
CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond

 , , , , , , , , , , , and
, , , , , , , , , , , and
Abstract
:1. Introduction
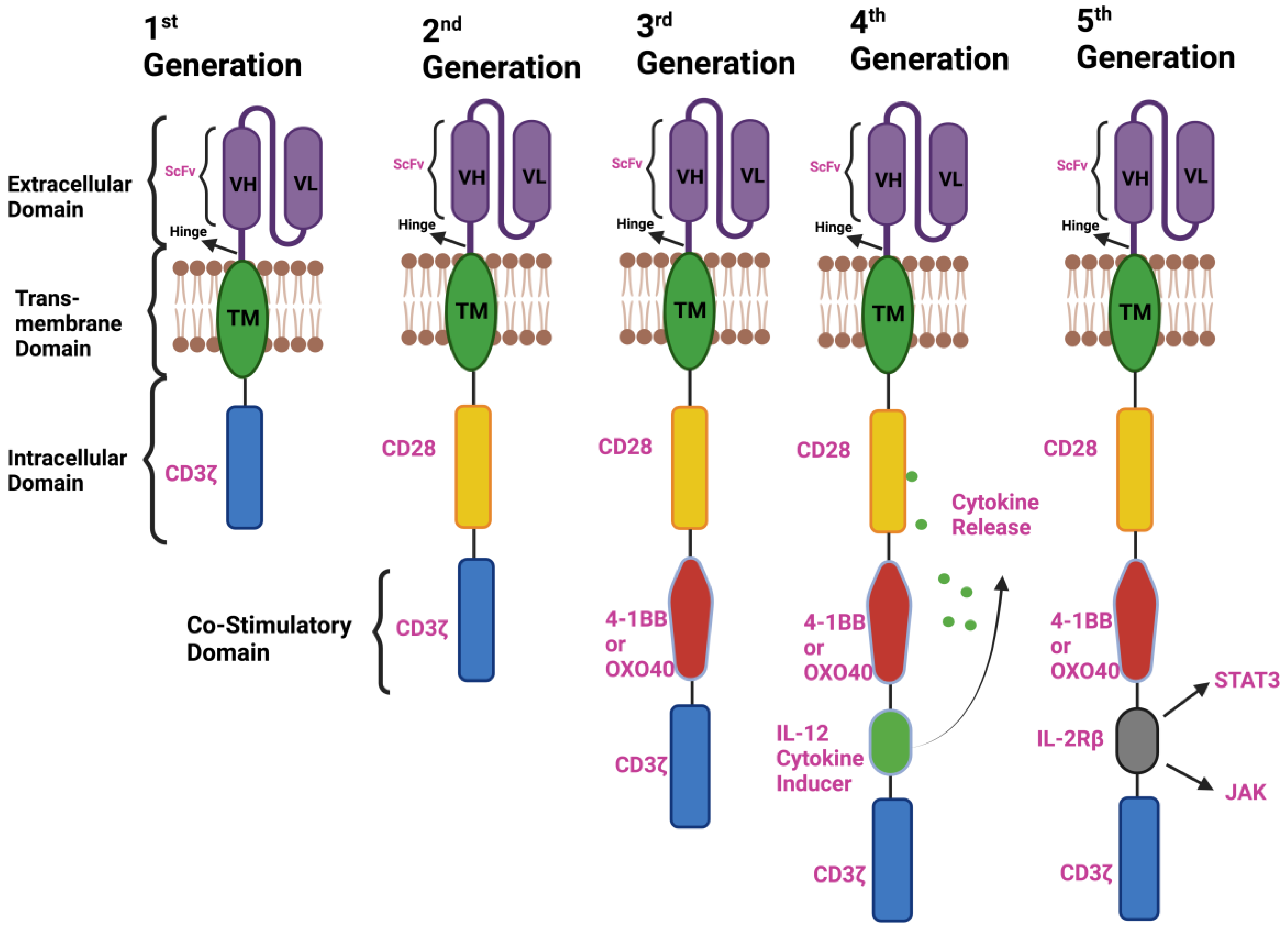
2. Components of CAR-T-Cell Therapy and the Emergence of a Different Generation of CARs
2.1. First-Generation CAR T Cells
2.2. Second-Generation CAR T Cells
2.3. Third-Generation CAR T Cells
2.4. Fourth-Generation CAR T Cells
2.5. Next or Fifth-Generation CAR T Cells
3. Emergence of B-Cell Maturation Antigen (BCMA) as a Promising Target for CAR-T-Cell Therapy in Multiple Myeloma
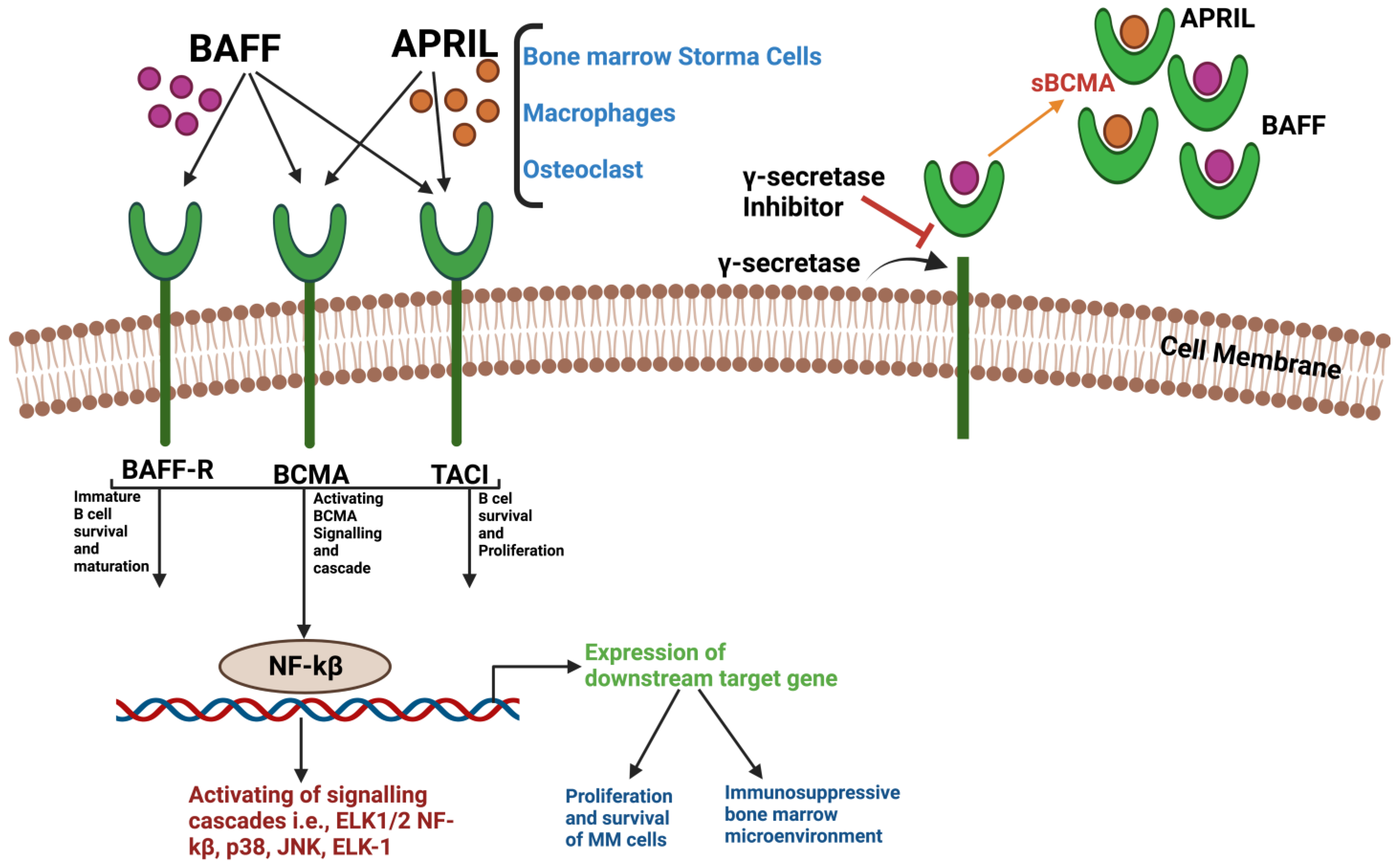
3.1. Structure and Interaction of BCMA
3.2. Idecabtagene Vicleucel (Ide-Cel, bb2121)
3.3. Ciltacabtagene Autoleucel
3.4. JCARH125 (Orva-Cel)
3.5. JNJ-4528 (LCAR-B38M)
4. Non-BCMA CAR-T-Cell Targets
4.1. GPRC5D
4.2. SLAMF7/CS1
4.3. CD38
4.4. CD19
4.5. CD138
4.6. NKG2D
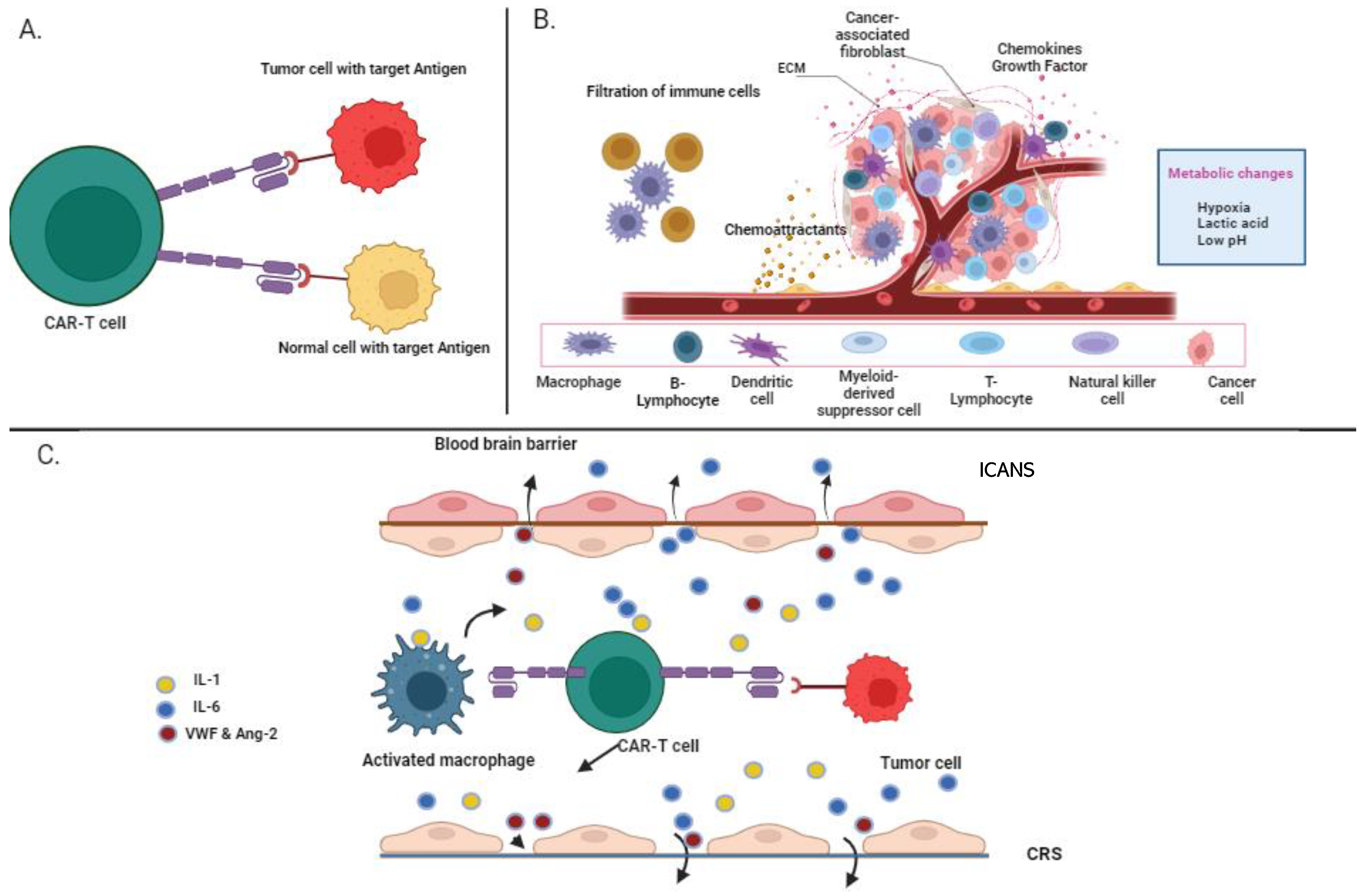
5. Limitations of CAR-T-Cell Therapy in Multiple Myeloma
5.1. On-Target, Off-Tumor
5.2. Cytokine Release Syndrome
5.3. Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)
5.4. Immunosuppressive Microenvironment
