, 77, 1859–1878. [Google Scholar] [CrossRef]
- Bhatt, K.; Salgame, P. Host innate immune response to Mycobacterium tuberculosis. J. Clin. Immunol. 2007, 27, 347–362. [Google Scholar] [CrossRef]
- Koeken, V.; Verrall, A.J.; Netea, M.G.; Hill, P.C.; van Crevel, R. Trained innate immunity and resistance to Mycobacterium tuberculosis infection. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2019, 25, 1468–1472. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Cell. Mol. Life Sci. CMLS 2015, 72, 4111–4126. [Google Scholar] [CrossRef]
- Gong, W.; Pan, C.; Cheng, P.; Wang, J.; Zhao, G.; Wu, X. Peptide-Based Vaccines for Tuberculosis. Front. Immunol. 2022, 13, 830497. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Liang, Y.; Wu, X. The current status, challenges, and future developments of new tuberculosis vaccines. Hum. Vaccines Immunother. 2018, 14, 1697–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Round, J.L.; Palm, N.W. Causal effects of the microbiota on immune-mediated diseases. Sci. Immunol. 2018, 3, eaao1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Yam, W.C.; Chen, Z. Mycobacterium tuberculosis infection and vaccine development. Tuberculosis 2016, 98, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Jagannath, C. Analysis of host-pathogen modulators of autophagy during Mycobacterium Tuberculosis infection and therapeutic repercussions. Int. Rev. Immunol. 2017, 36, 271–286. [Google Scholar] [CrossRef]
- De Martino, M.; Lodi, L.; Galli, L.; Chiappini, E. Immune Response to Mycobacterium tuberculosis: A Narrative Review. Front. Pediatr. 2019, 7, 350. [Google Scholar] [CrossRef] [Green Version]
- Guirado, E.; Schlesinger, L.S.; Kaplan, G. Macrophages in tuberculosis: Friend or foe. Semin. Immunopathol. 2013, 35, 563–583. [Google Scholar] [CrossRef] [Green Version]
- Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Breckpot, K. mRNA-based dendritic cell vaccines. Expert Rev. Vaccines 2015, 14, 161–176. [Google Scholar] [CrossRef]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.J.; Everts, B. Dendritic cell metabolism. Nat. Rev. Immunol. 2015, 15, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Junqueira-Kipnis, A.P.; Kipnis, A.; Jamieson, A.; Juarrero, M.G.; Diefenbach, A.; Raulet, D.H.; Turner, J.; Orme, I.M. NK cells respond to pulmonary infection with Mycobacterium tuberculosis, but play a minimal role in protection. J. Immunol. 2003, 171, 6039–6045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, C.; Fernández, A.S.; Carrillo, J.M.; Romero, P.; Molina, I.J.; Moreno, J.C.; Santamaría, M. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J. Leukoc. Biol. 2009, 86, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; Yang, X. Natural killer cells in host defense against veterinary pathogens. Vet. Immunol. Immunopathol. 2015, 168, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Choreño Parra, J.A.; Martínez Zúñiga, N.; Jiménez Zamudio, L.A.; Jiménez Álvarez, L.A.; Salinas Lara, C.; Zúñiga, J. Memory of Natural Killer Cells: A New Chance against Mycobacterium tuberculosis? Front. Immunol. 2017, 8, 967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.F.; Pi, J.; Xu, J.F. Emerging Role of Exosomes in Tuberculosis: From Immunity Regulations to Vaccine and Immunotherapy. Front. Immunol. 2021, 12, 628973. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Warren, E.; Teskey, G.; Venketaraman, V. Effector Mechanisms of Neutrophils within the Innate Immune System in Response to Mycobacterium tuberculosis Infection. J. Clin. Med. 2017, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Uzorka, J.W.; Bakker, J.A.; van Meijgaarden, K.E.; Leyten, E.M.S.; Delfos, N.M.; Hetem, D.J.; Kerremans, J.; Zwarts, M.; Cozijn, S.; Ottenhoff, T.H.M.; et al. Biomarkers to identify Mycobacterium tuberculosis infection among borderline QuantiFERON results. Eur. Respir. J. 2022, 60, 2102665. [Google Scholar] [CrossRef]
- Stevens, M.T.; Nagaria, B.D.; Britton, W.J.; Saunders, B.M. Macrophages of different tissue origin exhibit distinct inflammatory responses to mycobacterial infection. Immunol. Cell Biol. 2021, 99, 1085–1092. [Google Scholar] [CrossRef]
- Ankrah, A.O.; Glaudemans, A.; Maes, A.; Van de Wiele, C.; Dierckx, R.; Vorster, M.; Sathekge, M.M. Tuberculosis. Semin. Nucl. Med. 2018, 48, 108–130. [Google Scholar] [CrossRef]
- Ernst, J.D. Mechanisms of M. tuberculosis Immune Evasion as Challenges to TB Vaccine Design. Cell Host Microbe 2018, 24, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiya, T.; Yoshimura, A. In Vitro Th Differentiation Protocol. Methods Mol. Biol. 2016, 1344, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Abrahem, R.; Chiang, E.; Haquang, J.; Nham, A.; Ting, Y.S.; Venketaraman, V. The Role of Dendritic Cells in TB and HIV Infection. J. Clin. Med. 2020, 9, 2661. [Google Scholar] [CrossRef]
- Geginat, J.; Paroni, M.; Maglie, S.; Alfen, J.S.; Kastirr, I.; Gruarin, P.; De Simone, M.; Pagani, M.; Abrignani, S. Plasticity of human CD4 T cell subsets. Front. Immunol. 2014, 5, 630. [Google Scholar] [CrossRef] [Green Version]
- Cowley, S.C.; Elkins, K.L. CD4+ T cells mediate IFN-gamma-independent control of Mycobacterium tuberculosis infection both in vitro and in vivo. J. Immunol. 2003, 171, 4689–4699. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, A.; Umemura, M.; Tamura, T.; Kariyone, A.; Begum, M.D.; Kawakami, K.; Okamoto, Y.; Hamada, S.; Oshiro, K.; Kohama, H.; et al. Suppressed induction of mycobacterial antigen-specific Th1-type CD4+ T cells in the lung after pulmonary mycobacterial infection. Int. Immunol. 2010, 22, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Herbst, S.; Schaible, U.E.; Schneider, B.E. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS ONE 2011, 6, e19105. [Google Scholar] [CrossRef] [Green Version]
- Prezzemolo, T.; Guggino, G.; La Manna, M.P.; Di Liberto, D.; Dieli, F.; Caccamo, N. Functional Signatures of Human CD4 and CD8 T Cell Responses to Mycobacterium tuberculosis. Front. Immunol. 2014, 5, 180. [Google Scholar] [CrossRef] [Green Version]
- Barber, D.L.; Mayer-Barber, K.D.; Feng, C.G.; Sharpe, A.H.; Sher, A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J. Immunol. 2011, 186, 1598–1607. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, D.K.; Beasley, R.; Marks, G.B. The Immunological Mysteries of Tuberculosis. J. Allergy Clin. Immunol. Pract. 2019, 7, 649–650. [Google Scholar] [CrossRef]
- da Silva, M.V.; Massaro Junior, V.J.; Machado, J.R.; Silva, D.A.; Castellano, L.R.; Alexandre, P.B.; Rodrigues, D.B.; Rodrigues, V. Expression pattern of transcription factors and intracellular cytokines reveals that clinically cured tuberculosis is accompanied by an increase in Mycobacterium-specific Th1, Th2, and Th17 cells. Biomed Res. Int. 2015, 2015, 591237. [Google Scholar] [CrossRef]
- Namdeo, M.; Kandel, R.; Thakur, P.K.; Mohan, A.; Dey, A.B.; Mitra, D.K. Old age-associated enrichment of peripheral T regulatory cells and altered redox status in pulmonary tuberculosis patients. Eur. J. Immunol. 2020, 50, 1195–1208. [Google Scholar] [CrossRef]
- Pang, H.; Yu, Q.; Guo, B.; Jiang, Y.; Wan, L.; Li, J.; Wu, Y.; Wan, K. Frequency of regulatory T-cells in the peripheral blood of patients with pulmonary tuberculosis from shanxi province, china. PLoS ONE 2013, 8, e65496. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Xiang, Y.; Yang, L.; Liu, Y.; Chen, P.; Wang, L.; Feng, W.; Yin, K.; Fu, M.; Xu, Y.; et al. Impaired NK cells’ activity and increased numbers of CD4 + CD25+ regulatory T cells in multidrug-resistant Mycobacterium tuberculosis patients. Tuberculosis 2016, 98, 13–20. [Google Scholar] [CrossRef]
- Stringari, L.L.; Covre, L.P.; da Silva, F.D.C.; de Oliveira, V.L.; Campana, M.C.; Hadad, D.J.; Palaci, M.; Salgame, P.; Dietze, R.; Gomes, D.C.O.; et al. Increase of CD4+CD25highFoxP3+ cells impairs in vitro human microbicidal activity against Mycobacterium tuberculosis during latent and acute pulmonary tuberculosis. PLoS Neglected Trop. Dis. 2021, 15, e0009605. [Google Scholar] [CrossRef]
- Cardona, P.; Cardona, P.J. Regulatory T Cells in Mycobacterium tuberculosis Infection. Front. Immunol. 2019, 10, 2139. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.M.; Lonergan, R.; Costelloe, L.; Kinsella, K.; Moran, B.; O’Farrelly, C.; Tubridy, N.; Mills, K.H. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J. Immunol. 2009, 183, 7602–7610. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 Cell Responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, M.; He, M.; Cui, H.; Zhang, J.; Shi, L.; Wang, W.; Xu, W.; Gao, B.; Ding, J. Weak binder for MHC molecule is a potent Mycobacterium tuberculosis-specific CTL epitope in the context of HLA-A24 allele. Microb. Pathog. 2012, 53, 162–167. [Google Scholar] [CrossRef]
- Marashian, S.M.; Mortaz, E.; Jamaati, H.R.; Alavi-Moghaddam, M.; Kiani, A.; Abedini, A.; Garssen, J.; Adcock, I.M.; Velayati, A.A. Role of Innate Lymphoid Cells in Lung Disease. Iran. J. Allergy Asthma Immunol. 2015, 14, 346–360. [Google Scholar]
- Chen, Z.W. Protective immune responses of major Vγ2Vδ2 T-cell subset in M. tuberculosis infection. Curr. Opin. Immunol. 2016, 42, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.F.; Murray, H.W.; Wiebe, M.E.; Rubin, B.Y. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 1983, 158, 670–689. [Google Scholar] [CrossRef]
- Aqbi, H.F.; Wallace, M.; Sappal, S.; Payne, K.K.; Manjili, M.H. IFN-γ orchestrates tumor elimination, tumor dormancy, tumor escape, and progression. J. Leukoc. Biol. 2018, 103, 1219–1223. [Google Scholar] [CrossRef]
- Kramarska, E.; Squeglia, F.; De Maio, F.; Delogu, G.; Berisio, R. PE_PGRS33, an Important Virulence Factor of Mycobacterium tuberculosis and Potential Target of Host Humoral Immune Response. Cells 2021, 10, 161. [Google Scholar] [CrossRef]
- Rao, M.; Valentini, D.; Poiret, T.; Dodoo, E.; Parida, S.; Zumla, A.; Brighenti, S.; Maeurer, M. B in TB: B Cells as Mediators of Clinically Relevant Immune Responses in Tuberculosis. Clin. Infect. Dis. 2015, 61 (Suppl. S3), S225–S234. [Google Scholar] [CrossRef] [Green Version]
- DeFalco, J.; Harbell, M.; Manning-Bog, A.; Baia, G.; Scholz, A.; Millare, B.; Sumi, M.; Zhang, D.; Chu, F.; Dowd, C.; et al. Non-progressing cancer patients have persistent B Cell Responses expressing shared antibody paratopes that target public tumor antigens. Clin. Immunol. 2018, 187, 37–45. [Google Scholar] [CrossRef]
- Jagusztyn-Krynicka, E.K.; Roszczenko, P.; Grabowska, A. Impact of proteomics on anti-Mycobacterium tuberculosis (MTB) vaccine development. Pol. J. Microbiol. 2009, 58, 281–287. [Google Scholar]
- Ginsberg, A.M. What’s new in tuberculosis vaccines? Bull. World Health Organ. 2002, 80, 483–488. [Google Scholar]
- Sable, S.B.; Posey, J.E.; Scriba, T.J. Tuberculosis Vaccine Development: Progress in Clinical Evaluation. Clin. Microbiol. Rev. 2019, 33, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.; Triccas, J.A.; Petrovsky, N. Adjuvant Strategies for More Effective Tuberculosis Vaccine Immunity. Microorganisms 2019, 7, 255. [Google Scholar] [CrossRef] [Green Version]
- Cable, J.; Srikantiah, P.; Crowe, J.E., Jr.; Pulendran, B.; Hill, A.; Ginsberg, A.; Koff, W.; Mathew, A.; Ng, T.; Jansen, K.; et al. Vaccine innovations for emerging infectious diseases-a symposium report. Ann. N. Y. Acad. Sci. 2020, 1462, 14–26. [Google Scholar] [CrossRef]
- Romano, M.; Squeglia, F.; Kramarska, E.; Barra, G.; Choi, H.G.; Kim, H.J.; Ruggiero, A.; Berisio, R. A Structural View at Vaccine Development against M. tuberculosis. Cells 2023, 12, 317. [Google Scholar] [CrossRef]
