

Structural and Phylogenetic Analysis of CXCR4 Protein Reveals New Insights into Its Role in Emerging and Re-Emerging Diseases in Mammals
 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieval of CXCR4 Sequences
2.2. Phylogenetic Analysis of the CXCR4 Gene
2.3. Prediction and Validation of Human CXCR4 Structure
2.4. Disordered Analysis of Human CXCR4 Proteins
2.5. Analysis of Human CXCR4 Protein Ligands and Domain
2.6. Protein Interactions and Co-Expression Analysis
2.7. Consensus Sequence and Secondary Structure Prediction
3. Results
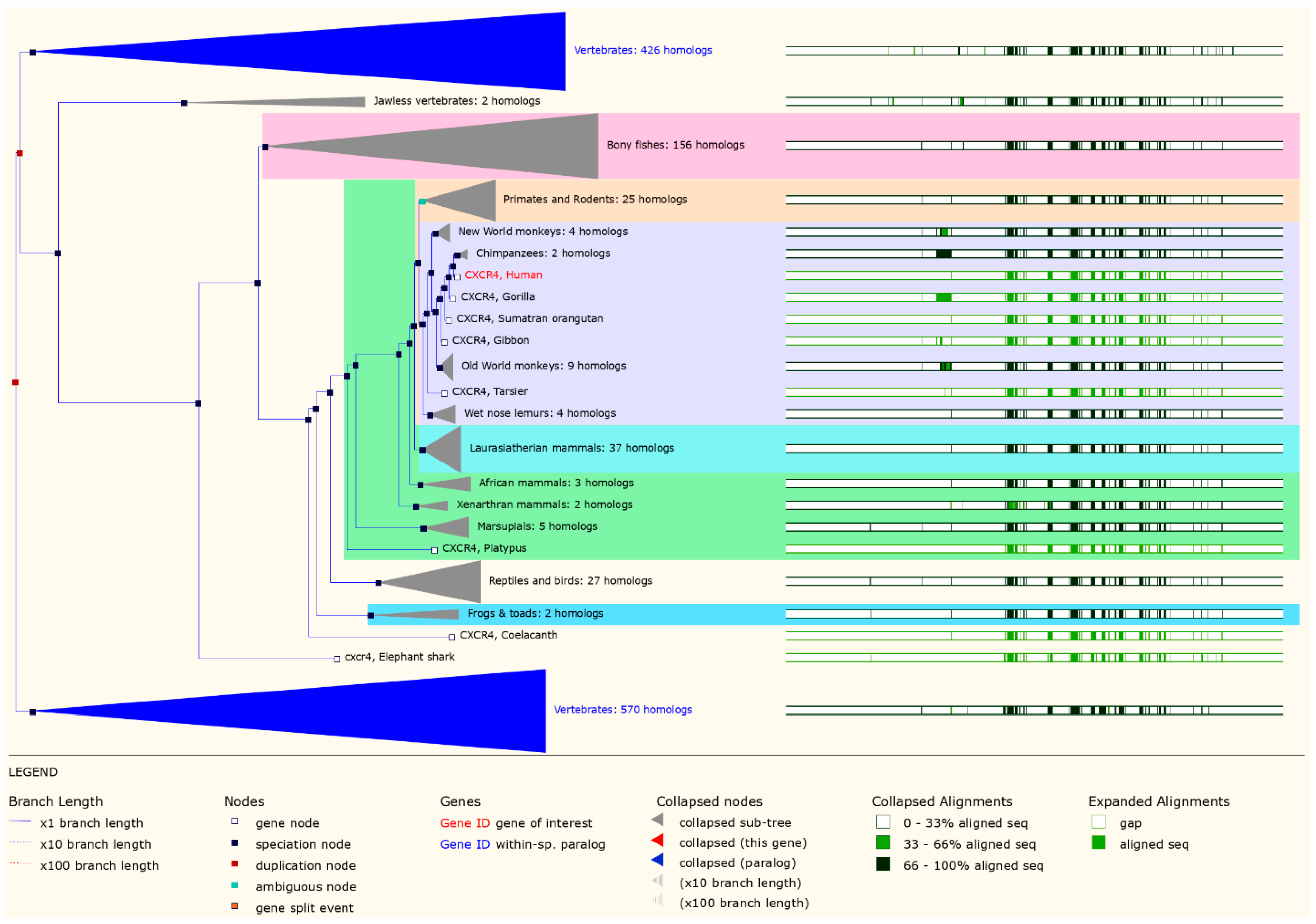
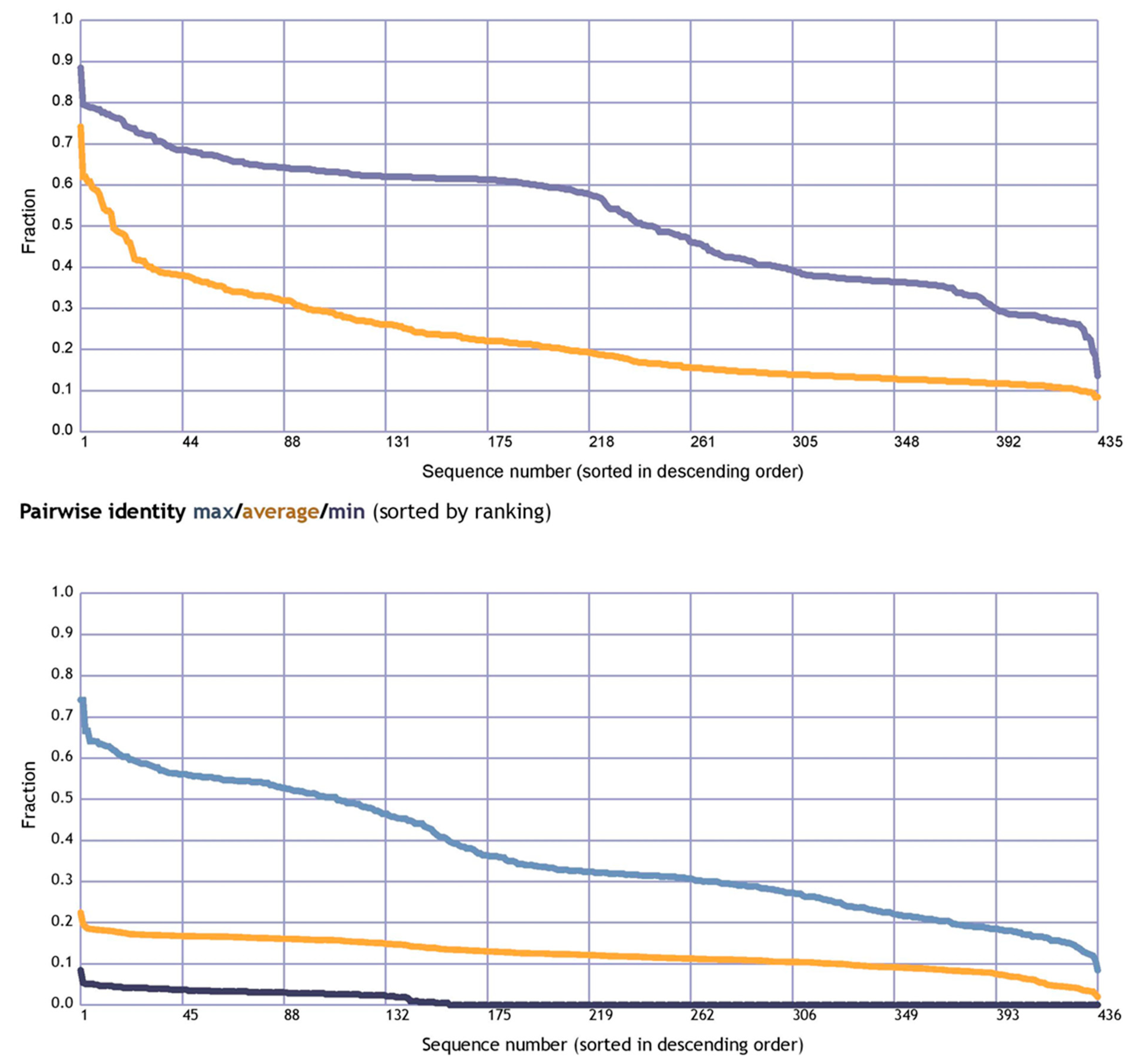
3.1. Evolutionary Analysis of the CXCR4 Gene
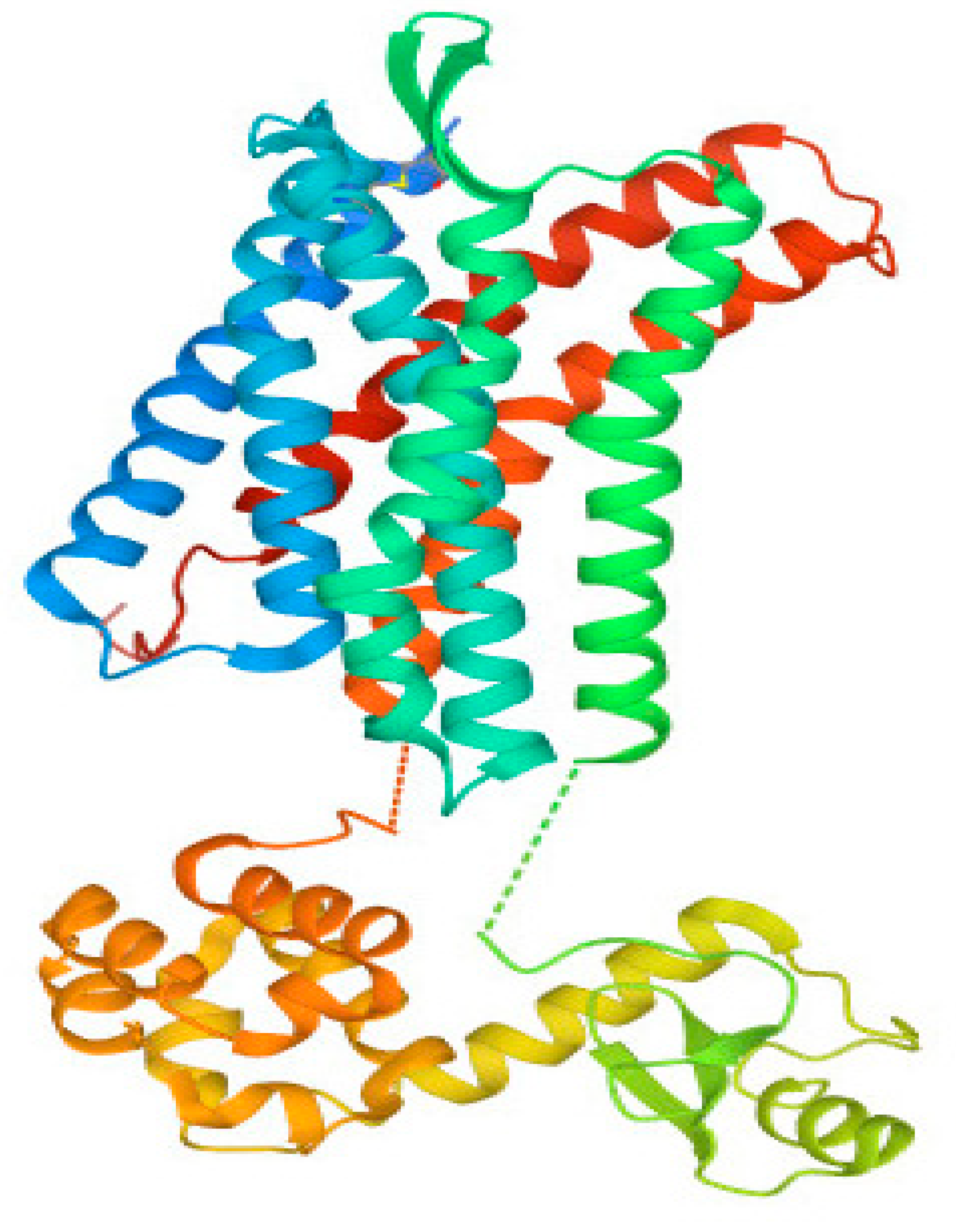
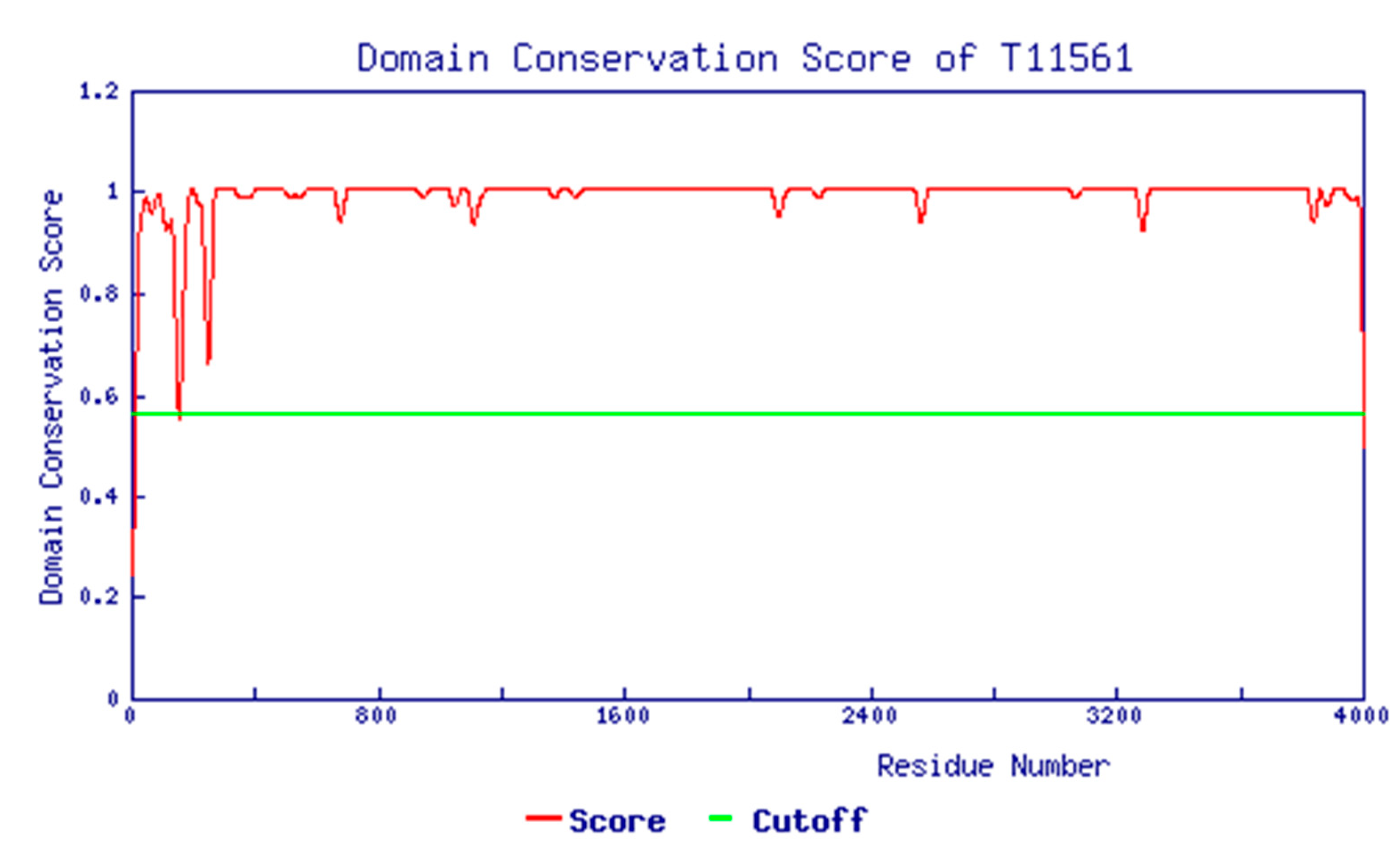
3.2. Structural Analysis of CXCR4 Protein
3.3. Functional Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palomino, D.C.T.; Marti, L.C. Chemokines and immunity. Einstein 2015, 13, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, K.; Hojo, S.; Akashi, T.; Yasumoto, K.; Saiki, I. Chemokine receptors in cancer metastasis and cancer cell-derived chemokines in host immune response. Cancer Sci. 2007, 98, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Cravens, P.D.; Lipsky, P.E. Dendritic cells, chemokine receptors and autoimmune inflammatory diseases. Immunol. Cell Biol. 2002, 80, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.; Antonangeli, F.; Bonanni, V.; Santoni, A. Dysregulation of chemokine/chemokine receptor axes and NK cell tissue localization during diseases. Front. Immunol. 2016, 7, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Laing, K.J.; Secombes, C.J. Chemokines. Dev. Comp. Immunol. 2004, 28, 443–460. [Google Scholar] [CrossRef]
- Latek, D.; Modzelewska, A.; Trzaskowski, B.; Palczewski, K.; Filipek, S. G protein-coupled receptors—Recent advances. Acta Biochim. Pol. 2012, 59, 515. [Google Scholar] [CrossRef] [Green Version]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and chemokine receptors: New targets for cancer immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [Green Version]