6. Recent Advances in BCMA Bispecific Antibodies
6.1. Comparison with BCMA CAR-T Therapy
6.2. Potential Synergies and Complementary Roles
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajkumar, S.V. Multiple Myeloma: 2022 Update on Diagnosis, Risk Stratification, and Management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef]
- Brown, R.D.; Pope, B.; Murray, A.; Esdale, W.; Sze, D.M.; Gibson, J.; Ho, P.J.; Hart, D.; Joshua, D. Dendritic Cells from Patients with Myeloma Are Numerically Normal but Functionally Defective as They Fail to Up-Regulate CD80 (B7-1) Expression after huCD40LT Stimulation Because of Inhibition by Transforming Growth Factor-Beta1 and Interleukin-10. Blood 2001, 98, 2992–2998. [Google Scholar] [CrossRef]
- Brimnes, M.K.; Svane, I.M.; Johnsen, H.E. Impaired Functionality and Phenotypic Profile of Dendritic Cells from Patients with Multiple Myeloma. Clin. Exp. Immunol. 2006, 144, 76–84. [Google Scholar] [CrossRef]
- Urashima, M.; Ogata, A.; Chauhan, D.; Hatziyanni, M.; Vidriales, M.B.; Dedera, D.A.; Schlossman, R.L.; Anderson, K.C. Transforming Growth Factor-Beta1: Differential Effects on Multiple Myeloma versus Normal B Cells. Blood 1996, 87, 1928–1938. [Google Scholar] [CrossRef]