- Zhu, B.; Dockrell, H.M.; Ottenhoff, T.H.M.; Evans, T.G.; Zhang, Y. Tuberculosis vaccines: Opportunities and challenges. Respirology 2018, 23, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hokey, D.A.; Ginsberg, A. The current state of tuberculosis vaccines. Hum. Vaccines Immunother. 2013, 9, 2142–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Meeren, O.; Hatherill, M.; Nduba, V.; Wilkinson, R.J.; Muyoyeta, M.; Van Brakel, E.; Ayles, H.M.; Henostroza, G.; Thienemann, F.; Scriba, T.J.; et al. Phase 2b Controlled Trial of M72/AS01(E) Vaccine to Prevent Tuberculosis. N. Engl. J. Med. 2018, 379, 1621–1634. [Google Scholar] [CrossRef]
- Tait, D.R.; Hatherill, M.; Van Der Meeren, O.; Ginsberg, A.M.; Van Brakel, E.; Salaun, B.; Scriba, T.J.; Akite, E.J.; Ayles, H.M.; Bollaerts, A.; et al. Final Analysis of a Trial of M72/AS01(E) Vaccine to Prevent Tuberculosis. N. Engl. J. Med. 2019, 381, 2429–2439. [Google Scholar] [CrossRef]
- Penn-Nicholson, A.; Geldenhuys, H.; Burny, W.; van der Most, R.; Day, C.L.; Jongert, E.; Moris, P.; Hatherill, M.; Ofori-Anyinam, O.; Hanekom, W.; et al. Safety and immunogenicity of candidate vaccine M72/AS01E in adolescents in a TB endemic setting. Vaccine 2015, 33, 4025–4034. [Google Scholar] [CrossRef] [Green Version]
- Vasina, D.V.; Kleymenov, D.A.; Manuylov, V.A.; Mazunina, E.P.; Koptev, E.Y.; Tukhovskaya, E.A.; Murashev, A.N.; Gintsburg, A.L.; Gushchin, V.A.; Tkachuk, A.P. First-In-Human Trials of GamTBvac, a Recombinant Subunit Tuberculosis Vaccine Candidate: Safety and Immunogenicity Assessment. Vaccines 2019, 7, 166. [Google Scholar] [CrossRef] [Green Version]
- Tkachuk, A.P.; Bykonia, E.N.; Popova, L.I.; Kleymenov, D.A.; Semashko, M.A.; Chulanov, V.P.; Fitilev, S.B.; Maksimov, S.L.; Smolyarchuk, E.A.; Manuylov, V.A.; et al. Safety and Immunogenicity of the GamTBvac, the Recombinant Subunit Tuberculosis Vaccine Candidate: A Phase II, Multi-Center, Double-Blind, Randomized, Placebo-Controlled Study. Vaccines 2020, 8, 652. [Google Scholar] [CrossRef] [PubMed]
- Luabeya, A.K.; Kagina, B.M.; Tameris, M.D.; Geldenhuys, H.; Hoff, S.T.; Shi, Z.; Kromann, I.; Hatherill, M.; Mahomed, H.; Hanekom, W.A.; et al. First-in-human trial of the post-exposure tuberculosis vaccine H56:IC31 in Mycobacterium tuberculosis infected and non-infected healthy adults. Vaccine 2015, 33, 4130–4140. [Google Scholar] [CrossRef]
- Bekker, L.G.; Dintwe, O.; Fiore-Gartland, A.; Middelkoop, K.; Hutter, J.; Williams, A.; Randhawa, A.K.; Ruhwald, M.; Kromann, I.; Andersen, P.L.; et al. A phase 1b randomized study of the safety and immunological responses to vaccination with H4:IC31, H56:IC31, and BCG revaccination in Mycobacterium tuberculosis-uninfected adolescents in Cape Town, South Africa. EClinicalMedicine 2020, 21, 100313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suliman, S.; Luabeya, A.K.K.; Geldenhuys, H.; Tameris, M.; Hoff, S.T.; Shi, Z.; Tait, D.; Kromann, I.; Ruhwald, M.; Rutkowski, K.T.; et al. Dose Optimization of H56:IC31 Vaccine for Tuberculosis-Endemic Populations. A Double-Blind, Placebo-controlled, Dose-Selection Trial. Am. J. Respir. Crit. Care Med. 2019, 199, 220–231. [Google Scholar] [CrossRef]
- Jenum, S.; Tonby, K.; Rueegg, C.S.; Rühwald, M.; Kristiansen, M.P.; Bang, P.; Olsen, I.C.; Sellæg, K.; Røstad, K.; Mustafa, T.; et al. A Phase I/II randomized trial of H56:IC31 vaccination and adjunctive cyclooxygenase-2-inhibitor treatment in tuberculosis patients. Nat. Commun. 2021, 12, 6774. [Google Scholar] [CrossRef]
- Norrby, M.; Vesikari, T.; Lindqvist, L.; Maeurer, M.; Ahmed, R.; Mahdavifar, S.; Bennett, S.; McClain, J.B.; Shepherd, B.M.; Li, D.; et al. Safety and immunogenicity of the novel H4:IC31 tuberculosis vaccine candidate in BCG-vaccinated adults: Two phase I dose escalation trials. Vaccine 2017, 35, 1652–1661. [Google Scholar] [CrossRef]
- Nemes, E.; Geldenhuys, H.; Rozot, V.; Rutkowski, K.T.; Ratangee, F.; Bilek, N.; Mabwe, S.; Makhethe, L.; Erasmus, M.; Toefy, A.; et al. Prevention of M. tuberculosis Infection with H4:IC31 Vaccine or BCG Revaccination. N. Engl. J. Med. 2018, 379, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Coler, R.N.; Day, T.A.; Ellis, R.; Piazza, F.M.; Beckmann, A.M.; Vergara, J.; Rolf, T.; Lu, L.; Alter, G.; Hokey, D.; et al. The TLR-4 agonist adjuvant, GLA-SE, improves magnitude and quality of immune responses elicited by the ID93 tuberculosis vaccine: First-in-human trial. NPJ Vaccines 2018, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Day, T.A.; Penn-Nicholson, A.; Luabeya, A.K.K.; Fiore-Gartland, A.; Du Plessis, N.; Loxton, A.G.; Vergara, J.; Rolf, T.A.; Reid, T.D.; Toefy, A.; et al. Safety and immunogenicity of the adjunct therapeutic vaccine ID93 + GLA-SE in adults who have completed treatment for tuberculosis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet Respir. Med. 2021, 9, 373–386. [Google Scholar] [CrossRef]
- Grode, L.; Ganoza, C.A.; Brohm, C.; Weiner, J., 3rd; Eisele, B.; Kaufmann, S.H. Safety and immunogenicity of the recombinant BCG vaccine VPM1002 in a phase 1 open-label randomized clinical trial. Vaccine 2013, 31, 1340–1348. [Google Scholar] [CrossRef]
- Loxton, A.G.; Knaul, J.K.; Grode, L.; Gutschmidt, A.; Meller, C.; Eisele, B.; Johnstone, H.; van der Spuy, G.; Maertzdorf, J.; Kaufmann, S.H.E.; et al. Safety and Immunogenicity of the Recombinant Mycobacterium bovis BCG Vaccine VPM1002 in HIV-Unexposed Newborn Infants in South Africa. Clin. Vaccine Immunol. 2017, 24, e00439-16. [Google Scholar] [CrossRef] [Green Version]
- Cotton, M.F.; Madhi, S.A.; Luabeya, A.K.; Tameris, M.; Hesseling, A.C.; Shenje, J.; Schoeman, E.; Hatherill, M.; Desai, S.; Kapse, D.; et al. Safety and immunogenicity of VPM1002 versus BCG in South African newborn babies: A randomised, phase 2 non-inferiority double-blind controlled trial. Lancet Infect. Dis. 2022, 22, 1472–1483. [Google Scholar] [CrossRef]
- Spertini, F.; Audran, R.; Chakour, R.; Karoui, O.; Steiner-Monard, V.; Thierry, A.C.; Mayor, C.E.; Rettby, N.; Jaton, K.; Vallotton, L.; et al. Safety of human immunisation with a live-attenuated Mycobacterium tuberculosis vaccine: A randomised, double-blind, controlled phase I trial. Lancet Respir. Med. 2015, 3, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Tameris, M.; Mearns, H.; Penn-Nicholson, A.; Gregg, Y.; Bilek, N.; Mabwe, S.; Geldenhuys, H.; Shenje, J.; Luabeya, A.K.K.; Murillo, I.; et al. Live-attenuated Mycobacterium tuberculosis vaccine MTBVAC versus BCG in adults and neonates: A randomised controlled, double-blind dose-escalation trial. Lancet Respir. Med. 2019, 7, 757–770. [Google Scholar] [CrossRef]
- Sharma, S.K.; Katoch, K.; Sarin, R.; Balambal, R.; Kumar Jain, N.; Patel, N.; Murthy, K.J.R.; Singla, N.; Saha, P.K.; Khanna, A.; et al. Efficacy and Safety of Mycobacterium indicus pranii as an adjunct therapy in Category II pulmonary tuberculosis in a randomized trial. Sci. Rep. 2017, 7, 3354. [Google Scholar] [CrossRef] [Green Version]
- Vilaplana, C.; Montané, E.; Pinto, S.; Barriocanal, A.M.; Domenech, G.; Torres, F.; Cardona, P.J.; Costa, J. Double-blind, randomized, placebo-controlled Phase I Clinical Trial of the therapeutical antituberculous vaccine RUTI. Vaccine 2010, 28, 1106–1116. [Google Scholar] [CrossRef]
- Nell, A.S.; D’Lom, E.; Bouic, P.; Sabate, M.; Bosser, R.; Picas, J.; Amat, M.; Churchyard, G.; Cardona, P.J. Safety, tolerability, and immunogenicity of the novel antituberculous vaccine RUTI: Randomized, placebo-controlled phase II clinical trial in patients with latent tuberculosis infection. PLoS ONE 2014, 9, e89612. [Google Scholar] [CrossRef]
- Masonou, T.; Hokey, D.A.; Lahey, T.; Halliday, A.; Berrocal-Almanza, L.C.; Wieland-Alter, W.F.; Arbeit, R.D.; Lalvani, A.; von Reyn, C.F. CD4+ T cell cytokine responses to the DAR-901 booster vaccine in BCG-primed adults: A randomized, placebo-controlled trial. PLoS ONE 2019, 14, e0217091. [Google Scholar] [CrossRef] [Green Version]
- Munseri, P.; Said, J.; Amour, M.; Magohe, A.; Matee, M.; Rees, C.A.; Mackenzie, T.; Tvaroha, S.; Bailey-Kellogg, C.; Maro, I.; et al. DAR-901 vaccine for the prevention of infection with Mycobacterium tuberculosis among BCG-immunized adolescents in Tanzania: A randomized controlled, double-blind phase 2b trial. Vaccine 2020, 38, 7239–7245. [Google Scholar] [CrossRef]
- Hawkridge, T.; Scriba, T.J.; Gelderbloem, S.; Smit, E.; Tameris, M.; Moyo, S.; Lang, T.; Veldsman, A.; Hatherill, M.; Merwe, L.; et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in healthy adults in South Africa. J. Infect. Dis. 2008, 198, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Nemes, E.; Hesseling, A.C.; Tameris, M.; Mauff, K.; Downing, K.; Mulenga, H.; Rose, P.; van der Zalm, M.; Mbaba, S.; Van As, D.; et al. Safety and Immunogenicity of Newborn MVA85A Vaccination and Selective, Delayed Bacille Calmette-Guerin for Infants of Human Immunodeficiency Virus-Infected Mothers: A Phase 2 Randomized, Controlled Trial. Clin. Infect. Dis. 2018, 66, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Tameris, M.D.; Hatherill, M.; Landry, B.S.; Scriba, T.J.; Snowden, M.A.; Lockhart, S.; Shea, J.E.; McClain, J.B.; Hussey, G.D.; Hanekom, W.A.; et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: A randomised, placebo-controlled phase 2b trial. Lancet 2013, 381, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Wilkie, M.; Satti, I.; Minhinnick, A.; Harris, S.; Riste, M.; Ramon, R.L.; Sheehan, S.; Thomas, Z.M.; Wright, D.; Stockdale, L.; et al. A phase I trial evaluating the safety and immunogenicity of a candidate tuberculosis vaccination regimen, ChAdOx1 85A prime—MVA85A boost in healthy UK adults. Vaccine 2020, 38, 779–789. [Google Scholar] [CrossRef]
- Jeyanathan, M.; Fritz, D.K.; Afkhami, S.; Aguirre, E.; Howie, K.J.; Zganiacz, A.; Dvorkin-Gheva, A.; Thompson, M.R.; Silver, R.F.; Cusack, R.P.; et al. Aerosol delivery, but not intramuscular injection, of adenovirus-vectored tuberculosis vaccine induces respiratory-mucosal immunity in humans. JCI Insight 2022, 7, e155655. [Google Scholar] [CrossRef]
- Saini, V.; Raghuvanshi, S.; Talwar, G.P.; Ahmed, N.; Khurana, J.P.; Hasnain, S.E.; Tyagi, A.K.; Tyagi, A.K. Polyphasic taxonomic analysis establishes Mycobacterium indicus pranii as a distinct species. PLoS ONE 2009, 4, e6263. [Google Scholar] [CrossRef] [Green Version]
- Guleria, I.; Mukherjee, R.; Kaufmann, S.H. In vivo depletion of CD4 and CD8 T lymphocytes impairs Mycobacterium w vaccine-induced protection against M. tuberculosis in mice. Med. Microbiol. Immunol. 1993, 182, 129–135. [Google Scholar] [CrossRef]
- Gupta, A.; Ahmad, F.J.; Ahmad, F.; Gupta, U.D.; Natarajan, M.; Katoch, V.M.; Bhaskar, S. Protective efficacy of Mycobacterium indicus pranii against tuberculosis and underlying local lung immune responses in guinea pig model. Vaccine 2012, 30, 6198–6209. [Google Scholar] [CrossRef]
- Das, S.; Chowdhury, B.P.; Goswami, A.; Parveen, S.; Jawed, J.; Pal, N.; Majumdar, S. Mycobacterium indicus pranii (MIP) mediated host protective intracellular mechanisms against tuberculosis infection: Involvement of TLR-4 mediated signaling. Tuberculosis 2016, 101, 201–209. [Google Scholar] [CrossRef]
- Katoch, K.; Singh, P.; Adhikari, T.; Benara, S.K.; Singh, H.B.; Chauhan, D.S.; Sharma, V.D.; Lavania, M.; Sachan, A.S.; Katoch, V.M. Potential of Mw as a prophylactic vaccine against pulmonary tuberculosis. Vaccine 2008, 26, 1228–1234. [Google Scholar] [CrossRef]
- Patel, N.; Deshpande, M.M.; Shah, M. Effect of an immunomodulator containing Mycobacterium w on sputum conversion in pulmonary tuberculosis. J. Indian Med. Assoc. 2002, 100, 191–193. [Google Scholar]
- Patel, N.; Trapathi, S.B. Improved cure rates in pulmonary tuberculosis category II (retreatment) with mycobacterium w. J. Indian Med. Assoc. 2003, 101, 680–682. [Google Scholar] [PubMed]