- Arimont, M.; Sun, S.-L.; Leurs, R.; Smit, M.; de Esch, I.J.; de Graaf, C. Structural analysis of chemokine receptor-ligand interactions. J. Med. Chem. 2017, 60, 4735–4779. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Nibbs, R.J.; Graham, G.J. Immune regulation by atypical chemokine receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef]
- Luster, A.D.; Alon, R.; von Andrian, U.H. Immune cell migration in inflammation: Present and future therapeutic targets. Nat. Immunol. 2005, 6, 1182–1190. [Google Scholar] [CrossRef]
- Brauner-Osborne, H.; Wellendorph, P.; Jensen, A.A. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr. Drug Targets 2007, 8, 169–184. [Google Scholar] [CrossRef]
- Becker, O.M.; Marantz, Y.; Shacham, S.; Inbal, B.; Heifetz, A.; Kalid, O.; Bar-Haim, S.; Warshaviak, D.; Fichman, M.; Noiman, S. G protein-coupled receptors: In silico drug discovery in 3D. Proc. Natl. Acad. Sci. USA 2004, 101, 11304–11309. [Google Scholar] [CrossRef] [Green Version]
- Lahti, J.L.; Tang, G.W.; Capriotti, E.; Liu, T.; Altman, R.B. Bioinformatics and variability in drug response: A protein structural perspective. J. R. Soc. Interface 2012, 9, 1409–1437. [Google Scholar] [CrossRef] [Green Version]
- Day, P.W.; Rasmussen, S.G.; Parnot, C.; Fung, J.J.; Masood, A.; Kobilka, T.S.; Yao, X.-J.; Choi, H.-J.; Weis, W.I.; Rohrer, D.K. A monoclonal antibody for G protein-coupled receptor crystallography. Nat. Methods 2007, 4, 927–929. [Google Scholar] [CrossRef]
- Wess, J.; Han, S.-J.; Kim, S.-K.; Jacobson, K.A.; Li, J.H. Conformational changes involved in G-protein-coupled-receptor activation. Trends Pharmacol. Sci. 2008, 29, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Wu, L.; Yuan, S.; Wu, M.; Xu, Y.; Sun, Q.; Li, S.; Zhao, S.; Hua, T.; Liu, Z.-J. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 2020, 585, 135–140. [Google Scholar] [CrossRef]