- Kazmi, S.M.; Nusrat, M.; Gunaydin, H.; Cornelison, A.M.; Shah, N.; Kebriaei, P.; Nieto, Y.; Parmar, S.; Popat, U.R.; Oran, B.; et al. Outcomes among High-Risk and Standard-Risk Multiple Myeloma Patients Treated with High-Dose Chemotherapy and Autologous Hematopoietic Stem-Cell Transplantation. Clin. Lymphoma Myeloma Leuk. 2015, 15, 687–693. [Google Scholar] [CrossRef]
- AlDallal, S.M. Yescarta: A New Era for Non-Hodgkin Lymphoma Patients. Cureus 2020, 12, e11504. [Google Scholar] [CrossRef]
- Ali, S.; Kjeken, R.; Niederlaender, C.; Markey, G.; Saunders, T.S.; Opsata, M.; Moltu, K.; Bremnes, B.; Grønevik, E.; Muusse, M.; et al. The European Medicines Agency Review of Kymriah (Tisagenlecleucel) for the Treatment of Acute Lymphoblastic Leukemia and Diffuse Large B-Cell Lymphoma. Oncologist 2020, 25, e321–e327. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research; FDA. Approves Brexucabtagene Autoleucel for Relapsed or Refractory Mantle Cell Lymphoma; FDA: Silver Spring, MD, USA, 2021.