- Cardona, P.J. RUTI: A new chance to shorten the treatment of latent tuberculosis infection. Tuberculosis 2006, 86, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K. New therapeutic approach for latent tuberculosis infection. Lung India 2011, 28, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Domingo, M.; Gil, O.; Serrano, E.; Guirado, E.; Nofrarias, M.; Grassa, M.; Cáceres, N.; Pérez, B.; Vilaplana, C.; Cardona, P.J. Effectiveness and safety of a treatment regimen based on isoniazid plus vaccination with Mycobacterium tuberculosis cells’ fragments: Field-study with naturally Mycobacterium caprae-infected goats. Scand. J. Immunol. 2009, 69, 500–507. [Google Scholar] [CrossRef]
- Cardona, P.-J.; Amat, I.; Gordillo, S.; Arcos, V.; Guirado, E.; Díaz, J.; Vilaplana, C.; Tapia, G.; Ausina, V. Immunotherapy with fragmented Mycobacterium tuberculosis cells increases the effectiveness of chemotherapy against a chronical infection in a murine model of tuberculosis. Vaccine 2005, 23, 1393–1398. [Google Scholar] [CrossRef]
- Boenickse, R.; Juhasz, E. Description of the New Species Mycobacterium Vaccae N. Sp. Zentralbl. Bakteriol. Orig. 1964, 192, 133–135. [Google Scholar]
- Tsukamura, M.; Mizuno, S.; Tsukamura, S. Classification of rapidly growing mycobacteria. Jpn. J. Microbiol. 1968, 12, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Wallis, R.S.; Johnson, J.L. Chapter 70—Immunotherapy of tuberculosis. In Tuberculosis; Schaaf, H.S., Zumla, A.I., Grange, J.M., Raviglione, M.C., Yew, W.W., Starke, J.R., Pai, M., Donald, P.R., Eds.; W.B. Saunders: Edinburgh, UK, 2009; pp. 718–726. [Google Scholar]
- Bahr, G.M.; Shaaban, M.A.; Gabriel, M.; al-Shimali, B.; Siddiqui, Z.; Chugh, T.D.; Denath, F.M.; Shahin, A.; Behbehani, K.; Chedid, L.; et al. Improved immunotherapy for pulmonary tuberculosis with Mycobacterium vaccae. Tubercle 1990, 71, 259–266. [Google Scholar] [CrossRef]
- Xu, L.J.; Wang, Y.Y.; Zheng, X.D.; Gui, X.D.; Tao, L.F.; Wei, H.M. Immunotherapeutical potential of Mycobacterium vaccae on M. tuberculosis infection in mice. Cell. Mol. Immunol. 2009, 6, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Onyebujoh, P.C.; Abdulmumini, T.; Robinson, S.; Rook, G.A.; Stanford, J.L. Immunotherapy with Mycobacterium vaccae as an addition to chemotherapy for the treatment of pulmonary tuberculosis under difficult conditions in Africa. Respir. Med. 1995, 89, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Corlan, E.; Marica, C.; Macavei, C.; Stanford, J.L.; Stanford, C.A. Immunotherapy with Mycobacterium vaccae in the treatment of tuberculosis in Romania. 2. Chronic or relapsed disease. Respir. Med. 1997, 91, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durban Immunotherapy Trial Group. Immunotherapy with Mycobacterium vaccae in patients with newly diagnosed pulmonary tuberculosis: A randomised controlled trial. Lancet 1999, 354, 116–119. [Google Scholar] [CrossRef]
- Mwinga, A.; Nunn, A.; Ngwira, B.; Chintu, C.; Warndorff, D.; Fine, P.; Darbyshire, J.; Zumla, A. Mycobacterium vaccae (SRL172) immunotherapy as an adjunct to standard antituberculosis treatment in HIV-infected adults with pulmonary tuberculosis: A randomised placebo-controlled trial. Lancet 2002, 360, 1050–1055. [Google Scholar] [CrossRef]
- Huang, C.Y.; Hsieh, W.Y. Efficacy of Mycobacterium vaccae immunotherapy for patients with tuberculosis: A systematic review and meta-analysis. Hum. Vaccines Immunother. 2017, 13, 1960–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, R.D.; Chintu, C.; Lein, A.D.; Zumla, A.; Karagas, M.R.; Baboo, K.S.; Habbema, J.D.; Tosteson, A.N.; Morin, P.; Tvaroha, S.; et al. Safety and immunogenicity of a five-dose series of inactivated Mycobacterium vaccae vaccination for the prevention of HIV-associated tuberculosis. Clin. Infect. Dis. 2000, 30 (Suppl. S3), S309–S315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuola, J.M.; Ristola, M.A.; Cole, B.; Järviluoma, A.; Tvaroha, S.; Rönkkö, T.; Rautio, O.; Arbeit, R.D.; von Reyn, C.F. Immunogenicity of an inactivated mycobacterial vaccine for the prevention of HIV-associated tuberculosis: A randomized, controlled trial. Aids 2003, 17, 2351–2355. [Google Scholar] [CrossRef] [PubMed]
- Von Reyn, C.F.; Mtei, L.; Arbeit, R.D.; Waddell, R.; Cole, B.; Mackenzie, T.; Matee, M.; Bakari, M.; Tvaroha, S.; Adams, L.V.; et al. Prevention of tuberculosis in Bacille Calmette-Guérin-primed, HIV-infected adults boosted with an inactivated whole-cell mycobacterial vaccine. Aids 2010, 24, 675–685. [Google Scholar] [CrossRef]
- De Bruyn, G.; Garner, P. Mycobacterium vaccae immunotherapy for treating tuberculosis. Cochrane Database Syst. Rev. 2003, 2003, Cd001166. [Google Scholar] [CrossRef]
- Lahey, T.; Laddy, D.; Hill, K.; Schaeffer, J.; Hogg, A.; Keeble, J.; Dagg, B.; Ho, M.M.; Arbeit, R.D.; von Reyn, C.F. Immunogenicity and Protective Efficacy of the DAR-901 Booster Vaccine in a Murine Model of Tuberculosis. PLoS ONE 2016, 11, e0168521. [Google Scholar] [CrossRef] [Green Version]
- Guirado, E.; Gil, O.; Cáceres, N.; Singh, M.; Vilaplana, C.; Cardona, P.J. Induction of a specific strong polyantigenic cellular immune response after short-term chemotherapy controls bacillary reactivation in murine and guinea pig experimental models of tuberculosis. Clin. Vaccine Immunol. 2008, 15, 1229–1237. [Google Scholar] [CrossRef] [Green Version]
- Von Reyn, C.F.; Lahey, T.; Arbeit, R.D.; Landry, B.; Kailani, L.; Adams, L.V.; Haynes, B.C.; Mackenzie, T.; Wieland-Alter, W.; Connor, R.I.; et al. Safety and immunogenicity of an inactivated whole cell tuberculosis vaccine booster in adults primed with BCG: A randomized, controlled trial of DAR-901. PLoS ONE 2017, 12, e0175215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbues, A.; Aguilo, J.I.; Gonzalo-Asensio, J.; Marinova, D.; Uranga, S.; Puentes, E.; Fernandez, C.; Parra, A.; Cardona, P.J.; Vilaplana, C.; et al. Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine 2013, 31, 4867–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinova, D.; Gonzalo-Asensio, J.; Aguilo, N.; Martin, C. MTBVAC from discovery to clinical trials in tuberculosis-endemic countries. Expert Rev. Vaccines 2017, 16, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, P.; Wadia, U.; Crawford, N.; Messina, N.L.; Kollmann, T.R.; Lucas, M.; Manning, L.; Richmond, P.; Pittet, L.F.; Curtis, N. Revaccination with Bacille Calmette-Guérin (BCG) is associated with an increased risk of abscess and lymphadenopathy. NPJ Vaccines 2022, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Bannister, S.; Sudbury, E.; Villanueva, P.; Perrett, K.; Curtis, N. The safety of BCG revaccination: A systematic review. Vaccine 2021, 39, 2736–2745. [Google Scholar] [CrossRef]
- Ohara, N.; Yamada, T. Recombinant BCG vaccines. Vaccine 2001, 19, 4089–4098. [Google Scholar] [CrossRef]
- Hess, J.; Miko, D.; Catic, A.; Lehmensiek, V.; Russell, D.G.; Kaufmann, S.H. Mycobacterium bovis Bacille Calmette-Guérin strains secreting listeriolysin of Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 1998, 95, 5299–5304. [Google Scholar] [CrossRef]
- Velmurugan, K.; Grode, L.; Chang, R.; Fitzpatrick, M.; Laddy, D.; Hokey, D.; Derrick, S.; Morris, S.; McCown, D.; Kidd, R.; et al. Nonclinical Development of BCG Replacement Vaccine Candidates. Vaccines 2013, 1, 120–138. [Google Scholar] [CrossRef] [Green Version]
- Farinacci, M.; Weber, S.; Kaufmann, S.H. The recombinant tuberculosis vaccine rBCG ΔureC::hly(+) induces apoptotic vesicles for improved priming of CD4(+) and CD8(+) T cells. Vaccine 2012, 30, 7608–7614. [Google Scholar] [CrossRef]
- Nieuwenhuizen, N.E.; Kulkarni, P.S.; Shaligram, U.; Cotton, M.F.; Rentsch, C.A.; Eisele, B.; Grode, L.; Kaufmann, S.H.E. The Recombinant Bacille Calmette-Guérin Vaccine VPM1002: Ready for Clinical Efficacy Testing. Front. Immunol. 2017, 8, 1147. [Google Scholar] [CrossRef]
- Rentsch, C.A.; Wetterauer, C.; Gsponer, J.R.; Püschel, H.; Bachmann, A.; De Blok, W.; Van Der, W.N.; Behrens, D.; Minhas, A.; Grode, L.; et al. 521 VPM1002—A recombinant BCG with favourable preclinical toxicity and immunogenicity for potential improvement of BCG immunotherapy for non-muscle invasive bladder cancer. Eur. Urol. Suppl. 2014, 13, e521. [Google Scholar] [CrossRef]
- Tkachuk, A.P.; Gushchin, V.A.; Potapov, V.D.; Demidenko, A.V.; Lunin, V.G.; Gintsburg, A.L. Multi-subunit BCG booster vaccine GamTBvac: Assessment of immunogenicity and protective efficacy in murine and guinea pig TB models. PLoS ONE 2017, 12, e0176784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.C.; Wang, C.; Chin, J.; Wong, S.L. A bio-coupling approach using a dextran-binding domain to immobilize an engineered streptavidin to Sephadex for easy preparation of affinity matrix. Sci. Rep. 2019, 9, 3359. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Hu, Q.; He, X.; Cui, X.; Liang, Z.; Wang, L.; Deng, X.; Zhang, Z.; Sheng, W.; Han, X.D. CD16 CAR-T cells enhance antitumor activity of CpG ODN-loaded nanoparticle-adjuvanted tumor antigen-derived vaccinevia ADCC approach. J. Nanobiotechnology 2023, 21, 159. [Google Scholar] [CrossRef]
- Aagaard, C.; Hoang, T.; Dietrich, J.; Cardona, P.J.; Izzo, A.; Dolganov, G.; Schoolnik, G.K.; Cassidy, J.P.; Billeskov, R.; Andersen, P. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat. Med. 2011, 17, 189–194. [Google Scholar] [CrossRef]
- Aboutorabian, S.; Hakimi, J.; Boudet, F.; Montano, S.; Dookie, A.; Roque, C.; Ausar, S.F.; Rahman, N.; Brookes, R.H. A high ratio of IC31(®) adjuvant to antigen is necessary for H4 TB vaccine immunomodulation. Hum. Vaccines Immunother. 2015, 11, 1449–1455. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Su, J.; Ming, M.; Bernardo, L.; Chen, T.; Gisonni-Lex, L.; Gajewska, B. Flow cytometry: An efficient method for antigenicity measurement and particle characterization on an adjuvanted vaccine candidate H4-IC31 for tuberculosis. J. Immunol. Methods 2018, 452, 39–45. [Google Scholar] [CrossRef]
- Bhargava, S.; Choubey, S.; Mishra, S. Vaccines against tuberculosis: A review. Indian J. Tuberc. 2016, 63, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, H.; Mearns, H.; Miles, D.J.; Tameris, M.; Hokey, D.; Shi, Z.; Bennett, S.; Andersen, P.; Kromann, I.; Hoff, S.T.; et al. The tuberculosis vaccine H4:IC31 is safe and induces a persistent polyfunctional CD4 T Cell Response in South African adults: A randomized controlled trial. Vaccine 2015, 33, 3592–3599. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.L.; Reese, V.A.; Huang, P.W.; Beebe, E.A.; Podell, B.K.; Reed, S.G.; Coler, R.N. Protection and Long-Lived Immunity Induced by the ID93/GLA-SE Vaccine Candidate against a Clinical Mycobacterium tuberculosis Isolate. Clin. Vaccine Immunol. 2016, 23, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, S.L.; Bertholet, S.; Reese, V.A.; Ching, L.K.; Reed, S.G.; Coler, R.N. The importance of adjuvant formulation in the development of a tuberculosis vaccine. J. Immunol. 2012, 188, 2189–2197. [Google Scholar] [CrossRef] [Green Version]
- Bertholet, S.; Ireton, G.C.; Ordway, D.J.; Windish, H.P.; Pine, S.O.; Kahn, M.; Phan, T.; Orme, I.M.; Vedvick, T.S.; Baldwin, S.L.; et al. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug-resistant Mycobacterium tuberculosis. Sci. Transl. Med. 2010, 2, 53ra74. [Google Scholar] [CrossRef] [Green Version]
- Penn-Nicholson, A.; Tameris, M.; Smit, E.; Day, T.A.; Musvosvi, M.; Jayashankar, L.; Vergara, J.; Mabwe, S.; Bilek, N.; Geldenhuys, H.; et al. Safety and immunogenicity of the novel tuberculosis vaccine ID93 + GLA-SE in BCG-vaccinated healthy adults in South Africa: A randomised, double-blind, placebo-controlled phase 1 trial. Lancet Respir. Med. 2018, 6, 287–298. [Google Scholar] [CrossRef]
