
CAR T-Cell Immunotherapy Treating T-ALL: Challenges and Opportunities

Abstract
:1. Introduction
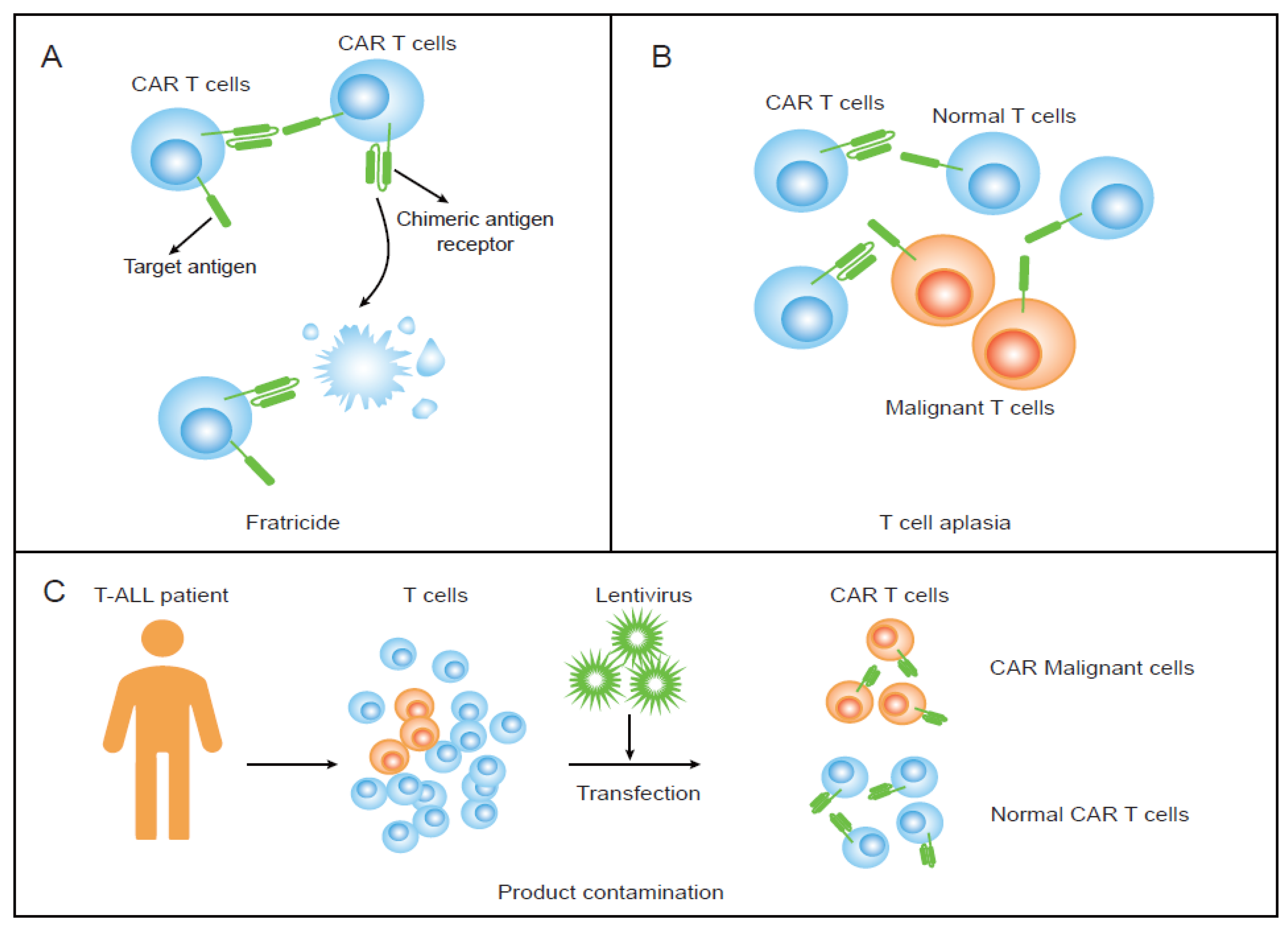
2. Fratricide and Promising Strategies
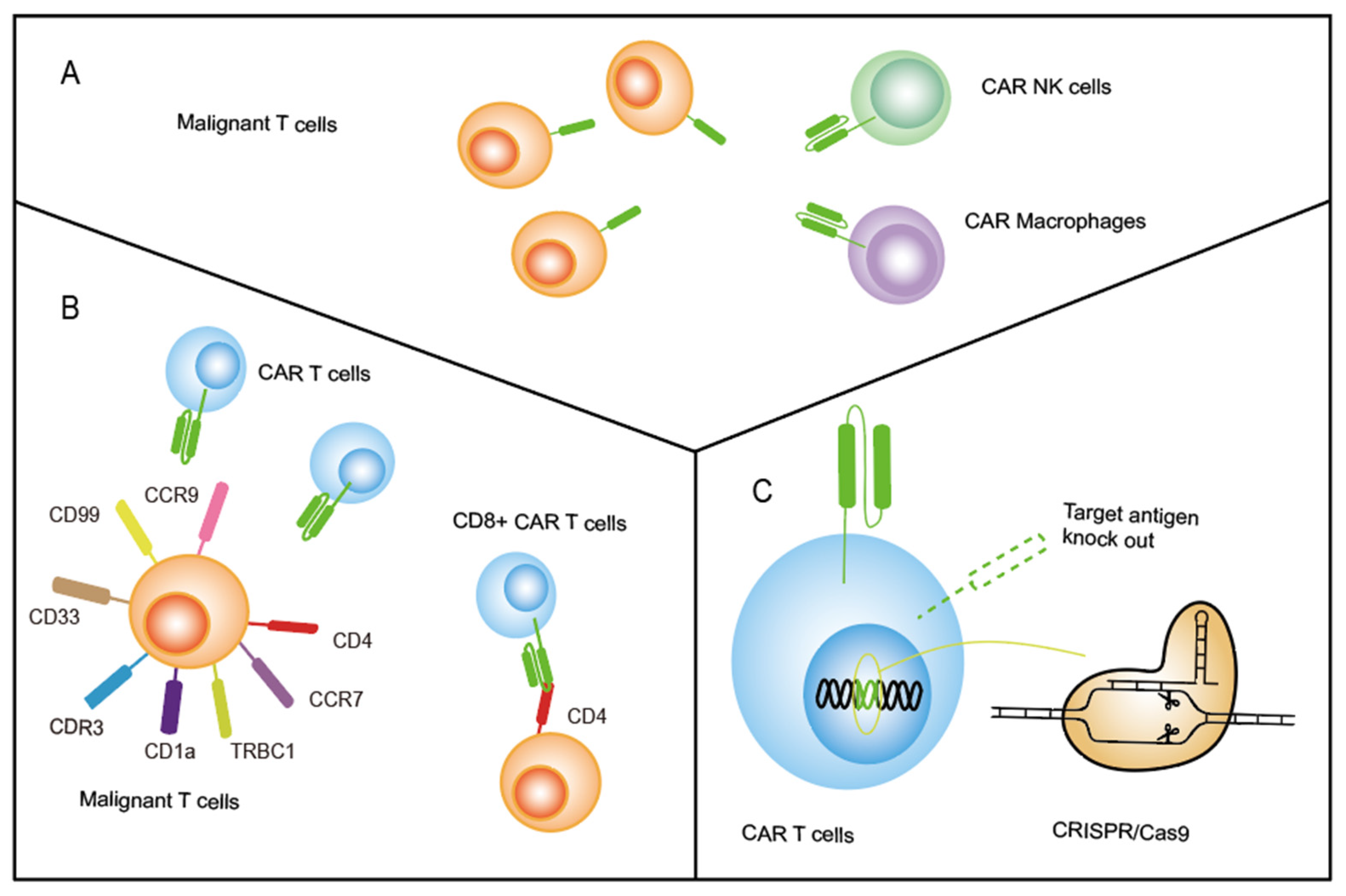
2.1. Transduce CAR beyond T Cells
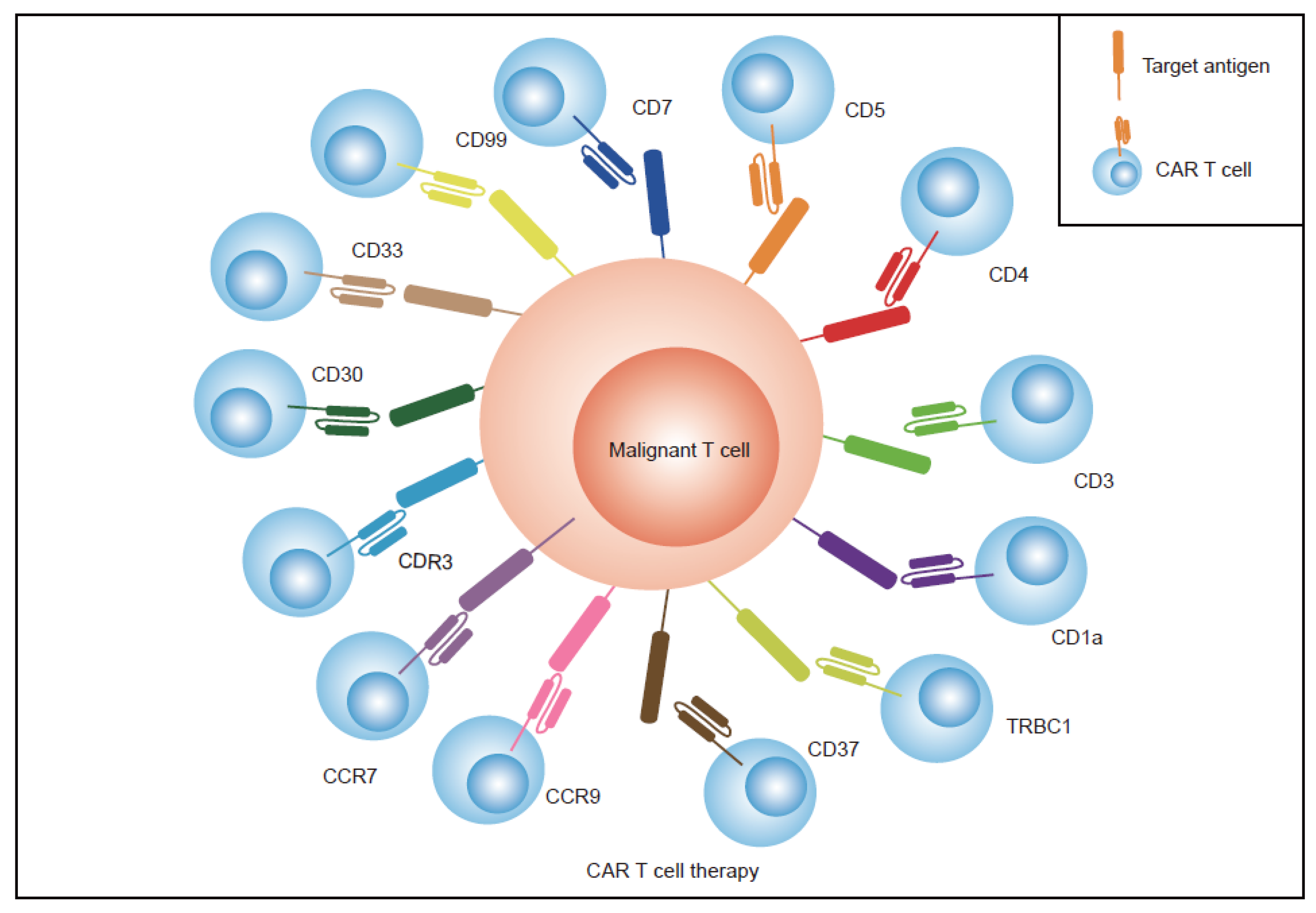
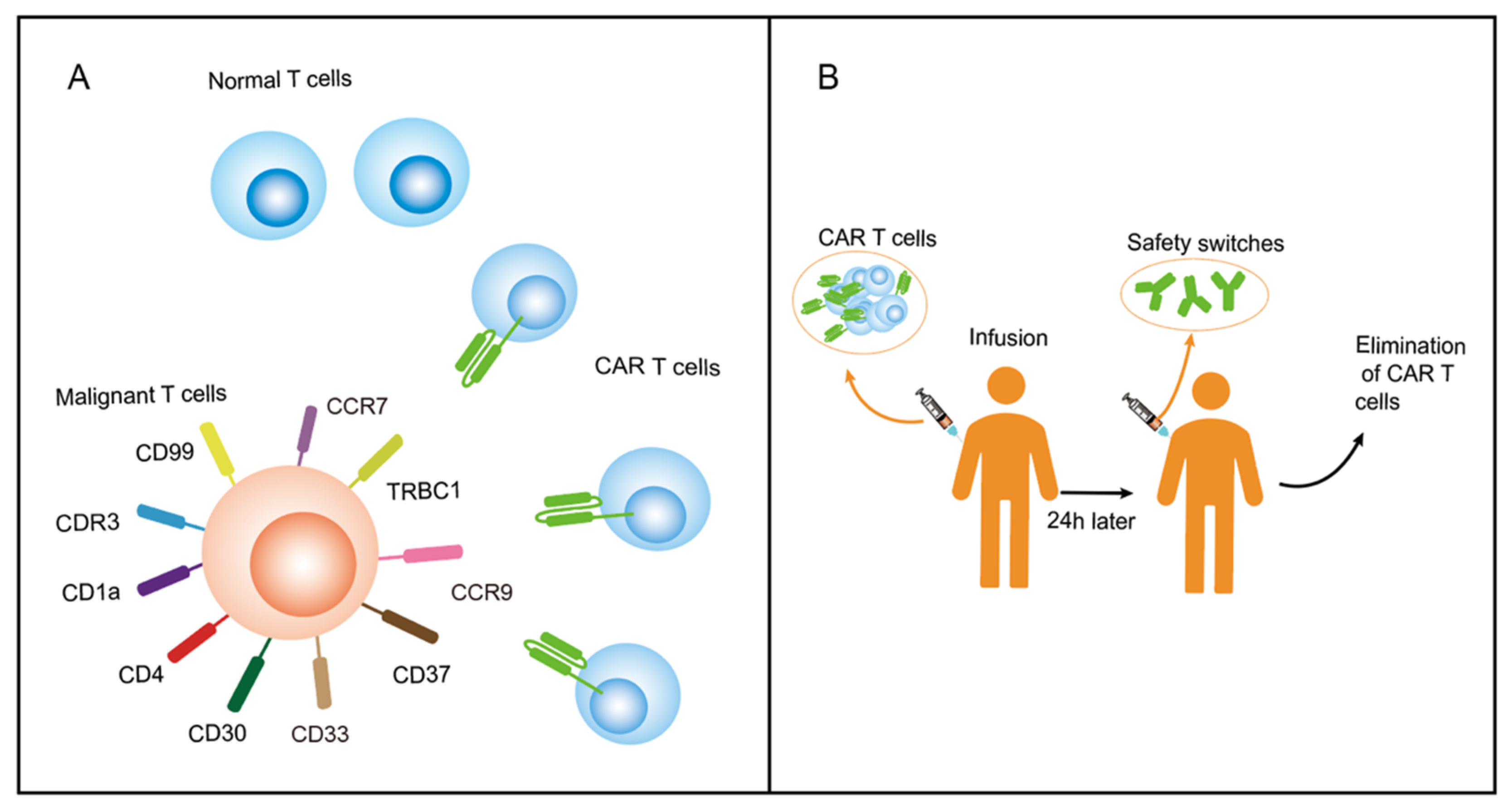
2.2. Find Specific Antigens Restricted Expression on T Cells
2.3. Knock-Out Pan-T-Cell Targeting Antigens on CAR T Cells by CRISPR-Cas9
3. T-Cell Aplasia and Proposed Solutions
4. Product Contamination and Potential Strategies
5. Discussion
6. Prospective Research Areas
- (1)
- Transducing CAR on other types of immune cells beyond T cells. NK cells, the NK-92 cell line, γδT cells, and macrophages have been used as CAR-transduced effector cells instead of T cells to treat tumors, and several clinical trials are ongoing to evaluate the effect in a variety of tumor types (NCT05007379, NCT04660929, NCT04702841). We believe that more therapy focused on applying γδT cells, macrophages, and induced pluripotent stem (iPS) cell-derived NK cells should be initiated to treat T-ALL;
- (2)
- Identifying more specific antigens limitedly expressed on normal T cells. TRBC1/2, CDR3, CD1a, CCR7, CCR9, CD33, CD30, and CD99 have been identified and tested as targets for CAR T therapy in the treatment of T-cell malignancies. These antigens have limited expression on normal T cells or only express one subtype of tumor T cells, which allows CAR T cells and normal T cells to proliferate normally. Previous research has largely focused on single antigen CAR T therapy in T-cell malignancies; however, dual antigens are superior and thus this area requires further investigation;
- (3)
- Optimizing cell editing technology controlling the antigens expressed on CAR T. TALEN and CRISPR/Cas9 have been used to knock out the target antigen expressed on CAR T cells, preventing CAR T-cell fratricide. Diorio et al. created cytosine base editor (CBE) technology, which was applied to create CD7-directed allogeneic CAR T using four simultaneous base edits [80]. This study suggests a promising potential method for genome editing of antigens in cellular products without the unpredictable and undesirable outcomes associated with CRISPR-cas9. Thus, the CBE technology could be used to knock out other antigens, and more optimized gene-editing techniques can be created to inhibit CAR T-cell fratricide in the future;
- (4)
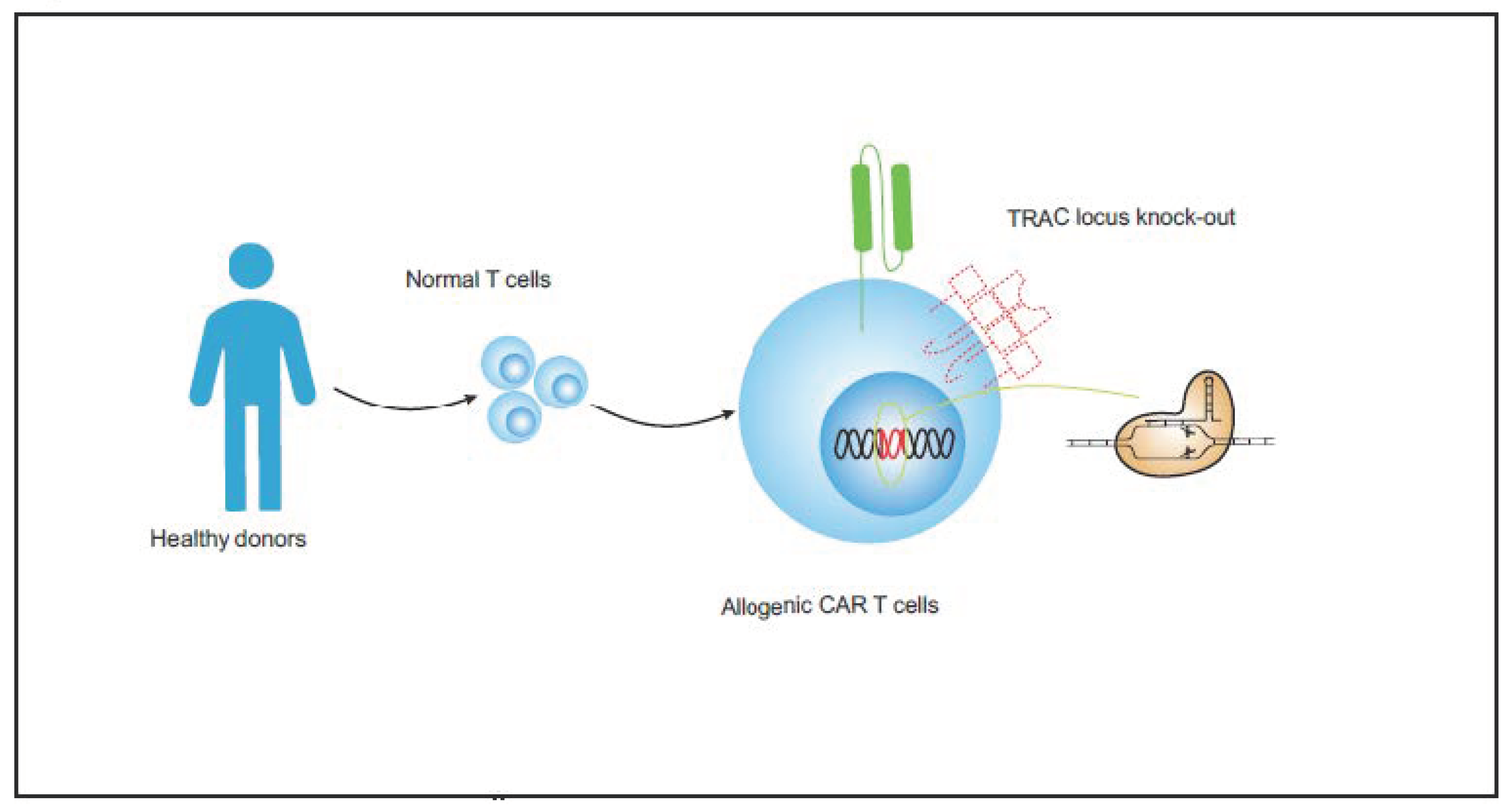
- Updating and developing the universal CAR T-cell therapy for T-cell malignancies. Allogeneic CAR T cells from healthy donors could help to prevent problems associated with poor CAR T-cell quality and product contamination. The combination of UCAR T cell and gene-editing techniques could represent a mature biological product and become the trend of future T-ALL treatments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Target | Clinical Trials | Eligible Disease | Solved Problems | References |
|---|---|---|---|---|---|
| CD7 CAR-T CD7 CAR-NK | CD7 | NCT03690011 NCT04934774 NCT04840875 NCT05059912 NCT04599556 NCT04689659 NCT04762485 NCT02742727 ChiCTR190002531 ChiCTR2000034762 ChiCTR2000038714 ChiCTR2000040641 ChiCTR2100042807 ChiCTR2100043252 ChiCTR2100045863 | T-ALL CD7+ PTCL | Png, Y.T. et al., 2017 [41], You, Y. et al., 2019 [42], Gomes-Silva, D. et al., 2017 [50], Zhang, M. et al., 2021 [81] | |
| CD5 CAR-T CD5 CAR-NK | CD5 | NCT03081910 NCT04594135 NCT05032599 ChiCTR2000039519 | CD5+ T-ALL CD5+ PTCL | Chen, K.H. et al., 2017 [39], Raikar, S.S. et al., 2018 [43] | |