- Ito, S.; Sato, T.; Maeta, T. Role and therapeutic targeting of SDF-1α/CXCR4 axis in multiple myeloma. Cancers 2021, 13, 1793. [Google Scholar] [CrossRef]
- Hara, T.; Tanegashima, K. CXCL14 antagonizes the CXCL12-CXCR4 signaling axis. Biomol. Concepts 2014, 5, 167–173. [Google Scholar] [CrossRef]
- Bajoghli, B. Evolution and function of chemokine receptors in the immune system of lower vertebrates. Eur. J. Immunol. 2013, 43, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotnik, A.; Yoshie, O.; Nomiyama, H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006, 7, 243. [Google Scholar] [CrossRef]
- Rajasekaran, D.; Fan, C.; Meng, W.; Pflugrath, J.W.; Lolis, E.J. Structural insight into the evolution of a new chemokine family from zebrafish. Proteins: Struct. Funct. Bioinform. 2014, 82, 708–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haskill, S.; Peace, A.; Morris, J.; Sporn, S.A.; Anisowicz, A.; Lee, S.W.; Smith, T.; Martin, G.; Ralph, P.; Sager, R. Identification of three related human GRO genes encoding cytokine functions. Proc. Natl. Acad. Sci. USA 1990, 87, 7732–7736. [Google Scholar] [CrossRef] [Green Version]
- Richmond, A.; Yang, J.; Su, Y. The good and the bad of chemokines/chemokine receptors in melanoma. Pigment Cell Melanoma Res. 2009, 22, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161. [Google Scholar] [CrossRef]
- Yen, Y.-C.; Schafer, C.T.; Gustavsson, M.; Eberle, S.A.; Dominik, P.K.; Deneka, D.; Zhang, P.; Schall, T.J.; Kossiakoff, A.A.; Tesmer, J.J. Structures of atypical chemokine receptor 3 reveal the basis for its promiscuity and signaling bias. Sci. Adv. 2022, 8, eabn8063. [Google Scholar] [CrossRef]
- Rodríguez, D.; Gutiérrez-de-Terán, H. Characterization of the homodimerization interface and functional hotspots of the CXCR4 chemokine receptor. Proteins Struct. Funct. Bioinform. 2012, 80, 1919–1928. [Google Scholar] [CrossRef]