- Lu, J.B.; Chen, B.W.; Wang, G.Z.; Fu, L.L.; Shen, X.B.; Su, C.; Du, W.X.; Yang, L.; Xu, M. Recombinant tuberculosis vaccine AEC/BC02 induces antigen-specific cellular responses in mice and protects guinea pigs in a model of latent infection. J. Microbiol. Immunol. Infect. 2015, 48, 597–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.B.; Cheng, B.W.; Deng, H.Q.; Su, C.; Shen, X.B.; Du, W.X.; Yang, L.; Wang, G.Z.; Xu, M. Analysis of Koch phenomenon of Mycobacterium tuberculosis-infected guinea pigs vaccinated with recombinant tuberculosis vaccine AEC/BC02. Chin. J. Tuberc. Respir. Dis. 2016, 39, 524–528. [Google Scholar] [CrossRef]
- Lu, J.; Guo, X.; Wang, C.; Du, W.; Shen, X.; Su, C.; Wu, Y.; Xu, M. Therapeutic Effect of Subunit Vaccine AEC/BC02 on Mycobacterium tuberculosis Post-Chemotherapy Relapse Using a Latent Infection Murine Model. Vaccines 2022, 10, 825. [Google Scholar] [CrossRef]
- Beverley, P. Selective presentation of MVA85A tuberculosis booster vaccine preclinical animal data. Int. J. Epidemiol. 2016, 45, 581–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashangura, R.; Sena, E.S.; Young, T.; Garner, P. Effects of MVA85A vaccine on tuberculosis challenge in animals: Systematic review. Int. J. Epidemiol. 2015, 44, 1970–1981. [Google Scholar] [CrossRef] [Green Version]
- Tameris, M.; Geldenhuys, H.; Luabeya, A.K.; Smit, E.; Hughes, J.E.; Vermaak, S.; Hanekom, W.A.; Hatherill, M.; Mahomed, H.; McShane, H.; et al. The candidate TB vaccine, MVA85A, induces highly durable Th1 responses. PLoS ONE 2014, 9, e87340. [Google Scholar] [CrossRef] [Green Version]
- Scriba, T.J.; Tameris, M.; Mansoor, N.; Smit, E.; van der Merwe, L.; Mauff, K.; Hughes, E.J.; Moyo, S.; Brittain, N.; Lawrie, A.; et al. Dose-finding study of the novel tuberculosis vaccine, MVA85A, in healthy BCG-vaccinated infants. J. Infect. Dis. 2011, 203, 1832–1843. [Google Scholar] [CrossRef] [Green Version]
- Stylianou, E.; Griffiths, K.L.; Poyntz, H.C.; Harrington-Kandt, R.; Dicks, M.D.; Stockdale, L.; Betts, G.; McShane, H. Improvement of BCG protective efficacy with a novel chimpanzee adenovirus and a modified vaccinia Ankara virus both expressing Ag85A. Vaccine 2015, 33, 6800–6808. [Google Scholar] [CrossRef] [Green Version]
- Pinpathomrat, N.; Bull, N.; Pasricha, J.; Harrington-Kandt, R.; McShane, H.; Stylianou, E. Using an effective TB vaccination regimen to identify immune responses associated with protection in the murine model. Vaccine 2021, 39, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Pandey, R.K.; Prajapati, V.K. Chapter 5—Vaccine delivery systems against tuberculosis. In Nanotechnology Based Approaches for Tuberculosis Treatment; Kesharwani, P., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 75–90. [Google Scholar]
- Stosman, K.; Sivak, K.; Aleksandrov, A.; Buzitskaya, Z.; Rassokha, T.; Stukova, M. Preclinical Safety Evaluation: Acute and Repeated-Dose Toxicity of a New Intranasal Recombinant Vector Vaccine TB/FLU-04L Against Tuberculosis. Drug Res. 2022, 72, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Buzitskaya, Z.; Stosman, K.; Khairullin, B.; Kassenov, M.; Nurpeisova, A.; Abylai Sansyzbay, A.; Shurygina, A.P.; Aleksandrov, A.; Sivak, K.; Stukova, M. A New Intranasal Influenza Vector-Based Vaccine TB/FLU-04L Against Tuberculosis: Preclinical Safety Studies. Drug Res. 2022, 72, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Weiner, J.; von Reyn, C.F. Novel approaches to tuberculosis vaccine development. Int. J. Infect. Dis. 2017, 56, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Thorson, L.; Stokes, R.W.; Santosuosso, M.; Huygen, K.; Zganiacz, A.; Hitt, M.; Xing, Z. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J. Immunol. 2004, 173, 6357–6365. [Google Scholar] [CrossRef] [Green Version]
- Santosuosso, M.; Zhang, X.; McCormick, S.; Wang, J.; Hitt, M.; Xing, Z. Mechanisms of mucosal and parenteral tuberculosis vaccinations: Adenoviral-based mucosal immunization preferentially elicits sustained accumulation of immune protective CD4 and CD8 T cells within the airway lumen. J. Immunol. 2005, 174, 7986–7994. [Google Scholar] [CrossRef] [Green Version]
- Xing, Z.; McFarland, C.T.; Sallenave, J.M.; Izzo, A.; Wang, J.; McMurray, D.N. Intranasal mucosal boosting with an adenovirus-vectored vaccine markedly enhances the protection of BCG-primed guinea pigs against pulmonary tuberculosis. PLoS ONE 2009, 4, e5856. [Google Scholar] [CrossRef]
- Pérez de Val, B.; Villarreal-Ramos, B.; Nofrarías, M.; López-Soria, S.; Romera, N.; Singh, M.; Abad, F.X.; Xing, Z.; Vordermeier, H.M.; Domingo, M. Goats primed with Mycobacterium bovis BCG and boosted with a recombinant adenovirus expressing Ag85A show enhanced protection against tuberculosis. Clin. Vaccine Immunol. 2012, 19, 1339–1347. [Google Scholar] [CrossRef] [Green Version]
- Jeyanathan, M.; Shao, Z.; Yu, X.; Harkness, R.; Jiang, R.; Li, J.; Xing, Z.; Zhu, T. AdHu5Ag85A Respiratory Mucosal Boost Immunization Enhances Protection against Pulmonary Tuberculosis in BCG-Primed Non-Human Primates. PLoS ONE 2015, 10, e0135009. [Google Scholar] [CrossRef]
- Betts, G.; Poyntz, H.; Stylianou, E.; Reyes-Sandoval, A.; Cottingham, M.; Hill, A.; McShane, H. Optimising immunogenicity with viral vectors: Mixing MVA and HAdV-5 expressing the mycobacterial antigen Ag85A in a single injection. PLoS ONE 2012, 7, e50447. [Google Scholar] [CrossRef] [Green Version]
- Vordermeier, H.M.; Huygen, K.; Singh, M.; Hewinson, R.G.; Xing, Z. Immune responses induced in cattle by vaccination with a recombinant adenovirus expressing Mycobacterial antigen 85A and Mycobacterium bovis BCG. Infect. Immun. 2006, 74, 1416–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, G.; Clifford, D.; Gilbert, S.; McShane, H.; Hewinson, R.G.; Vordermeier, H.M.; Villarreal-Ramos, B. Effect of dose and route of immunisation on the immune response induced in cattle by heterologous Bacille Calmette-Guerin priming and recombinant adenoviral vector boosting. Vet. Immunol. Immunopathol. 2014, 158, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Smaill, F.; Jeyanathan, M.; Smieja, M.; Medina, M.F.; Thanthrige-Don, N.; Zganiacz, A.; Yin, C.; Heriazon, A.; Damjanovic, D.; Puri, L.; et al. A human type 5 adenovirus-based tuberculosis vaccine induces robust T Cell Responses in humans despite preexisting anti-adenovirus immunity. Sci. Transl. Med. 2013, 5, 205ra134. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Healey, R.; Adly, F.G. Current trends in separation of plasmid DNA vaccines: A review. Anal. Chim. Acta 2013, 760, 1–15. [Google Scholar] [CrossRef]
- Weng, S.; Zhang, J.; Ma, H.; Zhou, J.; Jia, L.; Wan, Y.; Cui, P.; Ruan, Q.; Shao, L.; Wu, J.; et al. B21 DNA vaccine expressing ag85b, rv2029c, and rv1738 confers a robust therapeutic effect against latent Mycobacterium tuberculosis infection. Front. Immunol. 2022, 13, 1025931. [Google Scholar] [CrossRef]
- Wang, N.; Liang, Y.; Ma, Q.; Mi, J.; Xue, Y.; Yang, Y.; Wang, L.; Wu, X. Mechanisms of ag85a/b DNA vaccine conferred immunotherapy and recovery from Mycobacterium tuberculosis-induced injury. Immun. Inflamm. Dis. 2023, 11, e854. [Google Scholar] [CrossRef]
- Ingolotti, M.; Kawalekar, O.; Shedlock, D.J.; Muthumani, K.; Weiner, D.B. DNA vaccines for targeting bacterial infections. Expert Rev. Vaccines 2010, 9, 747–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutzler, M.A.; Weiner, D.B. DNA vaccines: Ready for prime time? Nat. Rev. Genet. 2008, 9, 776–788. [Google Scholar] [CrossRef]
- Mohan, T.; Verma, P.; Rao, D.N. Novel adjuvants & delivery vehicles for vaccines development: A road ahead. Indian J. Med. Res. 2013, 138, 779–795. [Google Scholar]
- Cai, Y.; Rodriguez, S.; Hebel, H. DNA vaccine manufacture: Scale and quality. Expert Rev. Vaccines 2009, 8, 1277–1291. [Google Scholar] [CrossRef] [PubMed]
- Shafaati, M.; Saidijam, M.; Soleimani, M.; Hazrati, F.; Mirzaei, R.; Amirheidari, B.; Tanzadehpanah, H.; Karampoor, S.; Kazemi, S.; Yavari, B.; et al. A brief review on DNA vaccines in the era of COVID-19. Future Virol. 2022, 17, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, Y.; Xia, L.; Wang, P.; Chen, J.; Xu, M.; Liu, X.; Xia, Y. The deposition of anti-DNA IgG contributes to the development of cutaneous lupus erythematosus. Immunol. Lett. 2017, 191, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, T.; Mayeux, J.M.; Ortega, S.B.; Karandikar, N.J.; Li, Q.Z.; Rakheja, D.; Zhou, X.J.; Satterthwaite, A.B. IL-21 promotes the production of anti-DNA IgG but is dispensable for kidney damage in lyn−/− mice. Eur. J. Immunol. 2013, 43, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Sardesai, N.Y.; Weiner, D.B. Electroporation delivery of DNA vaccines: Prospects for success. Curr. Opin. Immunol. 2011, 23, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Gong, W. Will Mutations in the Spike Protein of SARS-CoV-2 Lead to the Failure of COVID-19 Vaccines? J. Korean Med. Sci. 2021, 36, e124. [Google Scholar] [CrossRef]
- Gong, W.; Aspatwar, A.; Wang, S.; Parkkila, S.; Wu, X. COVID-19 pandemic: SARS-CoV-2 specific vaccines and challenges, protection via BCG trained immunity, and clinical trials. Expert Rev. Vaccines 2021, 20, 857–880. [Google Scholar] [CrossRef]
- Frick, M. Tuberculosis Vaccines Pipeline Report 2022; Treatment Action Group (TAG): New York, NY, USA, 2022; pp. 1–18. [Google Scholar]
- Peng, C.; Tang, F.; Wang, J.; Cheng, P.; Wang, L.; Gong, W. Immunoinformatic-Based Multi-Epitope Vaccine Design for Co-Infection of Mycobacterium tuberculosis and SARS-CoV-2. J. Pers. Med. 2023, 13, 116. [Google Scholar] [CrossRef]
- Jiang, F.; Peng, C.; Cheng, P.; Wang, J.; Lian, J.; Gong, W. PP19128R, a Multiepitope Vaccine Designed to Prevent Latent Tuberculosis Infection, Induced Immune Responses In Silico and In Vitro Assays. Vaccines 2023, 11, 856. [Google Scholar] [CrossRef]
- Jiang, F.; Liu, Y.; Xue, Y.; Cheng, P.; Wang, J.; Lian, J.; Gong, W. Developing a multiepitope vaccine for the prevention of SARS-CoV-2 and monkeypox virus co-infection: A reverse vaccinology analysis. Int. Immunopharmacol. 2023, 115, 109728. [Google Scholar] [CrossRef]
- Cheng, P.; Jiang, F.; Wang, G.; Wang, J.; Xue, Y.; Wang, L.; Gong, W. Bioinformatics analysis and consistency verification of a novel tuberculosis vaccine candidate HP13138PB. Front. Immunol. 2023, 14, 1102578. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Liang, Y.; Mi, J.; Jia, Z.; Xue, Y.; Wang, J.; Wang, L.; Zhou, Y.; Sun, S.; Wu, X. Peptides-Based Vaccine MP3RT Induced Protective Immunity Against Mycobacterium Tuberculosis Infection in a Humanized Mouse Model. Front. Immunol. 2021, 12, 666290. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H. Fact and fiction in tuberculosis vaccine research: 10 years later. Lancet Infect. Dis. 2011, 11, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Weiner, J., 3rd; Kaufmann, S.H. Recent advances towards tuberculosis control: Vaccines and biomarkers. J. Intern. Med. 2014, 275, 467–480. [Google Scholar] [CrossRef]
- Sugarman, J.; Colvin, C.; Moran, A.C.; Oxlade, O. Tuberculosis in pregnancy: An estimate of the global burden of disease. Lancet. Glob. Health 2014, 2, e710–e716. [Google Scholar] [CrossRef] [Green Version]
- Maugans, C.; Loveday, M.; Hlangu, S.; Waitt, C.; Van Schalkwyk, M.; van de Water, B.; Salazar-Austin, N.; McKenna, L.; Mathad, J.S.; Kalk, E.; et al. Best practices for the care of pregnant people living with TB. Int. J. Tuberc. Lung Dis. Off. J. Int. Union Against Tuberc. Lung Dis. 2023, 27, 357–366. [Google Scholar] [CrossRef]
- Mathad, J.S.; Gupta, A. Tuberculosis in pregnant and postpartum women: Epidemiology, management, and research gaps. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2012, 55, 1532–1549. [Google Scholar] [CrossRef]
- Mathad, J.S.; Yadav, S.; Vaidyanathan, A.; Gupta, A.; LaCourse, S.M. Tuberculosis Infection in Pregnant People: Current Practices and Research Priorities. Pathogens 2022, 11, 1481. [Google Scholar] [CrossRef]