| CD4 CAR-T CD4 CAR-NK | CD4 | NCT03829540 NCT04162340 NCT04712864 ChiCTR2100042782 | CD4+ T-ALL CD4+ PTCL | Fratricide | Pinz, K. et al., 2017 [52], Ma, G. et al., 2019 [84] |
| CD3 CAR-NK | CD3 | N/A | T-ALL | Chen et al., 2016 [47], Rasaiyaah, J. et al., 2018 [74] | |
| CD1a CAR-T | CD1a | N/A | Cortical T-ALL | Fratricide T-cell aplasia | Sánchez-Martínez, D. et al., 2019 [86] |
| AUTO (an autologous CAR-T product) | TRBC1 | NCT03590574 EudraCT2017-001965-26 | TRBC1+ T-NHL | Fratricide T-cell aplasia | Maciocia, P.M. et al., 2017 [65] |
| CD37 CAR-T | CD37 | N/A | CD37+ PTCL | Scarfò et al., 2018 [118] | |
| CDR3 CAR-T | CDR3 | N/A | T-cell leukemia/lymphoma | Fratricide T-cell aplasia | Huang, J. et al., 2019 [46] |
| CD30 CAR-T | CD30 | NCT01979536 NCT02729961 NCT02917083 NCT03383965 NCT04008394 NCT01316146 NCT01192464 NCT03049449 NCT02690545 NCT02958410 NCT02663297 NCT02259556 NCT03602157 ChiCTR-OPN-16009069 ChiCTR2000030843 ChiCTR2100046763 EudraCT2019-001263-70 | ALCL ALCL CD30+ PTCL ALCL CD30+ PTCL | Fratricide T-cell aplasia | Zhang, S. et al., 2022 [72], Zheng, W. et al., 2014 [87], Ramos, C.A. et al., 2020 [88] |
| CD99 CAR-T | CD99 | ChiCTR2000033989 ChiCTR2100046764 | T-ALL | Fratricide T-cell aplasia | Shi, J. et al., 2021 [59] |
| Therapy | Target | Clinical Trials | Eligible Disease | Solved Problems |
|---|---|---|---|---|
| CD7 U-CAR-T CD7 Allogeneic CAR-T | CD7 | NCT04264078 NCT05377827 NCT04984356 NCT05509855 NCT05127135 | CD7+ T-ALL T-cell leukemia/lymphoma | Product contamination |
| CD5 Allogeneic CAR-T | CD5 | NCT03081910 | CD5+ T-ALL CD5+ T-NHLs | Product contamination |
| CD30 Allogeneic CAR-T | CD30 | NCT04288726 NCT04952584 | CD30+ T-cell lymphoma | Product contamination |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Schmitz, N.; Lenz, G.; Stelljes, M. Allogeneic hematopoietic stem cell transplantation for T-cell lymphomas. Blood 2018, 132, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, A.J.; Lunning, M.A.; Horwitz, S.M. How I treat the peripheral T-cell lymphomas. Blood 2014, 123, 2636–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murawski, N.; Pfreundschuh, M. New drugs for aggressive B-cell and T-cell lymphomas. Lancet Oncol. 2010, 11, 1074–1085. [Google Scholar] [CrossRef]
- Mahadevan, D.; Fisher, R. Novel Therapeutics for Aggressive Non-Hodgkin’s Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 1876–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlevi, C.; Matsuki, E.; Brentjens, R.; Younes, A. Novel immunotherapies in lymphoid malignancies. Nat. Rev. Clin. Oncol. 2015, 13, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.-L. Targeted therapy in T-cell malignancies: Dysregulation of the cellular signaling pathways. Leukemia 2010, 24, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [Green Version]
- Sentís, I.; Gonzalez, S.; Genescà, E.; García-Hernández, V.; Muiños, F.; Gonzalez, C.; López-Arribillaga, E.; Gonzalez, J.; Fernandez-Ibarrondo, L.; Mularoni, L. The evolution of relapse of adult T cell acute lymphoblastic leukemia. Genome Biol. 2020, 21, 284. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2017, 15, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; Roth, S.; Kong, J.; Guerra, G.; Narasimhan, V.; Pereira, L.; Desai, J.; Heriot, A.; Ramsay, R. An Update on Immunotherapy for Solid Tumors: A Review. Ann. Surg. Oncol. 2018, 25, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Choi, S.H.; Shah, K. Multifunctional receptor-targeting antibodies for cancer therapy. Lancet Oncol. 2015, 16, e543–e554. [Google Scholar] [CrossRef]