- Culhane, A.C.; Schwarzl, T.; Sultana, R.; Picard, K.C.; Picard, S.C.; Lu, T.H.; Franklin, K.R.; French, S.J.; Papenhausen, G.; Correll, M. GeneSigDB—A curated database of gene expression signatures. Nucleic Acids Res. 2010, 38, D716–D725. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The single global macromolecular structure archive. In Protein Crystallography: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2017; pp. 627–641. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Vilella, A.J.; Severin, J.; Ureta-Vidal, A.; Heng, L.; Durbin, R.; Birney, E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009, 19, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, H.I.; Khan, F.A.; Khan, M.A.; Imran, S.; Akhtar, R.W.; Pandupuspitasari, N.S.; Negara, W.; Chen, J. Molecular Evolution of the Bactericidal/Permeability-Increasing Protein (BPIFA1) Regulating the Innate Immune Responses in Mammals. Genes 2022, 14, 15. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Rokas, A. Phylogenetic analysis of protein sequence data using the Randomized Axelerated Maximum Likelihood (RAXML) Program. Curr. Protoc. Mol. Biol. 2011, 96, 19.11.11–19.11.14. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Buckley, T.R. Model selection and model averaging in phylogenetics: Advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Conant, G.C.; Wagner, G.P.; Stadler, P.F. Modeling amino acid substitution patterns in orthologous and paralogous genes. Mol. Phylogenetics Evol. 2007, 42, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S. Computational Evolutionary Biology. In Advances in Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2021; pp. 83–100. [Google Scholar]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, H.I.; Asif, A.R.; Ahmad, M.J.; Jabbir, F.; Adnan, M.; Ahmed, S.; Afzal, G.; Saleem, A.H.; Li, L.; Jiang, H. Adaptive evolution of peptidoglycan recognition protein family regulates the innate signaling against microbial pathogens in vertebrates. Microb. Pathog. 