- Bourbon, E.; Ghesquières, H.; Bachy, E. CAR-T Cells, from Principle to Clinical Applications. Bull. Cancer 2021, 108, S4–S17. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Ramos, C.A.; Dotti, G. Chimeric Antigen Receptor (CAR)-Engineered Lymphocytes for Cancer Therapy. Expert. Opin. Biol. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T Cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Non-Signaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for in Vivo Antitumor Activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef]
- Chandran, S.S.; Klebanoff, C.A. T Cell Receptor-Based Cancer Immunotherapy: Emerging Efficacy and Pathways of Resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- Romeo, C.; Seed, B. Cellular Immunity to HIV Activated by CD4 Fused to T Cell or Fc Receptor Polypeptides. Cell 1991, 64, 1037–1046. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T-Cell Receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically Targeted T Cells Eradicate Systemic Acute Lymphoblastic Leukemia Xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef]
- Hwu, P.; Yang, J.C.; Cowherd, R.; Treisman, J.; Shafer, G.E.; Eshhar, Z.; Rosenberg, S.A. In Vivo Antitumor Activity of T Cells Redirected with Chimeric Antibody/T-Cell Receptor Genes. Cancer Res. 1995, 55, 3369–3373. [Google Scholar]
- Moritz, D.; Wels, W.; Mattern, J.; Groner, B. Cytotoxic T Lymphocytes with a Grafted Recognition Specificity for ERBB2-Expressing Tumor Cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4318–4322. [Google Scholar] [CrossRef]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-Mediated Signalling Co-Stimulates Murine T Cells and Prevents Induction of Anergy in T-Cell Clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric Receptors Providing Both Primary and Costimulatory Signaling in T Cells from a Single Gene Product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [CrossRef]
- Valiullina, A.K.; Zmievskaya, E.A.; Ganeeva, I.A.; Zhuravleva, M.N.; Garanina, E.E.; Rizvanov, A.A.; Petukhov, A.V.; Bulatov, E.R. Evaluation of CAR-T Cells’ Cytotoxicity against Modified Solid Tumor Cell Lines. Biomedicines 2023, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-Specific T Cell Activation by Recombinant Immunoreceptors: CD3 Zeta Signaling and CD28 Costimulation Are Simultaneously Required for Efficient IL-2 Secretion and Can Be Integrated into One Combined CD28/CD3 Zeta Signaling Receptor Molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric Receptors with 4-1BB Signaling Capacity Provoke Potent Cytotoxicity against Acute Lymphoblastic Leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Poels, R.; Ruiter, R.; van de Donk, N.W.C.J.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-Tuned Chimeric Antigen Receptor-Engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric Antigen Receptor (CAR) T Cells Engineered with an Inducible Cytokine to Modulate the Tumor Stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef]
- Zhang, Q.; Nowak, I.; Vonderheid, E.C.; Rook, A.H.; Kadin, M.E.; Nowell, P.C.; Shaw, L.M.; Wasik, M.A. Activation of Jak/STAT Proteins Involved in Signal Transduction Pathway Mediated by Receptor for Interleukin 2 in Malignant T Lymphocytes Derived from Cutaneous Anaplastic Large T-Cell Lymphoma and Sezary Syndrome. Proc. Natl. Acad. Sci. USA 1996, 93, 9148–9153. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.-L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA Is Essential for the Survival of Long-Lived Bone Marrow Plasma Cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Anderson, K.C. Targeting B-Cell Maturation Antigen in Multiple Myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef]
- Ma, T.; Shi, J.; Xiao, Y.; Bian, T.; Wang, J.; Hui, L.; Wang, M.; Liu, H. Study on the Relationship Between the Expression of B Cell Mature Antigen and the Classification, Stage, and Prognostic Factors of Multiple Myeloma. Front. Immunol. 2021, 12, 724411. [Google Scholar] [CrossRef]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T Cells Expressing an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Multiple Myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-F.; Anderson, K.C.; Tai, Y.-T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-Cell Maturation Antigen Is a Promising Target for Adoptive T-Cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA Promote Human Multiple Myeloma Growth and Immunosuppression in the Bone Marrow Microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. γ-Secretase Inhibition Increases Efficacy of BCMA-Specific Chimeric Antigen Receptor T Cells in Multiple Myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef]
- Celgene. CRB-401 A Phase 1 Study of Bb2121 in BCMA-Expressing Multiple Myeloma; clinicaltrials.gov: Bethesda, MD, USA, 2023.