- Hixon, J.A.; Andrews, C.; Kashi, L.; Kohnhorst, C.L.; Senkevitch, E.; Czarra, K.; Barata, J.T.; Li, W.; Schneider, J.P.; Walsh, S.T.R. New anti-IL-7Ralpha monoclonal antibodies show efficacy against T cell acute lymphoblastic leukemia in pre-clinical models. Leukemia 2020, 34, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Akkapeddi, P.; Fragoso, R.; Hixon, J.A.; Ramalho, A.S.; Oliveira, M.L.; Carvalho, T.; Gloger, A.; Matasci, M.; Corzana, F.; Durum, S.K. A fully human anti-IL-7Ralpha antibody promotes antitumor activity against T-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 2155–2168. [Google Scholar] [CrossRef]
- Murthy, G.S.G.; Kearl, T.J.; Cui, W.; Johnson, B.; Hoffmeister, K.; Szabo, A.; Aoki, K.; Harrington, A.M.; Carlson, K.; Michaelis, L.C. A Phase 1 Study of XmAb18968, a CD3–CD38 Bispecific Antibody for the Treatment of Patients with Relapsed/Refractory Acute Leukemia and T Cell Lymphoblastic Lymphoma. Blood 2021, 138 (Suppl. S1), 4401. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [Green Version]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef]
- Mumprecht, S.; Schürch, C.; Schwaller, J.; Solenthaler, M.; Ochsenbein, A.F. Programmed death 1 signaling on chronic myeloid leukemia–specific T cells results in T-cell exhaustion and disease progression. Blood 2009, 114, 1528–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wartewig, T.; Ruland, J. PD-1 Tumor Suppressor Signaling in T Cell Lymphomas. Trends Immunol. 2019, 40, 403–414. [Google Scholar] [CrossRef]
- Chen, X.; Liu, S.; Wang, L.; Zhang, W.; Ji, Y.; Ma, X. Clinical significance of B7-H1 (PD-L1) expression in human acute leukemia. Cancer Biol. Ther. 2008, 7, 622–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, L.; Waldmann, T.A.; Janakiram, M.; Brammer, J.E. Rapid Progression of Adult T-Cell Leukemia–Lymphoma after PD-1 Inhibitor Therapy. N. Engl. J. Med. 2018, 378, 1947–1948. [Google Scholar] [CrossRef] [PubMed]
- Wartewig, T.; Kurgyis, Z.; Keppler, S.; Pechloff, K.; Hameister, E.; Öllinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 2017, 552, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. FDA approves fourth CAR-T cell therapy. Nat. Rev. Drug Discov. 2021, 20, 166. [Google Scholar] [CrossRef]
- Cao, J.; Wang, G.; Cheng, H.; Wei, C.; Qi, K.; Sang, W.; Zhenyu, L.; Shi, M.; Li, H.; Qiao, J. Potent anti-leukemia activities of humanized CD19-targeted Chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am. J. Hematol. 2018, 93, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Voelker, R. CAR-T Therapy Is Approved for Mantle Cell Lymphoma. JAMA 2020, 324, 832. [Google Scholar] [CrossRef] [PubMed]
- Kozani, P.S.; Kozani, P.S.; Rahbarizadeh, F. CAR-T cell therapy in T-cell malignancies: Is success a low-hanging fruit? Stem Cell Res. Ther. 2021, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, M.; Tesio, M.; June, C.H.; Houot, R. CAR T-cells for T-cell malignancies: Challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia 2018, 32, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D. Persistence and Efficacy of Second Generation CAR T Cell Against the LeY Antigen in Acute Myeloid Leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Suck, G.; Odendahl, M.; Nowakowska, P.; Seidl, C.; Wels, W.S.; Klingemann, H.G.; Tonn, T. NK-92: An ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. [Google Scholar] [CrossRef]
- Xia, J.; Minamino, S.; Kuwabara, K. CAR-expressing NK cells for cancer therapy: A new hope. Biosci. Trends 2020, 14, 354–359. [Google Scholar] [CrossRef]