2020, 147, 104361. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, H.I.; Majeed, M.B.B.; Ahmad, M.Z.; Jabbar, A.; Maqbool, B.; Ahmed, S.; Mustafa, H.; Simirgiotis, M.J.; Chen, J. Comparative analysis of the mitochondrial proteins reveals complex structural and functional relationships in Fasciola species. Microb. Pathog. 2021, 152, 104754. [Google Scholar] [CrossRef]
- Cheng, J.; Zhen, Y.; Miksys, S.; Beyoğlu, D.; Krausz, K.W.; Tyndale, R.F.; Yu, A.; Idle, J.R.; Gonzalez, F.J. Potential role of CYP2D6 in the central nervous system. Xenobiotica 2013, 43, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Källberg, M.; Margaryan, G.; Wang, S.; Ma, J.; Xu, J. RaptorX server: A resource for template-based protein structure modeling. In Protein Structure Prediction; Springer: Berlin/Heidelberg, Germany, 2014; pp. 17–27. [Google Scholar]
- Wu, Q.; Peng, Z.; Zhang, Y.; Yang, J. COACH-D: Improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018, 46, W438–W442. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2012, 41, D1096–D1103. [Google Scholar] [CrossRef] [Green Version]
- Ngan, C.-H.; Hall, D.R.; Zerbe, B.; Grove, L.E.; Kozakov, D.; Vajda, S. FTSite: High accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics 2012, 28, 286–287. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, H.I.; Iqbal, A.; Ijaz, N.; Ullah, M.I.; Asif, A.R.; Rahman, A.; Mehmood, T.; Haider, G.; Ahmed, S.; Mahmoud, S.F. Molecular Evolution of the Activating Transcription Factors Shapes the Adaptive Cellular Responses to Oxidative Stress. Oxidative Med. Cell. Longev. 2022, 2022, 2153996. [Google Scholar] [CrossRef]
- Singh, H.; Srivastava, H.K.; Raghava, G.P. A web server for analysis, comparison and prediction of protein ligand binding sites. Biol. Direct 2016, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, H.I.; Afzal, G.; Jamal, A.; Kiran, S.; Khan, M.A.; Mehmood, K.; Kamran, Z.; Ahmed, I.; Ahmad, S.; Ahmad, A. In silico structural, functional, and phylogenetic analysis of cytochrome (CYPD) protein family. BioMed Res. Int. 2021, 2021, 5574789. [Google Scholar] [CrossRef]