- Zhong, G.; Zhuang, C.; Hu, X.; Chen, Q.; Bi, Z.; Jia, X.; Peng, S.; Li, Y.; Huang, Y.; Zhang, Q.; et al. Safety of hepatitis E vaccination for pregnancy: A post-hoc analysis of a randomized, double-blind, controlled phase 3 clinical trial. Emerg. Microbes Infect. 2023, 12, 2185456. [Google Scholar] [CrossRef]
- Amone, A.; Wavamunno, P.; Gabagaya, G.; Rukundo, G.; Namale-Matovu, J.; Malamba, S.S.; Lubega, I.; Homsy, J.; King, R.; Nakabiito, C.; et al. HIV genotypic resistance among pregnant women initiating ART in Uganda: A baseline evaluation of participants in the Option B+ clinical trial. Afr. Health Sci. 2022, 22, 428–434. [Google Scholar] [CrossRef]
- Jones, C.E.; Calvert, A.; Southern, J.; Matheson, M.; Andrews, N.; Khalil, A.; Cuthbertson, H.; Hallis, B.; England, A.; Heath, P.T.; et al. A phase IV, multi-centre, randomized clinical trial comparing two pertussis-containing vaccines in pregnant women in England and vaccine responses in their infants. BMC Med. 2021, 19, 138. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, J.; Liang, Q.; Pan, L.; Duan, H.; Yang, Y.; Li, H.; Guo, C.; Sun, Q.; Jia, H.; et al. Use of T-SPOT.TB for the diagnosis of unconventional pleural tuberculosis is superior to ADA in high prevalence areas: A prospective analysis of 601 cases. BMC Infect. Dis. 2021, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.E.; Ortiz-Brizuela, E.; Bastos, M.L.; Menzies, D. Comparing the Diagnostic Performance of QuantiFERON-TB Gold Plus to Other Tests of Latent Tuberculosis Infection: A Systematic Review and Meta-analysis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2021, 73, e1116–e1125. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef]
- Schijns, V.E.; Lavelle, E.C. Trends in vaccine adjuvants. Expert Rev. Vaccines 2011, 10, 539–550. [Google Scholar] [CrossRef]
- Shon, N.N.; Yarbrough, T.; Shah, A.D. Aluminum Hydroxide. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Kooijman, S.; Vrieling, H.; Verhagen, L.; de Ridder, J.; de Haan, A.; van Riet, E.; Heck, A.J.R.; Kersten, G.F.A.; Pennings, J.L.A.; Metz, B.; et al. Aluminum Hydroxide And Aluminum Phosphate Adjuvants Elicit A Different Innate Immune Response. J. Pharm. Sci. 2022, 111, 982–990. [Google Scholar] [CrossRef]
- Sato, S.; Oga, J.; Shirahata, A.; Ishida, Y. Clinical impact of a new method using a clear proctoscope to evaluate the therapeutic effect of sclerotherapy with aluminum potassium sulfate and tannic acid (ALTA) for internal hemorrhoids: A prospective cohort study. Quant. Imaging Med. Surg. 2023, 13, 441–448. [Google Scholar] [CrossRef]
- Sasaki, E.; Furuhata, K.; Mizukami, T.; Hamaguchi, I. An investigation and assessment of the muscle damage and inflammation at injection site of aluminum-adjuvanted vaccines in guinea pigs. J. Toxicol. Sci. 2022, 47, 439–451. [Google Scholar] [CrossRef]
- Baylor, N.W.; Egan, W.; Richman, P. Aluminum salts in vaccines--US perspective. Vaccine 2002, 20 (Suppl. S3), S18–S23. [Google Scholar] [CrossRef]
- Masson, J.D.; Thibaudon, M.; Bélec, L.; Crépeaux, G. Calcium phosphate: A substitute for aluminum adjuvants? Expert Rev. Vaccines 2017, 16, 289–299. [Google Scholar] [CrossRef]
- Jefferson, T.; Rudin, M.; Di Pietrantonj, C. Adverse events after immunisation with aluminium-containing DTP vaccines: Systematic review of the evidence. Lancet Infect. Dis. 2004, 4, 84–90. [Google Scholar] [CrossRef]
- Brewer, J.M.; Pollock, K.G.; Tetley, L.; Russell, D.G. Vesicle size influences the trafficking, processing, and presentation of antigens in lipid vesicles. J. Immunol. 2004, 173, 6143–6150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agger, E.M. Novel adjuvant formulations for delivery of anti-tuberculosis vaccine candidates. Adv. Drug Deliv. Rev. 2016, 102, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Chen, M.; Wang, T. Liposomes used as a vaccine adjuvant-delivery system: From basics to clinical immunization. J. Control. Release 2019, 303, 130–150. [Google Scholar] [CrossRef]
- Rahnfeld, L.; Thamm, J.; Steiniger, F.; van Hoogevest, P.; Luciani, P. Study on the in situ aggregation of liposomes with negatively charged phospholipids for use as injectable depot formulation. Colloids Surf. B Biointerfaces 2018, 168, 10–17. [Google Scholar] [CrossRef]
- Fox, C.B. Squalene emulsions for parenteral vaccine and drug delivery. Molecules 2009, 14, 3286–3312. [Google Scholar] [CrossRef] [Green Version]
- Coccia, M.; Collignon, C.; Hervé, C.; Chalon, A.; Welsby, I.; Detienne, S.; van Helden, M.J.; Dutta, S.; Genito, C.J.; Waters, N.C.; et al. Cellular and molecular synergy in AS01-adjuvanted vaccines results in an early IFNγ response promoting vaccine immunogenicity. NPJ Vaccines 2017, 2, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoya, J.; Solon, J.A.; Cunanan, S.R.; Acosta, L.; Bollaerts, A.; Moris, P.; Janssens, M.; Jongert, E.; Demoitié, M.A.; Mettens, P.; et al. A randomized, controlled dose-finding Phase II study of the M72/AS01 candidate tuberculosis vaccine in healthy PPD-positive adults. J. Clin. Immunol. 2013, 33, 1360–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumarasamy, N.; Poongulali, S.; Bollaerts, A.; Moris, P.; Beulah, F.E.; Ayuk, L.N.; Demoitié, M.A.; Jongert, E.; Ofori-Anyinam, O. A Randomized, Controlled Safety, and Immunogenicity Trial of the M72/AS01 Candidate Tuberculosis Vaccine in HIV-Positive Indian Adults. Medicine 2016, 95, e2459. [Google Scholar] [CrossRef]
- Orr, M.T.; Duthie, M.S.; Windish, H.P.; Lucas, E.A.; Guderian, J.A.; Hudson, T.E.; Shaverdian, N.; O’Donnell, J.; Desbien, A.L.; Reed, S.G.; et al. MyD88 and TRIF synergistic interaction is required for TH1-cell polarization with a synthetic TLR4 agonist adjuvant. Eur. J. Immunol. 2013, 43, 2398–2408. [Google Scholar] [CrossRef] [Green Version]
- Desbien, A.L.; Reed, S.J.; Bailor, H.R.; Dubois Cauwelaert, N.; Laurance, J.D.; Orr, M.T.; Fox, C.B.; Carter, D.; Reed, S.G.; Duthie, M.S. Squalene emulsion potentiates the adjuvant activity of the TLR4 agonist, GLA, via inflammatory caspases, IL-18, and IFN-γ. Eur. J. Immunol. 2015, 45, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Aichinger, M.C.; Ginzler, M.; Weghuber, J.; Zimmermann, L.; Riedl, K.; Schütz, G.; Nagy, E.; von Gabain, A.; Schweyen, R.; Henics, T. Adjuvating the adjuvant: Facilitated delivery of an immunomodulatory oligonucleotide to TLR9 by a cationic antimicrobial peptide in dendritic cells. Vaccine 2011, 29, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Schellack, C.; Prinz, K.; Egyed, A.; Fritz, J.H.; Wittmann, B.; Ginzler, M.; Swatosch, G.; Zauner, W.; Kast, C.; Akira, S.; et al. IC31, a novel adjuvant signaling via TLR9, induces potent cellular and humoral immune responses. Vaccine 2006, 24, 5461–5472. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Qian, R.; Liu, T.; Wu, T.; Wang, T. Nanoparticulate Carriers Used as Vaccine Adjuvant Delivery Systems. Crit. Rev. Ther. Drug Carr. Syst. 2019, 36, 449–484. [Google Scholar] [CrossRef]
- Francica, J.R.; Sheng, Z.; Zhang, Z.; Nishimura, Y.; Shingai, M.; Ramesh, A.; Keele, B.F.; Schmidt, S.D.; Flynn, B.J.; Darko, S.; et al. Analysis of immunoglobulin transcripts and hypermutation following SHIV(AD8) infection and protein-plus-adjuvant immunization. Nat. Commun. 2015, 6, 6565. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Zhu, H.; Xia, X.; Liang, Z.; Ma, X.; Sun, B. Vaccine adjuvants: Understanding the structure and mechanism of adjuvanticity. Vaccine 2019, 37, 3167–3178. [Google Scholar] [CrossRef]
- Orme, I.M. Preclinical testing of new vaccines for tuberculosis: A comprehensive review. Vaccine 2006, 24, 2–19. [Google Scholar] [CrossRef]
- Williams, A.; Hatch, G.J.; Clark, S.O.; Gooch, K.E.; Hatch, K.A.; Hall, G.A.; Huygen, K.; Ottenhoff, T.H.; Franken, K.L.; Andersen, P.; et al. Evaluation of vaccines in the EU TB Vaccine Cluster using a guinea pig aerosol infection model of tuberculosis. Tuberculosis 2005, 85, 29–38. [Google Scholar] [CrossRef]
- Baldwin, S.L.; D’Souza, C.; Roberts, A.D.; Kelly, B.P.; Frank, A.A.; Lui, M.A.; Ulmer, J.B.; Huygen, K.; McMurray, D.M.; Orme, I.M. Evaluation of new vaccines in the mouse and guinea pig model of tuberculosis. Infect. Immun. 1998, 66, 2951–2959. [Google Scholar] [CrossRef]
- Orme, I.M.; McMurray, D.N.; Belisle, J.T. Tuberculosis vaccine development: Recent progress. Trends Microbiol. 2001, 9, 115–118. [Google Scholar] [CrossRef]
- Gong, W.; Liang, Y.; Wu, X. Animal Models of Tuberculosis Vaccine Research: An Important Component in the Fight against Tuberculosis. BioMed Res. Int. 2020, 2020, 4263079. [Google Scholar] [CrossRef]
- McMurray, D.N.; Collins, F.M.; Dannenberg, A.M., Jr.; Smith, D.W. Pathogenesis of experimental tuberculosis in animal models. Curr. Top. Microbiol. Immunol. 1996, 215, 157–179. [Google Scholar] [CrossRef]
- Marinova, D.; Gonzalo-Asensio, J.; Aguilo, N.; Martin, C. Recent developments in tuberculosis vaccines. Expert Rev. Vaccines 2013, 12, 1431–1448. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, G.S.; Smith, E.E.; Shanley, C.A.; Ordway, D.J.; Orme, I.M.; Basaraba, R.J. Disseminated disease severity as a measure of virulence of Mycobacterium tuberculosis in the guinea pig model. Tuberculosis 2008, 88, 295–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basaraba, R.J. Experimental tuberculosis: The role of comparative pathology in the discovery of improved tuberculosis treatment strategies. Tuberculosis 2008, 88 (Suppl. S1), S35–S47. [Google Scholar] [CrossRef]
- Dharmadhikari, A.S.; Nardell, E.A. What animal models teach humans about tuberculosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 503–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenberg, A.M., Jr. Pathogenesis of pulmonary Mycobacterium bovis infection: Basic principles established by the rabbit model. Tuberculosis 2001, 81, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, A.M., Jr. Liquefaction and cavity formation in pulmonary TB: A simple method in rabbit skin to test inhibitors. Tuberculosis 2009, 89, 243–247. [Google Scholar] [CrossRef]
- Sun, H.; Ma, X.; Zhang, G.; Luo, Y.; Tang, K.; Lin, X.; Yu, H.; Zhang, Y.; Zhu, B. Effects of immunomodulators on liquefaction and ulceration in the rabbit skin model of tuberculosis. Tuberculosis 2012, 92, 345–350. [Google Scholar] [CrossRef]
- Bishai, W.R.; Dannenberg, A.M., Jr.; Parrish, N.; Ruiz, R.; Chen, P.; Zook, B.C.; Johnson, W.; Boles, J.W.; Pitt, M.L. Virulence of Mycobacterium tuberculosis CDC1551 and H37Rv in rabbits evaluated by Lurie’s pulmonary tubercle count method. Infect. Immun. 1999, 67, 4931–4934. [Google Scholar] [CrossRef]
- Tsenova, L.; Sokol, K.; Freedman, V.H.; Kaplan, G. A combination of thalidomide plus antibiotics protects rabbits from mycobacterial meningitis-associated death. J. Infect. Dis. 1998, 177, 1563–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorman, S.E.; Hatem, C.L.; Tyagi, S.; Aird, K.; Lopez-Molina, J.; Pitt, M.L.; Zook, B.C.; Dannenberg, A.M., Jr.; Bishai, W.R.; Manabe, Y.C. Susceptibility to tuberculosis: Clues from studies with inbred and outbred New Zealand White rabbits. Infect. Immun. 2004, 72, 1700–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, S.L.; Dascher, C.C.; Sambandamurthy, V.K.; Russell, R.G.; Jacobs, W.R., Jr.; Bloom, B.R.; Hondalus, M.K. Protection elicited by a double leucine and pantothenate auxotroph of Mycobacterium tuberculosis in guinea pigs. Infect. Immun. 2004, 72, 3031–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardona, P.J.; Williams, A. Experimental animal modelling for TB vaccine development. Int. J. Infect. Dis. 2017, 56, 268–273. [Google Scholar] [CrossRef]
- Flynn, J.L.; Capuano, S.V.; Croix, D.; Pawar, S.; Myers, A.; Zinovik, A.; Klein, E. Non-human primates: A model for tuberculosis research. Tuberculosis 2003, 83, 116–118. [Google Scholar] [CrossRef]