- Yi, Y.; Chai, X.; Zheng, L.; Zhang, Y.; Shen, J.; Hu, B.; Tao, G. CRISPR-edited CART with GM-CSF knockout and auto secretion of IL6 and IL1 blockers in patients with hematologic malignancy. Cell Discov. 2021, 7, 27. [Google Scholar] [CrossRef]
- Rabinowich, H.; Pricop, L.; Herberman, R.B.; Whiteside, T.L. Expression and function of CD7 molecule on human natural killer cells. J. Immunol. 1994, 152, 517–526. [Google Scholar] [CrossRef]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Lin, K.W.; Jares, A.; Firor, A.E.; Shuai, X.; Salman, H.; Golightly, M. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia 2017, 31, 2151–2160. [Google Scholar] [CrossRef] [Green Version]
- Dalloul, A. CD5: A safeguard against autoimmunity and a shield for cancer cells. Autoimmun. Rev. 2009, 8, 349–353. [Google Scholar] [CrossRef]
- Png, Y.T.; Vinanica, N.; Kamiya, T.; Shimasaki, N.; Coustan-Smith, E.; Campana, D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017, 1, 2348–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, F.; Wang, Y.; Jiang, L.; Zhu, X.; Chen, D.; Yuan, L.; An, G.; Meng, H.; Yang, L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 2019, 9, 64–78. [Google Scholar] [PubMed]
- Raikar, S.S.; Fleischer, L.C.; Moot, R.; Fedanov, A.; Paik, N.Y.; Knight, K.A.; Doering, C.B.; Spencer, H.T. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology 2017, 7, e1407898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2019, 472, 175–180. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. Ebiomedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Huang, J.; Alexey, S.; Li, J.; Jones, T.; Grande, G.; Douthit, L.; Xie, J.; Chen, D.; Wu, X.; Michael, M. Unique CDR3 epitope targeting by CAR-T cells is a viable approach for treating T-cell malignancies. Leukemia 2019, 33, 2315–2319. [Google Scholar] [CrossRef]
- Chen, K.H.; Wada, M.; Firor, A.; Pinz, K.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219–56232. [Google Scholar] [CrossRef] [Green Version]
- Voynova, E.; Hawk, N.; Flomerfelt, F.A.; Telford, W.G.; Gress, R.E.; Kanakry, J.A.; Kovalovsky, D. Increased Activity of a NK-Specific CAR-NK Framework Targeting CD3 and CD5 for T-Cell Leukemias. Cancers 2022, 14, 524. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019, 12, 49. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.M.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef]
- Montagner, I.; Penna, A.; Fracasso, G.; Carpanese, D.; Dalla Pietà, A.; Barbieri, V.; Zuccolotto, G. Anti-PSMA CAR-Engineered NK-92 Cells: An off-the-Shelf Cell Therapy for Prostate Cancer. Cells 2020, 9, 1382. [Google Scholar] [CrossRef] [PubMed]
- Pinz, K.; Yakaboski, E.; Jares, A.; Liu, H.; Firor, A.E.; Chen, K.H.; Wada, M.; Salman, H.; Tse, W.; Hagag, N. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget 2017, 8, 112783–112796. [Google Scholar] [CrossRef] [Green Version]
- Kweon, S.; Phan, M.T.T.; Chun, S.; Yu, H.; Kim, J.; Kim, S.; Lee, J.; Ali, A.K.; Lee, S.H.; Kim, S.K. Expansion of Human NK Cells Using K562 Cells Expressing OX40 Ligand and Short Exposure to IL-21. Front. Immunol. 2019, 10, 879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, M. Macrophages enter CAR immunotherapy. Nat. Methods 2020, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, L.; Su, H.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef]
- Maciocia, P.M.; Pule, M.A. Anti-CD1a CAR T cells to selectively target T-ALL. Blood 2019, 133, 2246–2247. [Google Scholar] [CrossRef] [Green Version]