- Von Mering, C.; Jensen, L.J.; Kuhn, M.; Chaffron, S.; Doerks, T.; Krüger, B.; Snel, B.; Bork, P. STRING 7—Recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007, 35, D358–D362. [Google Scholar] [CrossRef] [Green Version]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for visualization and analysis of biological networks. In Data Mining in Proteomics; Springer: Berlin/Heidelberg, Germany, 2011; pp. 291–303. [Google Scholar]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Gouet, P.; Courcelle, E. ENDscript: A workflow to display sequence and structure information. Bioinformatics 2002, 18, 767–768. [Google Scholar] [CrossRef] [Green Version]
- Buchan, D.W.; Jones, D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbir, F.; Irfan, M.; Mustafa, G.; Ahmad, H.I. Bioinformatics approaches to explore the phylogeny and role of BRCA1 in breast cancer. Crit. Rev. Eukaryot. Gene Expr. 2019, 29, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, T.; Verkade, H.; Heath, J.K.; Kuroiwa, A.; Kikuchi, Y. Sdf1/Cxcr4 signaling controls the dorsal migration of endodermal cells during zebrafish gastrulation. Development 2008, 135, 2521–2529. [Google Scholar] [CrossRef] [Green Version]
- Tulotta, C.; Stefanescu, C.; Chen, Q.; Torraca, V.; Meijer, A.; Snaar-Jagalska, B. CXCR4 signaling regulates metastatic onset by controlling neutrophil motility and response to malignant cells. Sci. Rep. 2019, 9, 2399. [Google Scholar] [CrossRef] [Green Version]
- Beutler, B.; Rehli, M. Evolution of the TIR, tolls and TLRs: Functional inferences from computational biology. In Toll-Like Receptor Family Members and Their Ligands; Springer: Berlin/Heidelberg, Germany, 2002; pp. 1–21. [Google Scholar]