- Gupta, U.D.; Katoch, V.M. Animal models of tuberculosis for vaccine development. Indian J. Med. Res. 2009, 129, 11–18. [Google Scholar]
- Myllymäki, H.; Niskanen, M.; Oksanen, K.E.; Rämet, M. Animal models in tuberculosis research—Where is the beef? Expert Opin. Drug Discov. 2015, 10, 871–883. [Google Scholar] [CrossRef]
- Dannenberg, A.M., Jr. Perspectives on clinical and preclinical testing of new tuberculosis vaccines. Clin. Microbiol. Rev. 2010, 23, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Gupta, U.D. Animal models of tuberculosis: Lesson learnt. Indian J. Med. Res. 2018, 147, 456–463. [Google Scholar] [CrossRef]
- Schmidt, B.; Hildebrandt, A. Deep learning in next-generation sequencing. Drug Discov. Today 2021, 26, 173–180. [Google Scholar] [CrossRef]
- Alharbi, W.S.; Rashid, M. A review of deep learning applications in human genomics using next-generation sequencing data. Hum. Genom. 2022, 16, 26. [Google Scholar] [CrossRef] [PubMed]
- Hederman, A.P.; Ackerman, M.E. Leveraging deep learning to improve vaccine design. Trends Immunol. 2023, 44, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Tsai, C.C.; Kuo, L.C.; Kuo, P.C.; Lee, M.R.; Wang, J.Y.; Ko, J.C.; Shih, J.Y.; Wang, H.C.; Yu, C.J. A deep learning model using chest X-ray for identifying TB and NTM-LD patients: A cross-sectional study. Insights Imaging 2023, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Wu, L.S.; Huang, G.M.; Huang, K.Y.; Lee, T.Y.; Weng, J.T. Gene expression profiling identifies candidate biomarkers for active and latent tuberculosis. BMC Bioinform. 2016, 17 (Suppl. S1), 3. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Wu, J.; Wang, H.; Wang, S.; Diao, N.; Wang, F.; Gao, Y.; Chen, J.; Shao, L.; Weng, X.; et al. Novel biomarkers distinguishing active tuberculosis from latent infection identified by gene expression profile of peripheral blood mononuclear cells. PLoS ONE 2011, 6, e24290. [Google Scholar] [CrossRef]
- Wang, S.; He, L.; Wu, J.; Zhou, Z.; Gao, Y.; Chen, J.; Shao, L.; Zhang, Y.; Zhang, W. Transcriptional Profiling of Human Peripheral Blood Mononuclear Cells Identifies Diagnostic Biomarkers That Distinguish Active and Latent Tuberculosis. Front. Immunol. 2019, 10, 2948. [Google Scholar] [CrossRef] [Green Version]
- Maertzdorf, J.; McEwen, G.; Weiner, J., 3rd; Tian, S.; Lader, E.; Schriek, U.; Mayanja-Kizza, H.; Ota, M.; Kenneth, J.; Kaufmann, S.H. Concise gene signature for point-of-care classification of tuberculosis. EMBO Mol. Med. 2016, 8, 86–95. [Google Scholar] [CrossRef]
- Cao, S.H.; Chen, Y.Q.; Sun, Y.; Liu, Y.; Zheng, S.H.; Zhang, Z.G.; Li, C.Y. Screening of Serum Biomarkers for Distinguishing between Latent and Active Tuberculosis Using Proteome Microarray. Biomed. Environ. Sci. BES 2018, 31, 515–526. [Google Scholar] [CrossRef]
- Li, Z.; Hu, J.; Liu, P.; Cui, D.; Di, H.; Wu, S. Microarray-based selection of a serum biomarker panel that can discriminate between latent and active pulmonary TB. Sci. Rep. 2021, 11, 14516. [Google Scholar] [CrossRef]
- Peng, Z.; Chen, L.; Zhang, H. Serum proteomic analysis of Mycobacterium tuberculosis antigens for discriminating active tuberculosis from latent infection. J. Int. Med. Res. 2020, 48, 300060520910042. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Bileschi, M.L.; Belanger, D.; Bryant, D.H.; Sanderson, T.; Carter, B.; Sculley, D.; Bateman, A.; DePristo, M.A.; Colwell, L.J. Using deep learning to annotate the protein universe. Nat. Biotechnol. 2022, 40, 932–937. [Google Scholar] [CrossRef]
- AlQuraishi, M. End-to-End Differentiable Learning of Protein Structure. Cell Syst. 2019, 8, 292–301.e293. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Sidney, J.; Dow, C.; Mothé, B.; Sette, A.; Peters, B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 2008, 4, e1000048. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Nielsen, M.; Lund, O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform. 2009, 10, 296. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Sturniolo, T.; Bono, E.; Ding, J.; Raddrizzani, L.; Tuereci, O.; Sahin, U.; Braxenthaler, M.; Gallazzi, F.; Protti, M.P.; Sinigaglia, F.; et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat. Biotechnol. 1999, 17, 555–561. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Peters, B.; Sette, A.; Justesen, S.; Buus, S.; Lund, O. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput. Biol. 2008, 4, e1000107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemøller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. A Publ. Protein Soc. 2003, 12, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Sette, A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 2005, 6, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidney, J.; Assarsson, E.; Moore, C.; Ngo, S.; Pinilla, C.; Sette, A.; Peters, B. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 2008, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.C.; Sidney, J.; Bui, H.H.; Grey, H.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-Cell Responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. [Google Scholar] [CrossRef]
- Hoof, I.; Peters, B.; Sidney, J.; Pedersen, L.E.; Sette, A.; Lund, O.; Buus, S.; Nielsen, M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009, 61, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pham, M.N.; Nguyen, T.N.; Tran, L.S.; Nguyen, Q.B.; Nguyen, T.H.; Pham, T.M.Q.; Nguyen, H.N.; Giang, H.; Phan, M.D.; Nguyen, V. epiTCR: A highly sensitive predictor for TCR-peptide binding. Bioinformatics 2023, 39, btad284. [Google Scholar] [CrossRef]
- Xu, Y.; Qian, X.; Tong, Y.; Li, F.; Wang, K.; Zhang, X.; Liu, T.; Wang, J. AttnTAP: A Dual-input Framework Incorporating the Attention Mechanism for Accurately Predicting TCR-peptide Binding. Front. Genet. 2022, 13, 942491. [Google Scholar] [CrossRef]
- Karosiene, E.; Lundegaard, C.; Lund, O.; Nielsen, M. NetMHCcons: A consensus method for the major histocompatibility complex class I predictions. Immunogenetics 2012, 64, 177–186. [Google Scholar] [CrossRef]
- Zhang, H.; Lund, O.; Nielsen, M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: Application to MHC-peptide binding. Bioinformatics 2009, 25, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.; Fenoy, E.; Harndahl, M.; Kristensen, A.B.; Nielsen, I.K.; Nielsen, M.; Buus, S. Pan-Specific Prediction of Peptide-MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. J. Immunol. 2016, 197, 1517–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, J.B.; Kaabinejadian, S.; Yari, H.; Peters, B.; Barra, C.; Gragert, L.; Hildebrand, W.; Nielsen, M. Machine learning reveals limited contribution of trans-only encoded variants to the HLA-DQ immunopeptidome. Commun. Biol. 2023, 6, 442. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, P.; Arun, A.; Meena, L.S. Development of peptide vaccine candidate using highly antigenic PE-PGRS family proteins to stimulate the host immune response against Mycobacterium tuberculosis H(37)Rv: An immuno-informatics approach. J. Biomol. Struct. Dyn. 2023, 41, 3382–3404. [Google Scholar] [CrossRef]
- Andongma, B.T.; Huang, Y.; Chen, F.; Tang, Q.; Yang, M.; Chou, S.H.; Li, X.; He, J. In silico design of a promiscuous chimeric multi-epitope vaccine against Mycobacterium tuberculosis. Comput. Struct. Biotechnol. J. 2023, 21, 991–1004. [Google Scholar] [CrossRef]
- Pitaloka, D.A.E.; Izzati, A.; Amirah, S.R.; Syakuran, L.A. Multi Epitope-Based Vaccine Design for Protection Against Mycobacterium tuberculosis and SARS-CoV-2 Coinfection. Adv. Appl. Bioinform. Chem. AABC 2022, 15, 43–57. [Google Scholar] [CrossRef]
- Moodley, A.; Fatoba, A.; Okpeku, M.; Emmanuel Chiliza, T.; Blessing Cedric Simelane, M.; Pooe, O.J. Reverse vaccinology approach to design a multi-epitope vaccine construct based on the Mycobacterium tuberculosis biomarker PE_PGRS17. Immunol. Res. 2022, 70, 501–517. [Google Scholar] [CrossRef]
- Ghandadi, M. An Immunoinformatic Strategy to Develop New Mycobacterium tuberculosis Multi-epitope Vaccine. Int. J. Pept. Res. Ther. 2022, 28, 99. [Google Scholar] [CrossRef]
- Wang, J.; Mi, J.; Liang, Y.; Wu, X.Q.; Zhang, J.X.; Liu, Y.P.; Wang, L.; Xue, Y.; Shi, Y.C.; Gong, W.P. Transcriptomic analysis of tuberculosis peptide-based vaccine MP3RT in humanized mice. Chin. J. Tuberc. Respir. Dis. 2022, 45, 894–903. [Google Scholar] [CrossRef]
- Cheng, P.; Xue, Y.; Wang, J.; Jia, Z.; Wang, L.; Gong, W. Evaluation of the consistence between the results of immunoinformatics predictions and real-world animal experiments of a new tuberculosis vaccine MP3RT. Front. Cell. Infect. Microbiol. 2022, 12, 1047306. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Liang, Y.; Mi, J.; Xue, Y.; Wang, J.; Wang, L.; Zhou, Y.; Sun, S.; Wu, X. A peptide-based vaccine ACP derived from antigens of Mycobacterium tuberculosis induced Th1 response but failed to enhance the protective efficacy of BCG in mice. Indian J. Tuberc. 2022, 69, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Iyer, B.; Prasath, V.B.S.; Ni, Y.; Salomonis, N. DeepImmuno: Deep learning-empowered prediction and generation of immunogenic peptides for T-cell immunity. Brief. Bioinform. 2021, 22, bbab160. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, L.; Wei, F.; Li, J. DeepNetBim: Deep learning model for predicting HLA-epitope interactions based on network analysis by harnessing binding and immunogenicity information. BMC Bioinform. 2021, 22, 231. [Google Scholar] [CrossRef]
- Sidhom, J.W.; Oliveira, G.; Ross-MacDonald, P.; Wind-Rotolo, M.; Wu, C.J.; Pardoll, D.M.; Baras, A.S. Deep learning reveals predictive sequence concepts within immune repertoires to immunotherapy. Sci. Adv. 2022, 8, eabq5089. [Google Scholar] [CrossRef]
- Pollard, C.; De Koker, S.; Saelens, X.; Vanham, G.; Grooten, J. Challenges and advances towards the rational design of mRNA vaccines. Trends Mol. Med. 2013, 19, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Linares-Fernández, S.; Lacroix, C.; Exposito, J.Y.; Verrier, B. Tailoring mRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol. Med. 2020, 26, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20. [Google Scholar] [CrossRef]
- Naik, R.; Peden, K. Regulatory Considerations on the Development of mRNA Vaccines. Curr. Top. Microbiol. Immunol. 2022, 440, 187–205. [Google Scholar] [CrossRef]
- Rice, A.M.; Castillo Morales, A.; Ho, A.T.; Mordstein, C.; Mühlhausen, S.; Watson, S.; Cano, L.; Young, B.; Kudla, G.; Hurst, L.D. Evidence for Strong Mutation Bias toward, and Selection against, U Content in SARS-CoV-2: Implications for Vaccine Design. Mol. Biol. Evol. 2021, 38, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.A.; Kommareddy, S.; Maione, D.; Uematsu, Y.; Giovani, C.; Berlanda Scorza, F.; Otten, G.R.; Yu, D.; Mandl, C.W.; Mason, P.W.; et al. Self-amplifying mRNA vaccines. Adv. Genet. 2015, 89, 179–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Z.; Luo, J.; Han, X.; Wei, Y.; Wei, X. mRNA vaccine: A potential therapeutic strategy. Mol. Cancer 2021, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020, 383, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Zhang, X.; Drelich, A.; Shi, J.; Hsu, J.C.; Luchsinger, L.; Hillyer, C.D.; Tseng, C.K.; Jiang, S.; Du, L. A novel receptor-binding domain (RBD)-based mRNA vaccine against SARS-CoV-2. Cell Res. 2020, 30, 932–935. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.D.; Shukla, S.; Chung, Y.H.; Beiss, V.; Chan, S.K.; Ortega-Rivera, O.A.; Wirth, D.M.; Chen, A.; Sack, M.; Pokorski, J.K.; et al. COVID-19 vaccine development and a potential nanomaterial path forward. Nat. Nanotechnol. 2020, 15, 646–655. [Google Scholar] [CrossRef]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. NPJ Vaccines 2020, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.; Stavropoulos, E.; Yang, M.; Ragno, S.; Vordermeier, M.; Chambers, M.; Hewinson, G.; Lowrie, D.B.; Colston, M.J.; Tascon, R.E. RNA encoding the MPT83 antigen induces protective immune responses against Mycobacterium tuberculosis infection. Infect. Immun. 2004, 72, 6324–6329. [Google Scholar] [CrossRef] [Green Version]