- Maciocia, P.M.; Wawrzyniecka, P.A.; Maciocia, N.C.; Burley, A.; Karpanasamy, T.; Devereaux, S.; Hoekx, M.; O’Connor, D.; Leon, T.; Rapoz-D’Silva, T. Anti-CCR9 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia. Blood 2022, 140, 25–37. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Z.; Cen, H.; Wu, H.; Zhang, S.; Liu, J.; Leng, Y.; Ren, A.; Liu, X.; Zhang, Z. CAR T cells targeting CD99 as an approach to eradicate T-cell acute lymphoblastic leukemia without normal blood cells toxicity. J. Hematol. Oncol. 2021, 14, 162. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol. 2018, 200, 459–468. [Google Scholar] [CrossRef]
- Ferrari, M.; Baldan, V.; Ghongane, P.; Nicholson, A.; Bughda, R.; Akbar, Z.; Wawrzyniecka, P.; Maciocia, P.; Cordoba, S.; Thomas, S. Abstract 2183: Targeting TRBC1 and 2 for the treatment of T cell lymphomas. Immunology 2020, 80, 2183. [Google Scholar] [CrossRef]
- Kim, M.Y.; Yu, K.R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453.e19. [Google Scholar] [CrossRef] [Green Version]
- Pinz, K.; Liu, H.; Golightly, M.; Jares, A.; Lan, F.; Zieve, G.W.; Hagag, N.; Schuster, M.; Firor, A.E.; Jiang, X. Preclinical targeting of human T-cell malignancies using CD4-specific chimeric antigen receptor (CAR)-engineered T cells. Leukemia 2015, 30, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Niehues, T.; Kapaun, P.; Harms, D.O.; Burdach, S.; Kramm, C.; Körholz, D.; Janka-Schaub, G.; Göbel, U. A classification based on T cell selection-related phenotypes identifies a subgroup of childhood T-ALL with favorable outcome in the COALL studies. Leukemia 1999, 13, 614–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciocia, P.M.; Wawrzyniecka, P.A.; Philip, B.; Ricciardelli, I.; Akarca, A.U.; Onuoha, S.C.; Legut, M.; Cole, D.K.; Sewell, A.K.; Gritti, G. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat. Med. 2017, 23, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.D.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Khogeer, H.; Rahman, H.; Jain, N.; Angelova, E.A.; Yang, H.; Quesada, A.; Ok, C.Y.; Sui, D.; Wei, P.; Al Fattani, A. Early T precursor acute lymphoblastic leukaemia/lymphoma shows differential immunophenotypic characteristics including frequent CD33 expression and in vitro response to targeted CD33 therapy. Br. J. Haematol. 2019, 186, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.; Graux, C.; Lhermitte, L.; Lara, D.; Cluzeau, T.; Leguay, T.; Cieslak, A.; Trinquand, A.; Pastoret, C.; Belhocine, M. Early Response–Based Therapy Stratification Improves Survival in Adult Early Thymic Precursor Acute Lymphoblastic Leukemia: A Group for Research on Adult Acute Lymphoblastic Leukemia Study. J. Clin. Oncol. 2017, 35, 2683–2691. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.J.; Bahmanyar, M.; Minden, M.D.; Chang, H. CD33, not early precursor T-cell phenotype, is associated with adverse outcome in adult T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2016, 172, 823–825. [Google Scholar] [CrossRef]
- Maciocia, P.M.; Wawrzyniecka, P.; Maciocia, N.C.; Burley, A.; O’Connor, D.; Leon, T.E.; Rapoz-D’Silva, T.; Karpanasamy, T.; Pule, M.; Mansour, M.R. Anti-CCR9 CAR-T Cells for T Acute Lymphoblastic Leukemia. Blood 2021, 138 (Suppl. S1), 903. [Google Scholar] [CrossRef]
- Zhang, S.; Gu, C.; Huang, L.; Wu, H.; Shi, J.; Zhang, Z.; Zhou, Y.; Zhou, J.; Gao, Y.; Liu, J. The third-generation anti-CD30 CAR T-cells specifically homing to the tumor and mediating powerful antitumor activity. Sci. Rep. 2022, 12, 10488. [Google Scholar] [CrossRef] [PubMed]