- DeVries, M.E.; Kelvin, A.A.; Xu, L.; Ran, L.; Robinson, J.; Kelvin, D.J. Defining the origins and evolution of the chemokine/chemokine receptor system. J. Immunol. 2006, 176, 401–415. [Google Scholar] [CrossRef] [Green Version]
- Murali, S.; Aradhyam, G.K. Structure-function relationship and physiological role of apelin and its G protein coupled receptor. Biophys. Rev. 2023, 1–17. [Google Scholar] [CrossRef]
- Liggett, S.B. Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci. Signal. 2011, 4, pe36. [Google Scholar] [CrossRef]
- Onuffer, J.J.; Horuk, R. Chemokines, chemokine receptors and small-molecule antagonists: Recent developments. Trends Pharmacol. Sci. 2002, 23, 459–467. [Google Scholar] [CrossRef]
- Nemoto, W.; Toh, H. Membrane interactive α-helices in GPCRs as a novel drug target. Curr. Protein Pept. Sci. 2006, 7, 561–575. [Google Scholar] [CrossRef]
- Kwon, H.R. Study of the Structure and Function of CXC Chemokine Receptor 2. Ph.D. Thesis, University of Tennessee, Knoxville, TN, USA, 2010. [Google Scholar]
- Nasser, M.W.; Raghuwanshi, S.K.; Malloy, K.M.; Gangavarapu, P.; Shim, J.-Y.; Rajarathnam, K.; Richardson, R.M. CXCR1 and CXCR2 activation and regulation: Role of aspartate 199 of the second extracellular loop of CXCR2 in CXCL8-mediated rapid receptor internalization. J. Biol. Chem. 2007, 282, 6906–6915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, M.; van Westen, G.; Li, Q.; IJzerman, A. Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol. Sci. 2011, 32, 35–42. [Google Scholar] [CrossRef]
- Hoare, S.R. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov. Today 2005, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. Intrinsically disordered proteins play diverse roles in cell signaling. Cell Commun. Signal. 2022, 20, 20. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO-Term | Description | Network | Strength | FDR |
|---|---|---|---|---|
| GO:1990763 | Arrestin family protein binding | 2 of 10 | 2.55 | 0.0056 |
| GO:0042379 | Chemokine receptor binding | 3 of 70 | 1.88 | 0.0052 |