- Ramshaw, I.A.; Ramsay, A.J. The prime-boost strategy: Exciting prospects for improved vaccination. Immunol. Today 2000, 21, 163–165. [Google Scholar] [CrossRef]
- Skinner, M.A.; Buddle, B.M.; Wedlock, D.N.; Keen, D.; de Lisle, G.W.; Tascon, R.E.; Ferraz, J.C.; Lowrie, D.B.; Cockle, P.J.; Vordermeier, H.M.; et al. A DNA prime-Mycobacterium bovis BCG boost vaccination strategy for cattle induces protection against bovine tuberculosis. Infect. Immun. 2003, 71, 4901–4907. [Google Scholar] [CrossRef] [Green Version]
- Shahrear, S.; Islam, A. Modeling of MT. P495, an mRNA-based vaccine against the phosphate-binding protein PstS1 of Mycobacterium tuberculosis. Mol. Divers. 2022, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: Preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnology 2021, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Kheirvari, M.; Liu, H.; Tumban, E. Virus-like Particle Vaccines and Platforms for Vaccine Development. Viruses 2023, 15, 1109. [Google Scholar] [CrossRef] [PubMed]
- Tariq, H.; Batool, S.; Asif, S.; Ali, M.; Abbasi, B.H. Virus-Like Particles: Revolutionary Platforms for Developing Vaccines Against Emerging Infectious Diseases. Front. Microbiol. 2021, 12, 790121. [Google Scholar] [CrossRef]
- Lee, G.H.; Lim, S.G. CpG-Adjuvanted Hepatitis B Vaccine (HEPLISAV-B®) Update. Expert Rev. Vaccines 2021, 20, 487–495. [Google Scholar] [CrossRef]
- van den Ende, C.; Marano, C.; van Ahee, A.; Bunge, E.M.; De Moerlooze, L. The immunogenicity of GSK’s recombinant hepatitis B vaccine in children: A systematic review of 30 years of experience. Expert Rev. Vaccines 2017, 16, 789–809. [Google Scholar] [CrossRef] [Green Version]
- Tele, S.A.; Martins, R.M.; Lopes, C.L.; dos Santos Carneiro, M.A.; Souza, K.P.; Yoshida, C.F. Immunogenicity of a recombinant hepatitis B vaccine (Euvax-B) in haemodialysis patients and staff. Eur. J. Epidemiol. 2001, 17, 145–149. [Google Scholar] [CrossRef]
- Hieu, N.T.; Kim, K.H.; Janowicz, Z.; Timmermans, I. Comparative efficacy, safety and immunogenicity of Hepavax-Gene and Engerix-B, recombinant hepatitis B vaccines, in infants born to HBsAg and HBeAg positive mothers in Vietnam: An assessment at 2 years. Vaccine 2002, 20, 1803–1808. [Google Scholar] [CrossRef]
- Assateerawatt, A.; Tanphaichitr, V.S.; Suvatte, V.; Yodthong, S. Immunogenicity and efficacy of a recombinant DNA hepatitis B vaccine, GenHevac B Pasteur in high risk neonates, school children and healthy adults. Asian Pac. J. Allergy Immunol. 1993, 11, 85–91. [Google Scholar]
- Mphahlele, M.J.; Burnett, R.J.; Kyaw, T.; Schoeman, H.S.; Aspinall, S. Immunogenicity and safety of yeast-derived recombinant hepatitis B vaccine (Heberbiovac HB) in South African children. South Afr. Med. J. 2004, 94, 280–281. [Google Scholar]
- Wang, J.; Xie, T.; Ullah, I.; Mi, Y.; Li, X.; Gong, Y.; He, P.; Liu, Y.; Li, F.; Li, J.; et al. A VLP-Based Vaccine Displaying HBHA and MTP Antigens of Mycobacterium tuberculosis Induces Protective Immune Responses in M. tuberculosis H37Ra Infected Mice. Vaccines 2023, 11, 941. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, H.; Wu, S.; Dong, D.; Zhang, J.; Fu, L.; Xu, J.; Chen, W. Hepatitis B virus core particles displaying Mycobacterium tuberculosis antigen ESAT-6 enhance ESAT-6-specific immune responses. Vaccine 2011, 29, 5645–5651. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Schinko, T.; Messner, P.; Palmberger, D.; Ferko, B.; Grabherr, R. Influenza virus-like particles as an antigen-carrier platform for the ESAT-6 epitope of Mycobacterium tuberculosis. J. Virol. Methods 2010, 167, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Dhanasooraj, D.; Kumar, R.A.; Mundayoor, S. Subunit Protein Vaccine Delivery System for Tuberculosis Based on Hepatitis B Virus Core VLP (HBc-VLP) Particles. Methods Mol. Biol. 2016, 1404, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Dhanasooraj, D.; Kumar, R.A.; Mundayoor, S. Vaccine delivery system for tuberculosis based on nano-sized hepatitis B virus core protein particles. Int. J. Nanomed. 2013, 8, 835–843. [Google Scholar] [CrossRef] [Green Version]






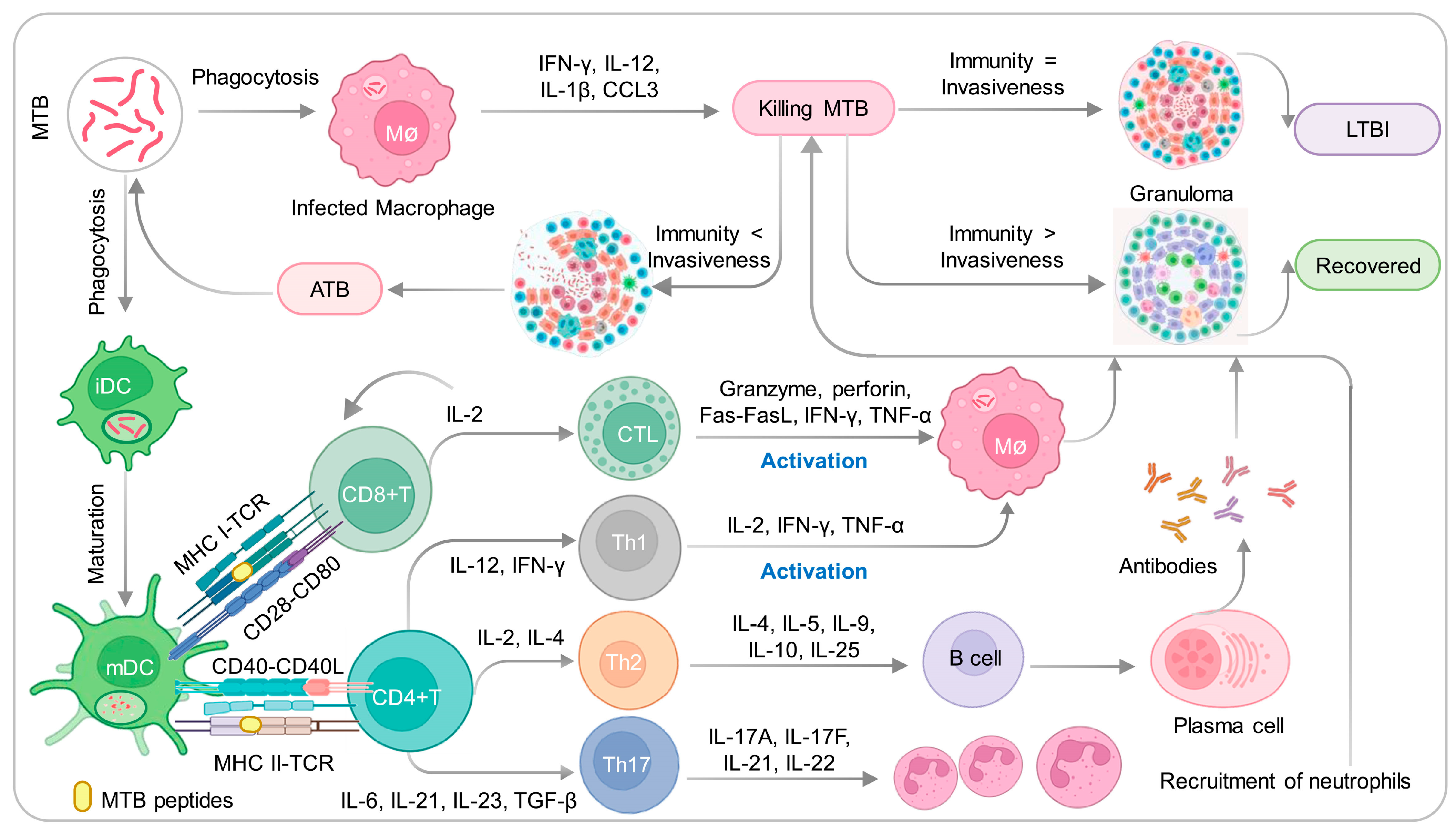{kind=link}
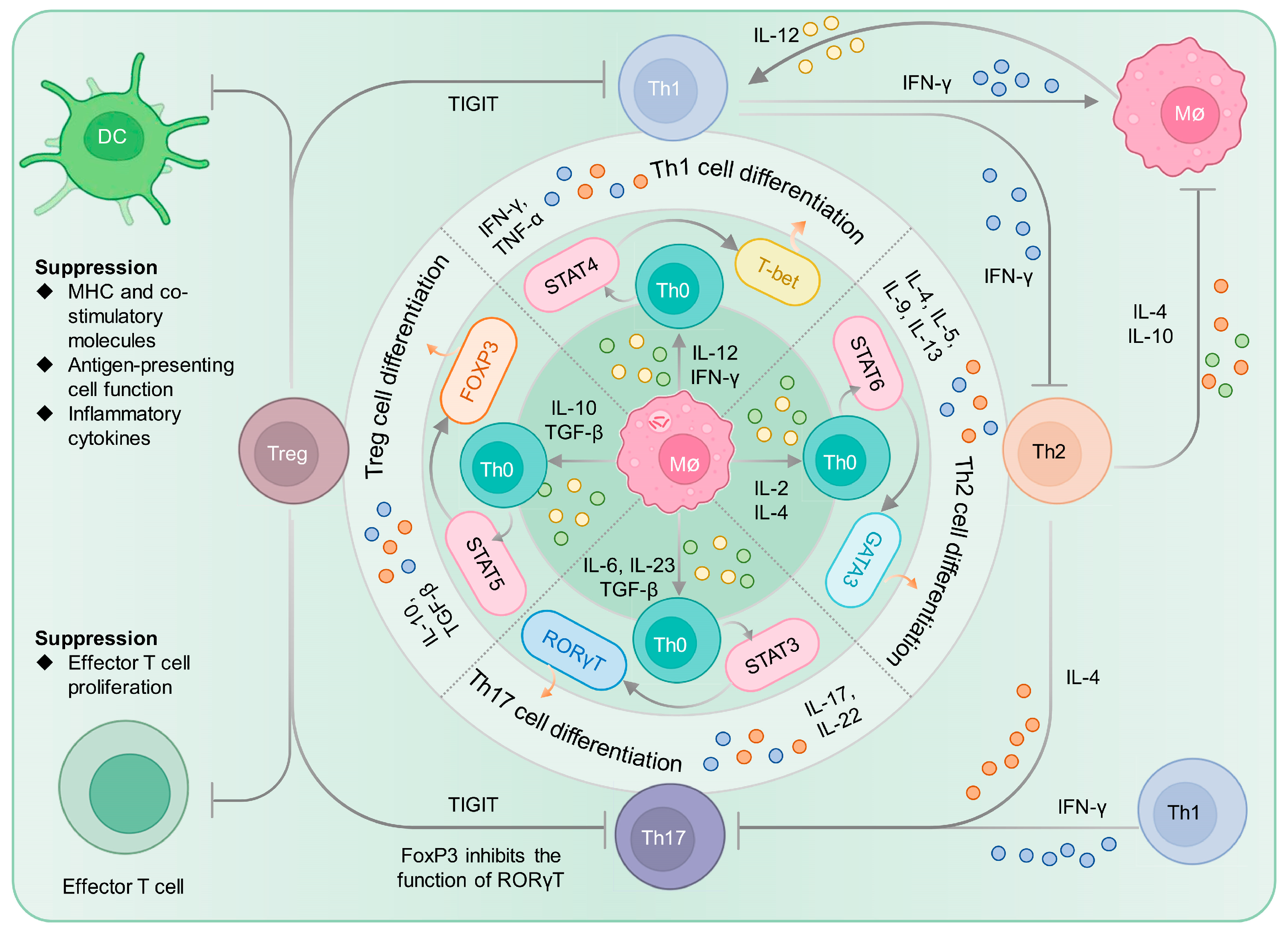{kind=link}
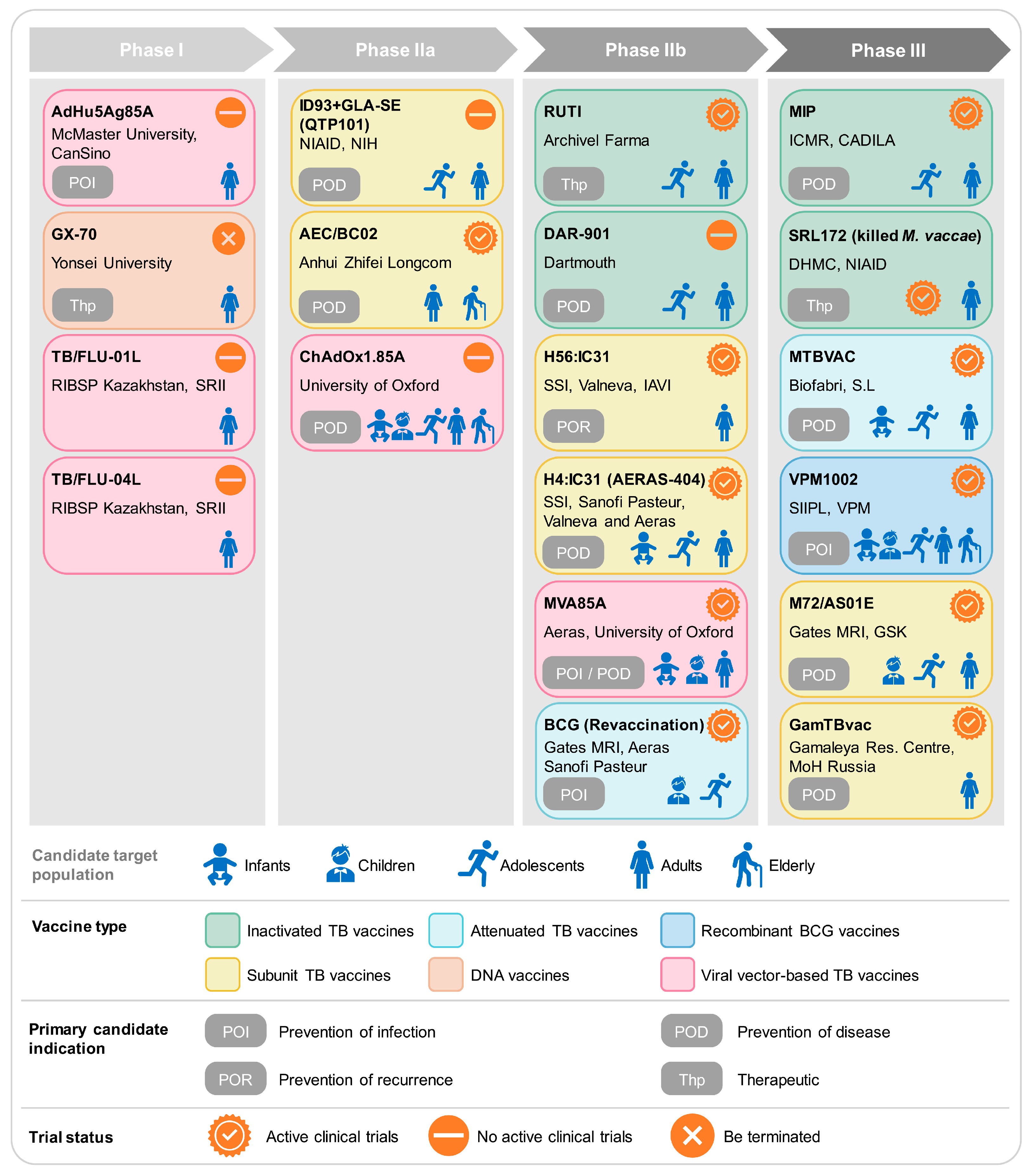{kind=link}
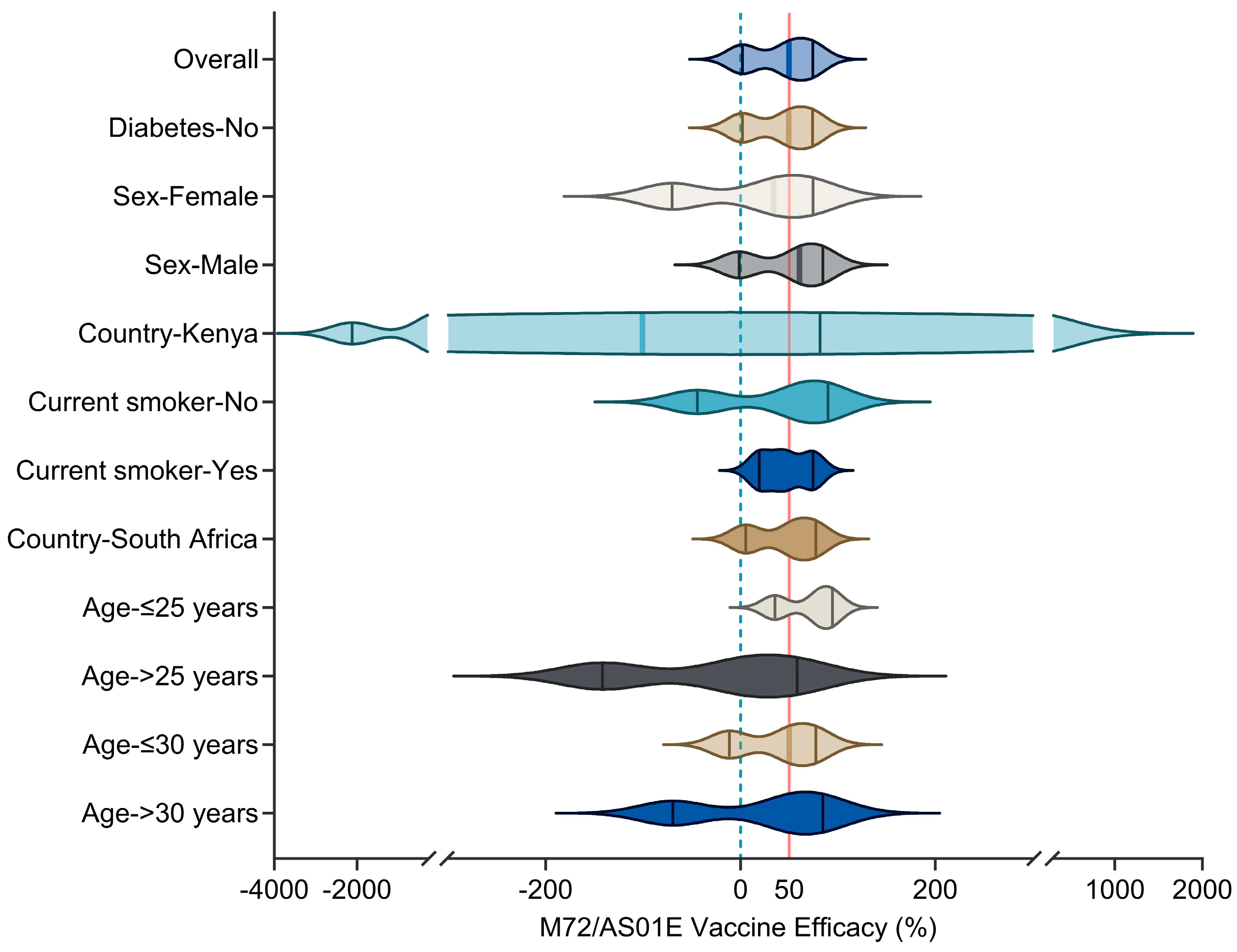{kind=link}
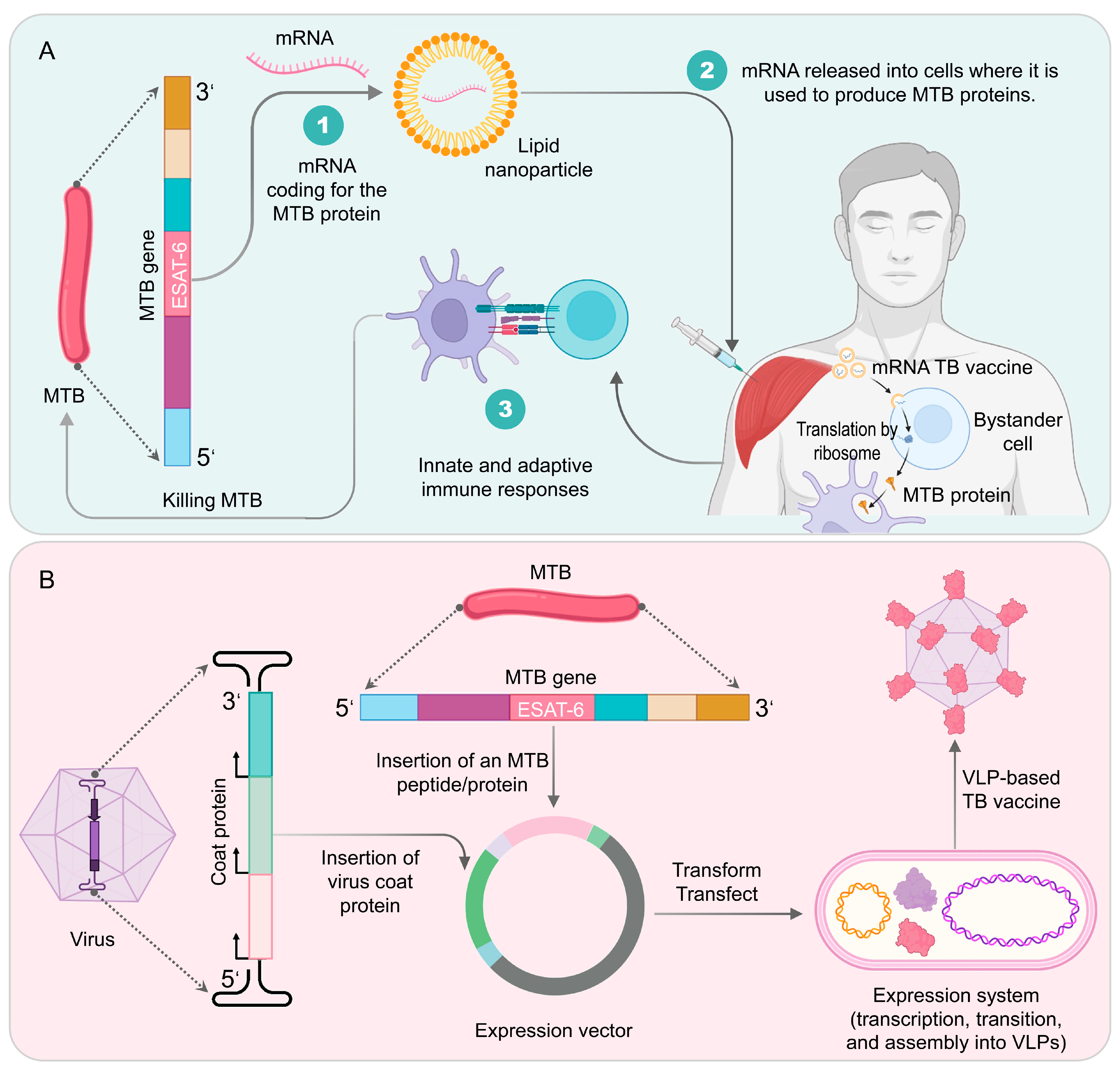{kind=link}
| Vaccine | NCT Number | Phase | Population | Sample Size | Arms and Interventions | Primary Outcome Measures | Results | Ref. |
|---|---|---|---|---|---|---|---|---|
| M72/AS01E | NCT01755598 | I | LTBI individuals | 3575 | Experiment: 2 doses of M72/AS01E (10 µg), spaced one month apart, i.m. in the arm triangle area. Placebo comparator: 2 doses of placebo with a 1-month interval, i.m. in the arm triangle area. | Incident rates of definite PTB disease, not associated with HIV-infection, meeting the case definition | LTBI population vaccinated with M72/AS01E can prevent latent infections from developing into TB, the vaccine efficacy in the 36th month was 49.7% | [78,79] |
| NCT00950612 | II | Adolescents | 60 | Experiment: 2 doses of M72/AS01E (10 µg), with an interval of one month, i.m. in the arm triangle area. Placebo comparator: 2 doses of placebo with a one-month interval, i.m. in the arm triangle area. | Number of subjects with solicited local symptoms and solicited general symptoms, unsolicited AEs and SAEs, different levels biochemical and hematological levels | M72/AS01E induced strong T cell and antibody responses, including NK cells and IFN- γ generation | [80] | |
| NCT04556981 | III | HIV-positive patients | 402 | Experiment: 2 doses of M72AS01E (10 µg), i.m., at days 1 and 29. Placebo comparator: control sodium chloride 0.9%, i.m., at days 1 and 29. | Number of participants with solicited AEs, unsolicited AEs, and SAEs | NA | ||
| GamTBvac | NCT03255278 | I | Healthy adults | 60 | Placebo comparator: s.c. 0.5 mL placebo. Safety and portable study group: s.c. 0.25 mL GamTBvac. Experimental 1: s.c. 0.25 mL GamTBvac. Experimental 2: s.c. 0.5 mL GamTBvac. Experimental 3: s.c. 1 mL GamTBvac. | The number of AEs | Different doses of vaccines were evaluated for immunogenicity, with half dose (0.5 mL) having the best effect | [81] |
| NCT03878004 | II | Healthy adults | 180 | Experiment: s.c. 2 doses of 0.5 mL GamTBvac, with an interval of 8 weeks. Placebo comparator: s.c. 2 doses of 0.5 mL placebo, with an interval of 8 weeks. | 1. Level of IFN-γ secretion in whole blood or PBMC fraction 2. Number of participants with AEs | The vaccine is well tolerated and induces specific and persistent Th1 and Humoral immunity responses | [82] | |
| NCT04975737 | III | Healthy adults | 7180 | Experiment: s.c. 2 doses of 0.5 mL GamTBvac, with an interval of 8 weeks. Placebo comparator: s.c. 2 doses of 0.5 mL placebo, with an interval of 8 weeks. | Preventive efficacy (Ep) | NA | ||
| H56:IC31 | NCT01967134 | I | LTBI/healthy adults | 25 | Experiment: LTBI negative, i.m. 3 doses of 15 µg/500 nmol H56:IC31 at days 0, 56 and 112. Experiment: LTBI positive, i.m., 3 doses of 50 µg/500 nmol H56:IC31, days 0, 56 and 112. | Number of participants with at least 1 AE until day 210 | H56:IC31 vaccine induces antigen-specific IgG response and CD4+ T cells expressing Th1 cytokines | [83] |
| NCT02378207 | Ib | Adolescent | 84 | Experiment: i.m. 0.5 mL H4:IC31 or H56:IC31 at days 0 and 56. Active comparator: i.m. 0.1 mL BCG at day 0. Placebo comparator: i.m. 0.5 mL NaCI at days 0 and 56. | 1. Number of participants with AEs 2. Percentage of participants with response rates to TB antigens as compared to baseline | Both H4: IC31 and H56: IC31 induce CD4+ T cell responses, and H4:IC31 and H56:IC31 induce serum IgG | [84] | |
| NCT01865487 | IIa | Healthy adults/LTBI | 98 | Placebo control: i.m. 2 doses of NaCl. Experimental groups: 5/500, 15/500, 50/500 H56ug/IC31nmol QFT-negative and/or -positive, 2 doses, days 0 and 56. | Number and percentage of unsolicited and solicited AEs | H56:IC31 induces a persistent antigen-specific Th1 response without being affected by MTB infection | [85] | |
| NCT02503839 | I/II | ATB patients | 51 | Experimental 1: 120 g of etoricoxib, po. at days 0 and 140. Experimental 2: 140 µg H56:IC31 and no etoricoxib i.m. at days 84 and 140. Placebo comparator: Standard TB treatment. Experimental 3: 120 g etoricoxib po. at days 0 and 140; 140 µg H56:IC31 i.m. at days 0 and 140. | 1. Participants with AEs 2. Immunogenicity of etoricoxib/H56:IC31 vaccine | H56:IC31 vaccination is safe and immunogenic in ATB patients | [86] | |
| NCT03512249 | IIb | ATB patients | 900 | Experiment: i.m. 2 doses of 0.5 mL H56:IC31 at days 0 and 56. Placebo comparator: i.m. 2 doses of 0.5 mL NaCI at days 0 and 56. | Rate of TB disease recurrence | NA | ||
| H4:IC31 | NCT02074956 NCT02066428 | I | Healthy adults | 60 | Experimental 1–4 groups: i.m. 5 µg, 15 µg, 50 µg, or 150 µg/500 nmol, H4:IC31 at days 0 and 56. Experimental 5: i.m. H4:IC31 (15 µg/100 nmol). Placebo: NaCl. | Safety profile of two injections of H4:IC31 at different dose levels; evaluate the safety of one or two injections of two H4:IC31 antigen amounts administered with three different amounts of adjuvant | The evidence provided by the dose escalation test indicates that the optimal antigen adjuvant dose combination is H4, 5, 15, or 50 µg and IC31 500 nmol | [87] |
| NCT02075203 | II | Adolescents | 989 | Experiment: i.d. 2 doses of H4:IC31 (15 mcgH4/500 nmol IC31) at days 0 and 56. Active comparator: i.d. BCG at day 0. Control group: i.d. 2 doses of 0.9% NaCl (0.9%) at day 0. | 1. Safety profile of H4:IC31 and BCG revaccination 2. Number of participants testing positive for MTB at day 84 | The immune efficacy of the H4: IC31 vaccine is 30.5% (p = 0.16). 44 (14.3%) H4:IC31 group experienced QFT conversion, 41 (13.1%) BCG group experienced QFT conversion, and 49 (15.8%) placebo group experienced QFT conversion | [88] | |
| ID93/GLA-SE | NCT01599897 | I | Healthy adults | 60 | Experimental 1–4: i.m. 2 µg ID93 + 2 µg GLA-SE, 10 µg ID93 + 2 µg GLA-SE, 2 µg ID93 + 5 µg GLA-SE, 10 µg ID93 + 5 µg GLA-SE at days 0, 28, and 56, respectively. Active comparator 1–2: i.m. 2 µg or 10 µg ID93 at days 0, 28, and 56. | Number of patients experiencing AEs | Showing a satisfactory safety profile and eliciting a functional humoral and T-helper 1 type cellular response | [89] |
| NCT02465216 | IIa | Healthy adults | 60 | Experimental 1–3: i.m. 2 μg ID93 + 2 μg GLA-SE, 10 μg ID93 + 2 μg GLA-SE, 2 μg ID93 + 5 μg GLA-SE at days 0 and 56, respectively. Experimental 4: i.m. 3 doses of 2 µg ID93+ 5 µg GLA-SE at days 0, 28, and 56. Active comparator: i.m., NaCI at days 0, 28, and 112. | Number of AEs | The antigen-specific IgG and CD4 T cell responses induced by a dose of 2 μg ID93 + 5 μg GLA-SE were significantly higher than those induced by placebo, and lasted for 6 months | [90] | |
| NCT03806686 | IIa | Healthy adults | 107 | Experimental 1–2: i.m. 2 μg ID93 + 5 µg GLA-SE, 5 μg ID93 +5 μg GLA-SE at days 0, 28, and 56, respectively. Active comparator: i.m. 0.5 mL NaCI at days 0, 28, and 56. | AEs | NA | ||
| AEC/BC02 | NCT04239313 | I | Healthy adults | 30 | Experiment: i.m. low-dose AEC/BC02; Active comparison: i.m. low-dose adjuvant; active comparator: i.m. placebo. | The number of AEs after i.m. | NA | |