- Maciocia, N.C.; Burley, A.; Nannini, F.; Wawrzyniecka, P.; Neves, M.; Karpanasamy, T.; Ferrari, M.; Marafioti, T.; Onuoha, S.; Khwaja, A. Anti-CD21 Chimeric Antigen Receptor (CAR)-T Cells for T Cell Acute Lymphoblastic Leukaemia (T-ALL). Blood 2021, 138 (Suppl. S1), 902. [Google Scholar] [CrossRef]
- Rasaiyaah, J.; Georgiadis, C.; Preece, R.; Mock, U.; Qasim, W. TCRαβ/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight 2018, 3, e99442. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.W.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2016, 27, 154–157. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, Y.; Zhang, M.; Zhao, H.; Wei, G.; Ge, W.; Cui, Q.; Mu, Q.; Chen, G.; Han, L. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed/refractory CD7-positive hematological malignancies: A phase I clinical study. Cell Res. 2022, 32, 995–1007. [Google Scholar] [CrossRef]
- Dai, Z.; Mu, W.; Zhao, Y.; Cheng, J.; Lin, H.; Ouyang, K.; Jia, X.; Liu, J.; Wei, Q.; Wang, M. T cells expressing CD5/CD7 bispecific chimeric antigen receptors with fully human heavy-chain-only domains mitigate tumor antigen escape. Signal Transduct. Target. Ther. 2022, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Mollanoori, H.; Shahraki, H.; Rahmati, Y.; Teimourian, S. CRISPR/Cas9 and CAR-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum. Immunol. 2018, 79, 876–882. [Google Scholar] [CrossRef]
- Mamonkin, M.; Mukherjee, M.; Srinivasan, M.; Sharma, S.; Gomes-Silva, D.; Mo, F.; Krenciute, G.; Orange, J.S.; Brenner, M.K. Reversible Transgene Expression Reduces Fratricide and Permits 4-1BB Costimulation of CAR T Cells Directed to T-cell Malignancies. Cancer Immunol. Res. 2017, 6, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Diorio, C.; Murray, R.; Naniong, M.; Barrera, L.; Camblin, A.; Chukinas, J.; Coholan, L.; Edwards, A.; Fuller, T.; Gonzales, C. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood 2022, 140, 619–629. [Google Scholar] [CrossRef]
- Zhang, M.; Fu, X.; Meng, H.; Wang, M.; Wang, Y.; Zhang, L.; Li, L.; Li, X.; Wang, X.; Sun, Z. A Single-Arm, Open-Label, Pilot Trial of Autologous CD7-CAR-T Cells for CD7 Positive Relapsed and Refractory T-Lymphoblastic Leukemia/Lymphoma. Blood 2021, 138, 3829. [Google Scholar]
- Pan, J.; Tan, Y.; Wang, G.; Deng, B.; Ling, Z.; Song, W.; Seery, S.; Zhang, Y.; Peng, S.; Xu, J. Donor-Derived CD7 Chimeric Antigen Receptor T Cells for T-Cell Acute Lymphoblastic Leukemia: First-in-Human, Phase I Trial. J. Clin. Oncol. 2021, 39, 3340–3351. [Google Scholar] [CrossRef]
- Daniël, K.; Gäken, J.; Farzaneh, F.; Kemeny, D.M. The Use of Intracellular Single-Chain Antibody Fragments to Specifically Inhibit Cytokine Secretion. Int. Arch. Allergy Immunol. 2001, 124, 216–217. [Google Scholar] [CrossRef]
- Ma, G.; Shen, J.; Pinz, K.; Wada, M.; Park, J.; Kim, S.; Togano, T.; Tse, W. Targeting T Cell Malignancies Using CD4CAR T-Cells and Implementing a Natural Safety Switch. Stem Cell Rev. Rep. 2019, 15, 443–447. [Google Scholar] [CrossRef]
- Boyiadzis, M.M.; Dhodapkar, M.V.; Brentjens, R.J.; Kochenderfer, J.N.; Neelapu, S.S.; Maus, M.V.; Porter, D.L.; Maloney, D.G.; Grupp, S.A.; Mackall, C.L. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: Clinical perspective and significance. J. Immunother. Cancer 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Martínez, D.; Baroni, M.L.; Gutierrez-Agüera, F.; Roca-Ho, H.; Blanch-Lombarte, O.; González-García, S.; Torrebadell, M.; Junca, J.; Ramírez-Orellana, M.A.; Velasco-Hernández, T.A.; et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood 2019, 133, 2291–2304. [Google Scholar] [CrossRef]