| GO:0045236 | CXCR chemokine receptor binding | 2 of 18 | 2.3 | 0.0127 |
| GO:0019955 | Cytokine binding | 3 of 134 | 1.6 | 0.0127 |
| GO:0004896 | Cytokine receptor activity | 3 of 97 | 1.74 | 0.0056 |
| GO:0005126 | Cytokine receptor binding | 4 of 264 | 1.43 | 0.0052 |
| GO:0019899 | Enzyme binding | 7 of 2239 | 0.75 | 0.0127 |
| GO:0001664 | G protein-coupled receptor binding | 6 of 294 | 1.56 | 1.73 × 10−5 |
| GO:0042289 | MHC class II protein binding | 2 of 6 | 2.77 | 0.0052 |
| GO:0005515 | Protein binding | 10 of 7026 | 0.4 | 0.0477 |
| GO:0044877 | Protein-containing complex binding | 7 of 1216 | 1.01 | 0.001 |
| GO:0005102 | Signaling receptor binding | 9 of 1581 | 1.01 | 1.73 × 10−5 |
| GO:0031702 | Type 1 angiotensin receptor binding | 2 of 7 | 2.71 | 0.0052 |
| GO:0031826 | Type 2a serotonin receptor binding | 2 of 3 | 3.07 | 0.0023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naheed, F.; Mumtaz, R.; Shabbir, S.; Jamil, A.; Asif, A.R.; Rahman, A.; Ahmad, H.I.; Essa, M.; Akhtar, H.; Mahmoud, S.F.; et al. Structural and Phylogenetic Analysis of CXCR4 Protein Reveals New Insights into Its Role in Emerging and Re-Emerging Diseases in Mammals. Vaccines 2023, 11, 671. https://doi.org/10.3390/vaccines11030671
Naheed F, Mumtaz R, Shabbir S, Jamil A, Asif AR, Rahman A, Ahmad HI, Essa M, Akhtar H, Mahmoud SF, et al. Structural and Phylogenetic Analysis of CXCR4 Protein Reveals New Insights into Its Role in Emerging and Re-Emerging Diseases in Mammals. Vaccines. 2023; 11(3):671. https://doi.org/10.3390/vaccines11030671
Chicago/Turabian StyleNaheed, Fouzia, Rabia Mumtaz, Sana Shabbir, Arshad Jamil, Akhtar Rasool Asif, Abdur Rahman, Hafiz Ishfaq Ahmad, Muhammad Essa, Hammad Akhtar, Samy F. Mahmoud, and et al. 2023. "Structural and Phylogenetic Analysis of CXCR4 Protein Reveals New Insights into Its Role in Emerging and Re-Emerging Diseases in Mammals" Vaccines 11, no. 3: 671. https://doi.org/10.3390/vaccines11030671