| NCT05284812 | II | LTBI individuals | 200 | Negative control group: not vaccinated. Sentinel group: 18–59-year-olds received low- or high-dose injections; ≥60 years old received low or high-dose injections, i.m. Experimental group 1: EC positive, i.m. low-dose AEC/BC02. Experimental group 2: EC positive, i.m. high-dose AEC/BC02. Experimental group 3: EC positive, i.m. high-dose AEC/BC02 into 1,3, and 6 doses; i.m. 2, 4, 5 doses of placebo. Placebo group: EC positive, i.m. placebo. Adjuvant group: high-dose adjuvant for AEC/BC02. | The number of AEs after i.m. injection | NA | ||
| VPM1002 | NCT00749034 | I | Healthy male | 80 | Experiment: i.d. 0.05 mL VPM1002 (5 × 103, 5 × 104, 5 × 105 CFUs). Active comparator: i.d. 0.05 mL BCG (2–8 × 105 CFUs). | Physical examination, vital signs, ECG, liver sonography, chest X-ray, laboratory safety parameters, tolerability, recording of concomitant medication and AEs | VPM1002 has safety and immunogenicity in response to B and T cells | [91] |
| NCT01479972 | II | Newborn infants | 48 | Experiment: i.d. 0.05 mL VPM1002. Active comparator: i.d. 0.05 mL BCG. | 1. Safety 2. Immunogenicity | Inoculation with VPM1002 can induce multifunctional CD+ 4 and CD8+ T cells | [92] | |
| NCT02391415 | II | Newborn infants | 416 | Experiment: HIV-unexposed infants, i.d. 0.05 mL VPM1002. Control group: HIV-unexposed infants, i.d. 0.05 mL BCG. Experiment: HIV-unexposed infants, i.d. 0.05 mL VPM1002 (Hyg+). Control group: HIV-exposed infants, i.d. 0.05 mL BCG. Experiment: HIV-exposed infants, i.d. 0.05 mL VPM1002. | The difference between the VPM1002 and BCG vaccination groups in the incidence of grade 3 and 4 adverse drug reactions and IMP-related ipsilateral or generalized lymphadenopathy of 10 mm or greater (diameter) | VPM1002 is safe for both HIV-exposed and -unexposed infants; both VPM1002 and BCG have immunogenicity, and from the 6th week onwards, the immune response intensity induced by BCG is greater than that of VPM1002 | [93] | |
| NCT03152903 | III | Healthy adults | 2000 | Experiment: i.d. 0.05 mL VPM1002. Active comparator: i.d. 0.05 mL placebo. | Percentage of bacteriologically confirmed TB recurrence cases | NA | ||
| NCT04351685 | III | Newborn infants | 6940 | Experimental group: i.d. 0.05 mL VPM1002. Active comparator: i.d. 0.05 mL BCG. | Incident cases of QFT conversion | NA | ||
| MTBVAC | NCT02013245 | I | Healthy adults | 34 | Experimental 1: i.d. 0.1 mL MTBVAC (5 × 103 CFUs). Experimental 2: i.d. 0.1 mL MTBVAC (5 × 104 CFUs). Experimental 3: i.d. 0.1 mL MTBVAC (5 × 105 CFUs). Active comparator: i.d. 0.1 mL BCG (5 dose 5 × 105 CFU). | Number of participants with AEs up to 210 days after vaccination | MTBVAC exhibits good safety in healthy adults, similar to BCG | [94] |
| NCT02729571 | Ib | Newborn infants | 54 | Experimental 1: i.d. 0.1 mL MTBVAC (5 × 105 CFUs)/0.1 mL BCG SII (5 × 105 CFUs) adults. Experimental 2: i.d. 0.05 mL MTBVAC (2.5 × 103 CFUs/2.5 × 104 CFUs/2.5 × 105 CFUs) infants. Experimental 3: i.d. 0.05 mL BCG (2.5 × 105 CFUs) infants. | Safety and reactogenicity in infants and adults | MTBVAC has good safety and immunogenicity, and can induce long-lasting CD4 cell responses in infants | [95] | |
| NCT02933281 | Ib/IIa | Healthy adults | 144 | Experimental 1–4: QFT negative, i.d. MTBVAC 5 × 103, 5 × 104, 5 × 105, or 5 × 106 CFUs at day 0, respectively. Experimental 5–8: QFT positive, i.d. MTBVAC 5 × 103, 5 × 104, 5 × 105, or 5 × 106 CFUs at day 0, respectively. Active comparator: i.d. BCG 5 × 105 CFUs at day 0. | Safety and reactogenicity of MTBVAC at escalating dose levels compared to BCG vaccine by assessing number of participants with AEs and SAEs | NA | ||
| NCT03536117 | IIa | Newborn infants | 99 | Experimental 1–3: i.d. 0.05 mL MTBVAC (2.5 × 104, 2.5 × 105, or 2.5 × 106 CFUs), respectively. Active comparator: i.d. 0.05 mL BCG (2.5 × 105 CFUs). | 1. Number of participants with treatment-related AEs as defined in protocol 2. Immunogenicity analysis in infants | NA | ||
| NCT04975178 | III | Newborn infants | 6960 | Experiment: i.d. 0.05 mL MTBVAC. Active comparator: i.d. 0.05 mL BCG. | Prevention of TB disease in healthy HIV-uninfected and HIV-exposed uninfected neonates | NA | ||
| BCG (revaccination) | NCT02378207 | IIb | Adolescents | 84 | Active comparator: BCG (2–8 × 105 CFUS), i.m. as 0.1 mL in either deltoid muscle at day 0. Placebo comparator: control sodium chloride 0.9%, i.m. as 0.5 mL in alternating deltoid muscle at days 0 and 56. | Number of participants with AEs; percentage of participants with response rates to TB antigens as compared to baseline | BCG revaccination had acceptable safety and induced robust, multifunctional BCG-specific CD4+ T cells | [84] |
| NCT02075203 | II | Adolescents | 989 | Active comparator: BCG SSI vaccine, 1 dose on day 0; placebo comparator: placebo, 2 doses on days 0 and 56. | Safety profile of BCG revaccination; number of participants testing positive for MTB at day 84 | BCG revaccination was immunogenic and reduced the rate of sustained QFT conversion, with an efficacy of 45.4% (p = 0.03) | [88] | |
| NCT04152161 | IIb | Children and adolescents | 1820 | Experiment: a single 0.1 mL of BCG vaccine SSI, i.d. in deltoid region of the upper arm. Placebo: a single 0.1 mL of normal saline, i.d. in deltoid region of the upper arm. | Number of participants with sustained QFT conversion | NA | ||
| MIP | NCT00341328 | III | Category-I PTB patients | 300 | Experimental group: i.d., 6 doses MIP vaccine given 0.2 mL at baseline, then 0.1 mL every 2 weeks for 8 weeks. Control group: i.d., 6 doses placebo given 0.2 mL at baseline, then 0.1 mL every 2 weeks for 8 weeks. | The time of sputum conversion, the cure rate; the relapse in patients of category-I pulmonary TB, and any clinical adverse reactions | NA | |
| NCT00265226 | III | Category-II PTB patients | 1020 | Experimental group: intradermal administration of mycobacterium W vaccine and category II ATT as per RNTCP guidelines. Control group: placebo administration and category II ATT drugs as per RNTCP guidelines. | 1. Sputum conversion time 2. Cure rate 3. Clinical adverse reactions | At the 39th week follow-up, sputum culture conversion rate was 94.2% (309/328) in the MIP group and 89.17% (280/314) in the placebo group | [96] | |
| RUTI | NCT00546273 | I | Healthy adults | 24 | Experimental groups 1–4: RUTI (5 µg, 25 µg, 100 µg, or 200 µg) i.d., at days 0 and 28. Placebo comparator: placebo i.d. at days 0 and 28. | 1. VAS Pain Score 2. Local and systemic events occurrence, intensity, and relationship to vaccination 3. Number of clinically relevant abnormalities detected | RUTI has been proven to trigger specific reactions against MTB | [97] |
| NCT01136161 | IIa | LTBI individuals | 95 | Experimental groups 1–5: HIV-negative, 5 µg, 25 µg, 100 µg, 200 µg RUTI or placebo i.d. at days 28 and 56, respectively. Experimental groups 6–10: HIV-positive, 5 µg, 25 µg, 100 µg, 200 µg RUTI or Placebo i.d. at days 28 and 56, respectively. | 1. Local tolerability, focal tolerability 2. Systemic tolerability, vital signs and physical examination, ECG, and laboratory tests | The best cellular multi-antigen response was achieved when administered at 25 µg RUTI | [98] | |
| NCT04919239 | IIb | ATB patients | 140 | Experiment: 25 µg RUTI, i.d. Placebo comparator: 25 µg placebo, i.d. | Percentage of patients with sputum culture negative | NA | ||
| NCT05455112 | IIb | ATB patients | 44 | Experiment: 25 µg RUTI, i.d. Placebo comparator: 25 µg NaCI, i.d. | 1. Early bactericidal activity (EBA) 0–14 2. AEs and grade 3–4 AEs | NA | ||
| DAR-901 | NCT02063555 | I | Healthy adults | 59 | Experiment: 0.1 mL DAR-901 i.d., at 0, 2 and 4 months. Active comparator: 0.1 mL NaCI i.d., at 2 and 4 months; 0.1 mL BCG i.d., at 4 months. Placebo comparator: 0.1 mL NaCI, i.d. at 0, 2, and 4 months. | Safety | DAR-901 induces low-intensity multifunctional memory CD4+ T cell response and reaching peak in 7 days | [99] |
| NCT02712424 | IIb | Adolescents | 625 | Experimental group: 0.1 mL DAR-901, i.d. Placebo comparator: 0.1 mL NaCI, i.d. | New infection with MTB | The three-dose series of DAR-901 is safe and well tolerated, but cannot prevent initial or sustained IGRA conversion | [100] | |
| MVA85A | NCT00460590 | I | Healthy adults | 24 | Experimental groups 1–3: adults vaccinated with BCG, adults without BCG vaccination, or teenagers i.d. 5 × 107 pfu MVA85A, respectively. | The safety of a single injection of MVA85A in healthy subjects by collecting data on AEs | MVA85A vaccine induces the production of long-lasting, multifunctional, Ag85A specific CD4+ T cells | [101] |
| NCT01650389 | II | HIV-exposed infants | 248 | Experimental group: 1 × 108 pfu MVA85A vaccine within 96 h of birth, i.d. Placebo comparator: equal volume intradermal administration within 96 h of birth i.d. | Safety | MVA85A immunization in newborns exposed to HIV is safe and induces a moderate antigen-specific immune response without affecting BCG vaccination | [102] | |
| NCT00953927 | II | Infants | 2797 | Experiment: 1 × 108 pfu MVA85A vaccine, i.d. Placebo comparator: 1 × 108 pfu candida skin test antigen, i.d. | Safety profile of MVA85A in BCG-vaccinated, HIV-negative infants | 32 out of 1399 MVA85A subjects (2%) achieved the primary efficacy endpoint 39 out of 1395 controls (3%) achieved the primary efficacy endpoint | [103] | |
| ChAdOx1.85A | NCT01829490 | I | Healthy adults | 42 | Experimental groups 1–2: 1 dose of 5 × 109 or 2.5 × 1010 vp of ChAdOx1.85A, i.m., respectively. Experimental group 3: 1 dose of 2.5 × 1010 vp of ChAdOx1.85A, followed by a boost dose of 1 × 108 pfu of MVA85A, at 56 days later, i.m. Experimental group 4: 2 doses of 2.5 × 1010 vp of ChAdOx1.85A i.m., at days 0 and 28, followed by a boost dose of 1 × 108 pfu of MVA85A, at day 119, i.m. | Safety of ChAdOx1 85A vaccination with and without MVA85A boost vaccination in healthy, BCG-vaccinated adults | ChAdOx1.85A induces Ag85A specific CD4+ T and CD8+ T cell responses | [104] |
| NCT03681860 | IIa | Healthy adults | 72 | Experiment: i.m. 5 × 109 vp ChAdOx1.85A. Experimental 2: 3 adults i.m. 2.5 × 1010 vp ChAdOx1.85A. Experimental 3: 3 adolescents i.m. 5 × 109 vp ChAdOx1.85A. Experimental 4: 3 adolescents i.m. 2.5 × 1010 vpChAdOx1.85A. Placebo comparator: 30 EMaBS adolescents i.m. 2.5 × 1010 vp ChAdOx1.85A, further strengthened MVA85A, other adolescents BCG revaccinated. | Safety and immunogenicity | NA | [104] | |
| AdHu5Ag85A | NCT02337270 | I | Healthy adults | 36 | Experiment: aerosol 106 Ad5Ag85A at day 0. Experiment: aerosol 2 × 106 Ad5Ag85A at day 0. Experiment: i.m. 108 Ad5Ag85A at day 0. | Number of participants reporting AEs | Compared with i.m., aerosol delivered AdHu5Ag85A vaccine has advantages in inducing respiratory epithelium immunity | [105] |
| TB/FLU-01L | NCT03017378 | I | Healthy adults | 36 | Active comparator: TB/FLU-01L (intranasal application). Active comparator: TB/FLU-01L (sublingual application). | AEs, solicited local and systemic reactions, unsolicited AEs, SAEs, including abnormal laboratory findings | NA | |
| TB/FLU-04L | NCT02501421 | I | Healthy adults | 44 | Experiment: TB/FLU-04L, at days 1 and 21. Placebo comparator: placebo, at days 1 and 21. | Immediate reactions, solicited local and systemic reactions, unsolicited events and abnormal laboratory findings, SAEs | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuang, L.; Ye, Z.; Li, L.; Yang, L.; Gong, W. Next-Generation TB Vaccines: Progress, Challenges, and Prospects. Vaccines 2023, 11, 1304. https://doi.org/10.3390/vaccines11081304
Zhuang L, Ye Z, Li L, Yang L, Gong W. Next-Generation TB Vaccines: Progress, Challenges, and Prospects. Vaccines. 2023; 11(8):1304. https://doi.org/10.3390/vaccines11081304
Chicago/Turabian StyleZhuang, Li, Zhaoyang Ye, Linsheng Li, Ling Yang, and Wenping Gong. 2023. "Next-Generation TB Vaccines: Progress, Challenges, and Prospects" Vaccines 11, no. 8: 1304. https://doi.org/10.3390/vaccines11081304