- Zheng, W.; Medeiros, L.J.; Young, K.H.; Goswami, M.; Powers, L.; Kantarjian, H.H.; Thomas, D.A.; Cortes, J.E.; Wang, S.A. CD30 expression in acute lymphoblastic leukemia as assessed by flow cytometry analysis. Leuk. Lymphoma 2013, 55, 624–627. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.A.; Grover, N.S.; Beaven, A.W.; Lulla, P.D.; Wu, M.F.; Ivanova, A.; Wang, T.; Shea, T.C.; Rooney, C.M.; Dittus, C. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J. Clin. Oncol. 2020, 38, 3794–3804. [Google Scholar] [CrossRef]
- Jacoby, E. The role of allogeneic HSCT after CAR T cells for acute lymphoblastic leukemia. Bone Marrow Transplant. 2019, 54, 810–814. [Google Scholar] [CrossRef]
- Hotblack, A.; Kokalaki, E.K.; Palton, M.J.; Cheung, G.W.K.; Williams, I.P.; Manzoor, S.; Grothier, T.I.; Piapi, A.; Fiaccadori, V.; Wawrzyniecka, P. Tunable control of CAR T cell activity through tetracycline mediated disruption of protein–protein interaction. Sci. Rep. 2021, 11, 21902. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef]
- Takahashi, N.; Miura, I.; Saitoh, K.; Miura, A.B. Lineage Involvement of Stem Cells Bearing the Philadelphia Chromosome in Chronic Myeloid Leukemia in the Chronic Phase as Shown by a Combination of Fluorescence-Activated Cell Sorting and Fluorescence In Situ Hybridization. Blood 1998, 92, 4758–4763. [Google Scholar] [CrossRef] [PubMed]
- Handgretinger, R.; Lang, P.; Schumm, M.; Taylor, G.; Neu, S.; Koscielnak, E.; Niethammer, D.; Klingebiel, T. Isolation and transplantation of autologous peripheral CD34+ progenitor cells highly purified by magnetic-activated cell sorting. Bone Marrow Transplant. 1998, 21, 987–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, D.I.; Paietta, E.M.; Moorman, A.V.; Richards, S.M.; Buck, G.; DeWald, G.; Ferrando, A.; Fielding, A.K.; Goldstone, A.H.; Ketterling, R.P. T-cell acute lymphoblastic leukemia in adults: Clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood 2009, 114, 5136–5145. [Google Scholar]
- Litzow, M.R.; Ferrando, A.A. How I treat T-cell acute lymphoblastic leukemia in adults. Blood 2015, 126, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Longo, D.; Hunger, S.; Mullighan, C. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar]
- Legut, M.; Cole, D.K.; Sewell, A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015, 12, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, B.; Serre, K.; Norell, H. γδ T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef]
- Harrer, D.C.; Simon, B.; Fujii, S.I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: A safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; Lee, D.A.; Champlin, R.E. Bispecific T-cells Expressing Polyclonal Repertoire of Endogenous δ T-cell Receptors and Introduced CD19-specific Chimeric Antigen Receptor. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.; Kaminskiy, Y.; Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Rakhmatullina, A.; Petukhov, A.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Knowns and Unknowns about CAR-T Cell Dysfunction. Cancers 2022, 14, 1078. [Google Scholar] [CrossRef]
- Taraban, V.Y.; Rowley, T.F.; O’Brien, L.; Chan, H.T.C.; Haswell, L.E.; Green, M.H.A.; Tutt, A.L.; Glennie, M.J.; Al-Shamkhani, A. Expression and costimulatory effects of the TNF receptor superfamily members CD134 (OX40) and CD137 (4–1BB), and their role in the generation of anti-tumor immune responses. Eur. J. Immunol. 2002, 32, 3617–3627. [Google Scholar] [CrossRef]