- Sanoyan, D.A.; Seipel, K.; Bacher, U.; Kronig, M.-N.; Porret, N.; Wiedemann, G.; Daskalakis, M.; Pabst, T. Real-Life Experiences with CAR T-Cell Therapy with Idecabtagene Vicleucel (Ide-Cel) for Triple-Class Exposed Relapsed/Refractory Multiple Myeloma Patients. BMC Cancer 2023, 23, 345. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Mailankody, S.; Jakubowiak, A.J.; Htut, M.; Costa, L.J.; Lee, K.; Ganguly, S.; Kaufman, J.L.; Siegel, D.S.D.; Bensinger, W.; Cota, M.; et al. Orvacabtagene Autoleucel (Orva-Cel), a B-Cell Maturation Antigen (BCMA)-Directed CAR T Cell Therapy for Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Update of the Phase 1/2 EVOLVE Study (NCT03430011). J. Clin. Oncol. 2020, 38, 8504. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Liu, J.; Wang, B.-Y.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B.; et al. A Phase 1, Open-Label Study of LCAR-B38M, a Chimeric Antigen Receptor T Cell Therapy Directed against B Cell Maturation Antigen, in Patients with Relapsed or Refractory Multiple Myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Wang, B.-Y.; Chen, L.-J.; Fu, W.-J.; Xu, J.; Liu, J.; Jin, S.-W.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; et al. Four-Year Follow-up of LCAR-B38M in Relapsed or Refractory Multiple Myeloma: A Phase 1, Single-Arm, Open-Label, Multicenter Study in China (LEGEND-2). J. Hematol. Oncol. 2022, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mahendravada, A.; Ballard, B.; Kale, B.; Ramos, C.; West, J.; Maguire, T.; McKay, K.; Lichtman, E.; Tuchman, S.; et al. Safety and Efficacy of Targeting CD138 with a Chimeric Antigen Receptor for the Treatment of Multiple Myeloma. Oncotarget 2019, 10, 2369–2383. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D Is a Target for the Immunotherapy of Multiple Myeloma with Rationally Designed CAR T Cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- Jiang, D.; Huang, H.; Qin, H.; Tang, K.; Shi, X.; Zhu, T.; Gao, Y.; Zhang, Y.; Tian, X.; Fu, J.; et al. Chimeric Antigen Receptor T Cells Targeting FcRH5 Provide Robust Tumour-Specific Responses in Murine Xenograft Models of Multiple Myeloma. Nat. Commun. 2023, 14, 3642. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, M.; Usmani, S.Z. GPRC5D: The Next Frontier for Immunotherapy in Multiple Myeloma. Hematologist 2023, 20. [Google Scholar] [CrossRef]
- Mailankody, S.; Devlin, S.M.; Landa, J.; Nath, K.; Diamonte, C.; Carstens, E.J.; Russo, D.; Auclair, R.; Fitzgerald, L.; Cadzin, B.; et al. GPRC5D-Targeted CAR T Cells for Myeloma. N. Engl. J. Med. 2022, 387, 1196–1206. [Google Scholar] [CrossRef]
- Bal, S.; Kocoglu, M.H.; Nadeem, O.; Htut, M.; Gregory, T.; Anderson, L.D., Jr.; Costa, L.J.; Buchholz, T.J.; Ziyad, S.; Li, M.; et al. Clinical Activity of BMS-986393 (CC-95266), a G Protein-Coupled Receptor Class C Group 5 Member D (GPRC5D)-Targeted Chimeric Antigen Receptor (CAR) T Cell Therapy, in Patients with Relapsed and/or Refractory (R/R) Multiple Myeloma (MM): First Results from a Phase 1, Multicenter, Open-Label Study. Blood 2022, 140, 883–885. [Google Scholar] [CrossRef]
- Xia, J.; Li, H.; Yan, Z.; Zhou, D.; Wang, Y.; Qi, Y.; Cao, J.; Li, D.; Cheng, H.; Sang, W.; et al. Anti–G Protein–Coupled Receptor, Class C Group 5 Member D Chimeric Antigen Receptor T Cells in Patients with Relapsed or Refractory Multiple Myeloma: A Single-Arm, Phase Ⅱ Trial. J. Clin. Oncol. 2023, 41, 2583–2593. [Google Scholar] [CrossRef]
- Zhang, M.; Wei, G.; Zhou, L.; Zhou, J.; Chen, S.; Zhang, W.; Wang, D.; Luo, X.; Cui, J.; Huang, S.; et al. GPRC5D CAR T Cells (OriCAR-017) in Patients with Relapsed or Refractory Multiple Myeloma (POLARIS): A First-in-Human, Single-Centre, Single-Arm, Phase 1 Trial. Lancet Haematol. 2023, 10, e107–e116. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Safety and Efficacy of Anti-BCMA/GPRC5D CAR-T Cell Therapy in Treating Relapsed and Refractory Multiple Myeloma(Rr/MM). Available online: https://clinicaltrials.gov/ct2/show/NCT05509530 (accessed on 26 June 2023).
- ClinicalTrials.Gov. A Study of CAR-T Cells Targeting Both BCMA and GPRC5D in Treatment of Relapsed or Refractory Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT05325801 (accessed on 26 June 2023).
- ClinicalTrials.Gov. A Study of MCARH109 and MCARH125 in People with Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT05431608 (accessed on 26 June 2023).
- Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L. SLAM Family Receptors and SAP Adaptors in Immunity. Annu. Rev. Immunol. 2011, 29, 665–705. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Granell, M.; Oriol, A.; Martinez-Lopez, J.; Blade, J.; Hernandez, M.T.; Martín, J.; Gironella, M.; Lynch, M.; Bleickardt, E.; et al. Elotuzumab in Combination with Thalidomide and Low-Dose Dexamethasone: A Phase 2 Single-Arm Safety Study in Patients with Relapsed/Refractory Multiple Myeloma. Br. J. Haematol. 2016, 175, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T Cells Eliminate Myeloma and Confer Selective Fratricide of SLAMF7+ Normal Lymphocytes. Blood 2017, 130, 2838–2847. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-Specific Chimeric Antigen Receptor (CAR)-Engineered Natural Killer Cells Enhance in Vitro and in Vivo Antitumor Activity against Human Multiple Myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2021, 29, 702–717. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Off-the-Shelf CAR T Cells for Multiple Myeloma. Nat. Med. 2023, 29, 303–304. [Google Scholar] [CrossRef]
- Korst, C.L.B.M.; Bruins, W.S.C.; Cosovic, M.; Verkleij, C.P.M.; Twickler, I.; Le Clerre, D.; Chion-Sotinel, I.; Zweegman, S.; Galetto, R.; Mutis, T.; et al. Preclinical Activity of Allogeneic CS1-Specific CAR T-Cells (UCARTCS1) in Multiple Myeloma. Blood 2022, 140, 4215–4216. [Google Scholar] [CrossRef]
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically Optimized BCMA/CS1 Bispecific CAR-T Cells Robustly Control Heterogeneous Multiple Myeloma. Nat. Commun. 2020, 11, 2283. [Google Scholar] [CrossRef]
- Prommersberger, S.; Reiser, M.; Beckmann, J.; Danhof, S.; Amberger, M.; Quade-Lyssy, P.; Einsele, H.; Hudecek, M.; Bonig, H.; Ivics, Z. CARAMBA: A First-in-Human Clinical Trial with SLAMF7 CAR-T Cells Prepared by Virus-Free Sleeping Beauty Gene Transfer to Treat Multiple Myeloma. Gene Ther. 2021, 28, 560–571. [Google Scholar] [CrossRef]
- Leleu, X.; Martin, T.; Weisel, K.; Schjesvold, F.; Iida, S.; Malavasi, F.; Manier, S.; Min, C.-K.; Ocio, E.M.; Pawlyn, C.; et al. Anti-CD38 Antibody Therapy for Patients with Relapsed/Refractory Multiple Myeloma: Differential Mechanisms of Action and Recent Clinical Trial Outcomes. Ann. Hematol. 2022, 101, 2123–2137. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Nahi, H.; Plesner, T.; Weiss, B.M.; Bahlis, N.J.; Belch, A.; Voorhees, P.M.; Laubach, J.P.; van de Donk, N.W.; Ahmadi, T.; et al. Daratumumab Monotherapy in Patients with Heavily Pretreated Relapsed or Refractory Multiple Myeloma: Final Results from the Phase 2 GEN501 and SIRIUS Trials. Lancet Haematol. 2020, 76, e447–e455. Available online: https://pubmed.ncbi.nlm.nih.gov/32470437/ (accessed on 13 October 2023). [CrossRef]
- Ghosh, A.; Mailankody, S.; Giralt, S.A.; Landgren, C.O.; Smith, E.L.; Brentjens, R.J. CAR T Cell Therapy for Multiple Myeloma: Where Are We Now and Where Are We Headed? Leuk. Lymphoma 2018, 59, 2056–2067. [Google Scholar] [CrossRef]
- Drent, E.; Groen, R.; Noort, W.; van Bueren, J.L.; Parren, P.; Kuball, J.; Sebestyen, Z.; van de Donk, N.; Martens, A.; Lokhorst, H.; et al. Preclinical Evaluation of CD38 Chimeric Antigen Receptor Engineered T Cells for the Treatment of Multiple Myeloma. J. ImmunoTherapy Cancer 2015, 3, P13. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Study to Evaluate the Safety and Efficacy of Anti-CD38 CAR-T in Relapsed or Refractory Multiple Myeloma Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03464916 (accessed on 5 July 2023).
- Feng, Y.; Liu, X.; Li, X.; Zhou, Y.; Song, Z.; Zhang, J.; Shi, B.; Wang, J. Novel BCMA-OR-CD38 Tandem-Dual Chimeric Antigen Receptor T Cells Robustly Control Multiple Myeloma. OncoImmunology 2021, 10, 1959102. [Google Scholar] [CrossRef]