- Curran, M.A.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J.P. Combination CTLA-4 blockade and 4–1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS ONE 2011, 6, e19499. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Wang, X.; Cheng, D.; Xia, Z.; Luan, M.; Zhang, S. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS ONE 2014, 9, e89350. [Google Scholar] [CrossRef]
- Agarwal, S.; Hanauer, J.D.S.; Frank, A.M.; Riechert, V.; Thalheimer, F.B.; Buchholz, C.J. In Vivo Generation of CAR T Cells Selectively in Human CD4+ Lymphocytes. Mol. Ther. 2020, 28, 1783–1794. [Google Scholar] [CrossRef]
- Liadi, I.; Singh, H.; Romain, G.; Rey-Villamizar, N.; Merouane, A.; Adolacion, J.R.T.; Kebriaei, P.; Huls, H.; Qiu, P.; Roysam, B. Individual Motile CD4+ T Cells Can Participate in Efficient Multikilling through Conjugation to Multiple Tumor Cells. Cancer Immunol. Res. 2015, 3, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Mikuła-Pietrasik, J.; Witucka, A.; Pakuła, M.; Uruski, P.; Begier-Krasińska, B.; Niklas, A.; Tykarski, A.; Książek, K. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell. Mol. Life Sci. 2019, 76, 681–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrolia, P.J.; Wynn, R.; Hough, R.; Vora, A.; Bonney, D.; Veys, P.; Rao, K.; Chiesa, R.; Al-Hajj, M.; Cordoba, S.P. Simultaneous Targeting of CD19 and CD22: Phase I Study of AUTO3, a Bicistronic Chimeric Antigen Receptor (CAR) T-Cell Therapy, in Pediatric Patients with Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia (r/r B-ALL): Amelia Study. Blood 2018, 132 (Suppl. S1), 279. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.; Boissel, N.; Cassaday, R.D.; Forcade, E.; Bishop, M.R.; Topp, M.S.; Tzachanis, D. Phase 2 results of the ZUMA-3 study evaluating KTE-X19, an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, in adult patients (pts) with relapsed/refractory B-cell acute lymphoblastic leukemia (R/R B-ALL). J. Clin. Oncol. 2021, 39 (Suppl. S15), 7002. [Google Scholar] [CrossRef]
- Curran, K.J.; Sauter, C.S.; Kernan, N.A.; Prockop, S.E.; Boulad, F.; Perales, M.; Giralt, S.A.; Riviere, I.; Wang, X.; Boelens, J.J. Durable Remission Following “off-the-Shelf” Chimeric Antigen Receptor (CAR) T-Cells in Patients with Relapse/Refractory (R/R) B-Cell Malignancies. Biol. Blood Marrow Transplant. 2020, 26, S89. [Google Scholar] [CrossRef]
- Fleischer, L.C.; Spencer, H.T.; Raikar, S.A.-O. Targeting T cell malignancies using CAR-based immunotherapy: Challenges and potential solutions. J. Hematol. Oncol. 2019, 12, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Scarfò, I.; Ormhøj, M.; Frigault, M.J.; Castano, A.P.; Lorrey, S.; Bouffard, A.A.; Van Scoyk, A.; Rodig, S.J.; Shay, A.J.; Aster, J.C.; et al. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood 2018, 132, 1495–1506. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, A.; Tong, X.; Xu, N.; Zhang, T.; Zhou, F.; Zhu, H. CAR T-Cell Immunotherapy Treating T-ALL: Challenges and Opportunities. Vaccines 2023, 11, 165. https://doi.org/10.3390/vaccines11010165
Ren A, Tong X, Xu N, Zhang T, Zhou F, Zhu H. CAR T-Cell Immunotherapy Treating T-ALL: Challenges and Opportunities. Vaccines. 2023; 11(1):165. https://doi.org/10.3390/vaccines11010165
Chicago/Turabian StyleRen, Anqi, Xiqin Tong, Na Xu, Tongcun Zhang, Fuling Zhou, and Haichuan Zhu. 2023. "CAR T-Cell Immunotherapy Treating T-ALL: Challenges and Opportunities" Vaccines 11, no. 1: 165. https://doi.org/10.3390/vaccines11010165