- Tang, Y.; Yin, H.; Zhao, X.; Jin, D.; Liang, Y.; Xiong, T.; Li, L.; Tang, W.; Zhang, J.; Liu, M.; et al. High Efficacy and Safety of CD38 and BCMA Bispecific CAR-T in Relapsed or Refractory Multiple Myeloma. J. Exp. Clin. Cancer Res. 2022, 41, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, M.; Xiao, X.; Lv, H.; Jiang, Y.; Li, X.; Yuan, T.; Zhao, M. A Combination of Humanized Anti-BCMA and Murine Anti-CD38 CAR-T Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma. Leuk. Lymphoma 2022, 63, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.W.C.J.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Timmers, M.; Roex, G.; Wang, Y.; Campillo-Davo, D.; Van Tendeloo, V.F.I.; Chu, Y.; Berneman, Z.N.; Luo, F.; Van Acker, H.H.; Anguille, S. Chimeric Antigen Receptor-Modified T Cell Therapy in Multiple Myeloma: Beyond B Cell Maturation Antigen. Front. Immunol. 2019, 10, 1613. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Stadtmauer, E.A.; Hwang, W.-T.; Lacey, S.F.; Melenhorst, J.J.; Krevvata, M.; Carroll, M.P.; Matsui, W.H.; Wang, Q.; Dhodapkar, M.V.; et al. Anti-CD19 CAR T Cells with High-Dose Melphalan and Autologous Stem Cell Transplantation for Refractory Multiple Myeloma. JCI Insight 2018, 3, e120505. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, J.; Gu, W.; Shi, M.; Lan, J.; Yan, Z.; Jin, L.; Xia, J.; Ma, S.; Liu, Y.; et al. Long-Term Follow-Up of Combination of B-Cell Maturation Antigen and CD19 Chimeric Antigen Receptor T Cells in Multiple Myeloma. J. Clin. Oncol. 2022, 40, 2246–2256. [Google Scholar] [CrossRef]
- Shi, X.; Yan, L.; Shang, J.; Kang, L.; Yan, Z.; Jin, S.; Zhu, M.; Chang, H.; Gong, F.; Zhou, J.; et al. Anti-CD19 and Anti-BCMA CAR T Cell Therapy Followed by Lenalidomide Maintenance after Autologous Stem-Cell Transplantation for High-Risk Newly Diagnosed Multiple Myeloma. Am. J. Hematol. 2022, 97, 537–547. [Google Scholar] [CrossRef]
- Gouard, S.; Pallardy, A.; Gaschet, J.; Faivre-Chauvet, A.; Bruchertseifer, F.; Morgenstern, A.; Maurel, C.; Matous, E.; Kraeber-Bodéré, F.; Davodeau, F.; et al. Comparative Analysis of Multiple Myeloma Treatment by CD138 Antigen Targeting with Bismuth-213 and Melphalan Chemotherapy. Nucl. Med. Biol. 2014, 41, e30–e35. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Treatment of Chemotherapy Refractory Multiple Myeloma by CART-138. Available online: https://clinicaltrials.gov/ct2/show/NCT01886976 (accessed on 5 July 2023).
- Guo, B.; Chen, M.; Han, Q.; Hui, F.; Dai, H.; Zhang, W.; Zhang, Y.; Wang, Y.; Zhu, H.; Han, W. CD138-Directed Adoptive Immunotherapy of Chimeric Antigen Receptor (CAR)-Modified T Cells for Multiple Myeloma. J. Cell. Immunother. 2016, 2, 28–35. [Google Scholar] [CrossRef]
- Leivas, A.; Valeri, A.; Córdoba, L.; García-Ortiz, A.; Ortiz, A.; Sánchez-Vega, L.; Graña-Castro, O.; Fernández, L.; Carreño-Tarragona, G.; Pérez, M.; et al. NKG2D-CAR-Transduced Natural Killer Cells Efficiently Target Multiple Myeloma. Blood Cancer J. 2021, 11, 146. [Google Scholar] [CrossRef]
- Spear, P.; Barber, A.; Rynda-Apple, A.; Sentman, C.L. NKG2D CAR T-Cell Therapy Inhibits the Growth of NKG2D Ligand Heterogeneous Tumors. Immunol. Cell Biol. 2013, 91, 435–440. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase 1 Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1– and CTLA-4–Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T Cells Expressing a Mesothelin—Specific Chimeric Antibody Receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book. 2019, 39, 433–444. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine Release Syndrome and Associated Neurotoxicity in Cancer Immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef]
- Pehlivan, K.C.; Duncan, B.B.; Lee, D.W. CAR-T Cell Therapy for Acute Lymphoblastic Leukemia: Transforming the Treatment of Relapsed and Refractory Disease. Curr. Hematol. Malig. Rep. 2018, 13, 396–406. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Czajka-Francuz, P.; Prendes, M.J.; Mankan, A.; Quintana, Á.; Pabla, S.; Ramkissoon, S.; Jensen, T.J.; Peiró, S.; Severson, E.A.; Achyut, B.R.; et al. Mechanisms of Immune Modulation in the Tumor Microenvironment and Implications for Targeted Therapy. Front. Oncol. 2023, 13, 1200646. [Google Scholar] [CrossRef]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Cipkar, C.; Chen, C.; Trudel, S. Antibodies and Bispecifics for Multiple Myeloma: Effective Effector Therapy. Hematol. Am. Soc. Hematol. Educ. Program. 2022, 2022, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Li, C.; Jiang, H.; Zhao, X.; Huang, Z.; Jin, D.; Guo, T.; Kou, H.; Liu, L.; Tang, L.; et al. A Bispecific CAR-T Cell Therapy Targeting BCMA and CD38 in Relapsed or Refractory Multiple Myeloma. J. Hematol. Oncol. 2021, 14, 161. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| (A) BCMA Targeted CAR-T for MM | |||||
| Clinical Trial Name | Antigen Target | CAR Name | Sponsor | Phase | Current Status |
| NCT05393804 | BCMA | Idecabtagene Vicleucel (ide-cel, bb2121) | Memorial Sloan Kettering Cancer Cente | 2 | Recruiting |
| NCT05347485 | BCMA | Ciltacabtagene Autoleucel | Janssen Scientific Affairs, LLC | 2 | Recruiting |
| NCT04181827 | BCMA | JCARH125 | Janseen Research & Development | 3 | Active/Non-recruiting |
| NCT03090659 | BCMA | LEGEND-2 | Nanjing Legend Biotech Co. Second Affiliated Hospital of Xi’an Jiaotong University Ruijin Hospital Jiangsu Provincial People’s Hospital Shanghai Changzheng Hospital | 1/2 | Active, not recruiting |
| NCT03548207 | BCMA | CARTITUDE-1 | Janssen Research & Development, LLC | 1/2 | Completed |
| NCT02954445 | BCMA | NA | Shiqi Li, Southwest Hospital, China (Responsible Party) Southwest Hospital, China | 1/2 | Unknown |
| NCT05860036 | BCMA | NA | Institute of Hematology & Blood Diseases Hospital Gang An, Institute of Hematology & Blood Diseases Hospital (Responsible Party) | 1 | Recruiting |
| NCT05740891 | BCMA | NA | Zhejiang University He Huang, Zhejiang University | 1 | Recruiting |
| NCT03716856 | BCMA | NA | First Affiliated Hospital of Zhejiang University | 1 | Unknown |
| NCT05594797 | BCMA | NA | Hrain Biotechnology Co., Ltd. | 2 | Recruiting |
| NCT04003168 | BCMA | NA | Hrain Biotechnology Co., Ltd. | 1 | Recruiting |
| NCT03338972 | BCMA | NA | Fred Hutchinson Cancer Center | 1 | Completed |
| NCT04727008 | BCMA | CXCR4 | Sichuan University Ting Niu, West China Hospital | Early Phase 1 | Not Yet Recruiting |
| NCT05698303 | BCMA | NA | Nanjing IASO Biotherapeutics Co., Ltd. | 1 | Not Yet Recruiting |
| NCT04637269 | Xinqiao Hospital of Chongqing Xi Zhang, MD, Xinqiao Hospital of Chongqing | Early Phase 1 | Recruiting | ||
| NCT04322292 | BCMA | C-CAR088 | Institute of Hematology & Blood Diseases Hospital AnGang, Institute of Hematology & Blood Diseases Hospital | 1 | Unknown |
| (B) Non-BCMA Targeted CAR-T for MM | |||||
| Clinical Trial Name | Antigen Target | CAR Name | Sponsor | Phase | Current Status |
| NCT04674813 | GPRC5D | CC-95266 | Juno Therapeutics, a Subsidiary of Celgene | 1 | Active, Recruiting |
| NCT05016778 | GPRC5D | POLARIS | Zhejiang University & OriCell Therapeutics Co., Ltd. | Early Phase 1 | Active, not recruiting |
| NCT05431608 | GPRC5D | NA | Memorial Sloan Kettering Cancer Center | 1 | Active, Recruiting |
| NCT05325801 | GPRC5D & BCMA | OriC321 | Zhejiang University | 1 | Recruiting |
| NCT05509530 | GPRC5D & BCMA | NA | Xuzhou Medical University | 2 | Recruiting |
| NCT03958656 | SLAMF7 | NA | National Cancer Institute (NCI) | 1 | Completed |
| NCT03710421 | CS1 | NA | City of Hope Medical Center | 1 | Active, Recruiting |
| NCT04499339 | SLAMF7 | CARAMBA-1 | European Commission and Wuerzburg University Hospital | 1/2 | Active, Recruiting |
| NCT04142619 | SLAMF7 | UCARTCS1 | Cellectis S.A | 1 | Active, Recruiting |
| NCT03464916 | CD38 | CAR2 Anti-CD38 A2 | Sorrento Therapeutics, Inc. | 1 | Completed |
| NCT03767751 | CD38 | NA | Chinese PLA General Hospital | 1/2 | Unknown |
| NCT02135406 | CD19 | CTL019/tisagenlecleucel | University of Pennsylvania | 1 | Completed |
| NCT03672318 | CD138 | ATLCAR.CD138 | UNC Lineberger Comprehensive Cancer Center | 1 | Active, Recruiting |
| NCT01886976 | CD138 | CART-138 | Chinese PLA General Hospital | 1/2 | Unknown |
| NCT04182581 | BCMA/CD19 | BCMA/CD19 Dual-Target | Xijing Hospital | Early Phase1 | Unknown |
| NCT03706547 | CD19 | NA | Peng Liu Peng Liu, Shanghai Zhongshan Hospital | 1 | Unknown |
| NCT04714827 | CD19 | CD19-CD22 | Shanxi Province Cancer Hospital | 1/2 | Recruiting |
| NCT04541368 | SLAMF7 | CS1 | Zhejiang University He Huang, Zhejiang University | Early Phase 1 | Not Yet Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, A.K.; Gupta, A.; Dagar, G.; Das, D.; Chakraborty, A.; Haque, S.; Prasad, C.P.; Singh, A.; Bhat, A.A.; Macha, M.A.; et al. CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond. Vaccines 2023, 11, 1721. https://doi.org/10.3390/vaccines11111721
Mishra AK, Gupta A, Dagar G, Das D, Chakraborty A, Haque S, Prasad CP, Singh A, Bhat AA, Macha MA, et al. CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond. Vaccines. 2023; 11(11):1721. https://doi.org/10.3390/vaccines11111721
Chicago/Turabian StyleMishra, Abhinava K., Ashna Gupta, Gunjan Dagar, Dayasagar Das, Abhijit Chakraborty, Shabirul Haque, Chandra Prakash Prasad, Archana Singh, Ajaz A. Bhat, Muzafar A. Macha, and et al. 2023. "CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond" Vaccines 11, no. 11: 1721. https://doi.org/10.3390/vaccines11111721