
Epigenetics in Tuberculosis: Immunomodulation of Host Immune Response
 , ,
, , 

Abstract
:1. Introduction
2. Immuno-Pathophysiology of Mycobacterium tuberculosis
3. Tuberculosis and Epigenetic Regulations and Modifications
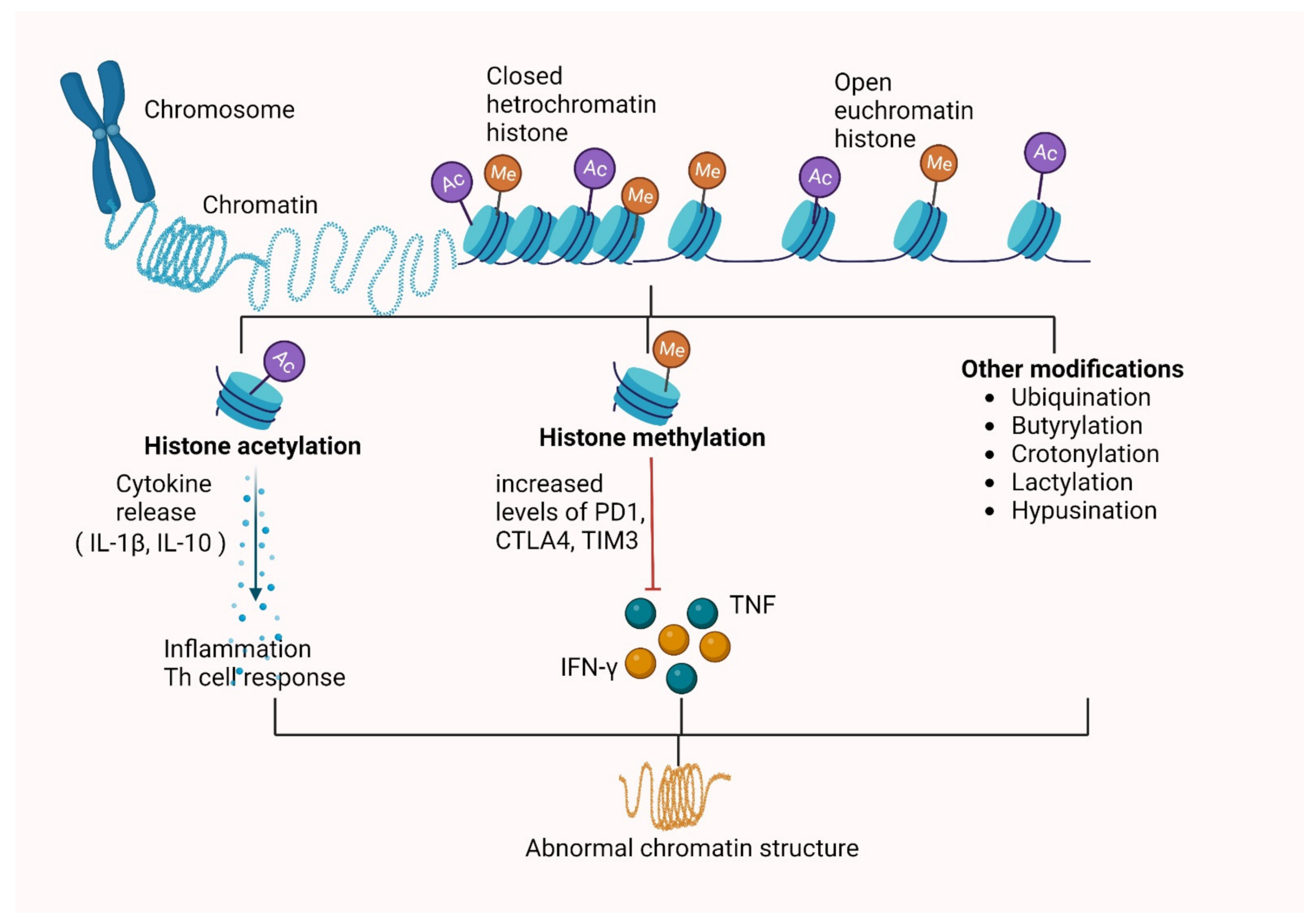
3.1. Histone Modifications
3.1.1. Histone Methylation
3.1.2. Histone Acetylation
3.2. Alteration in Expression of Non-Coding RNAs
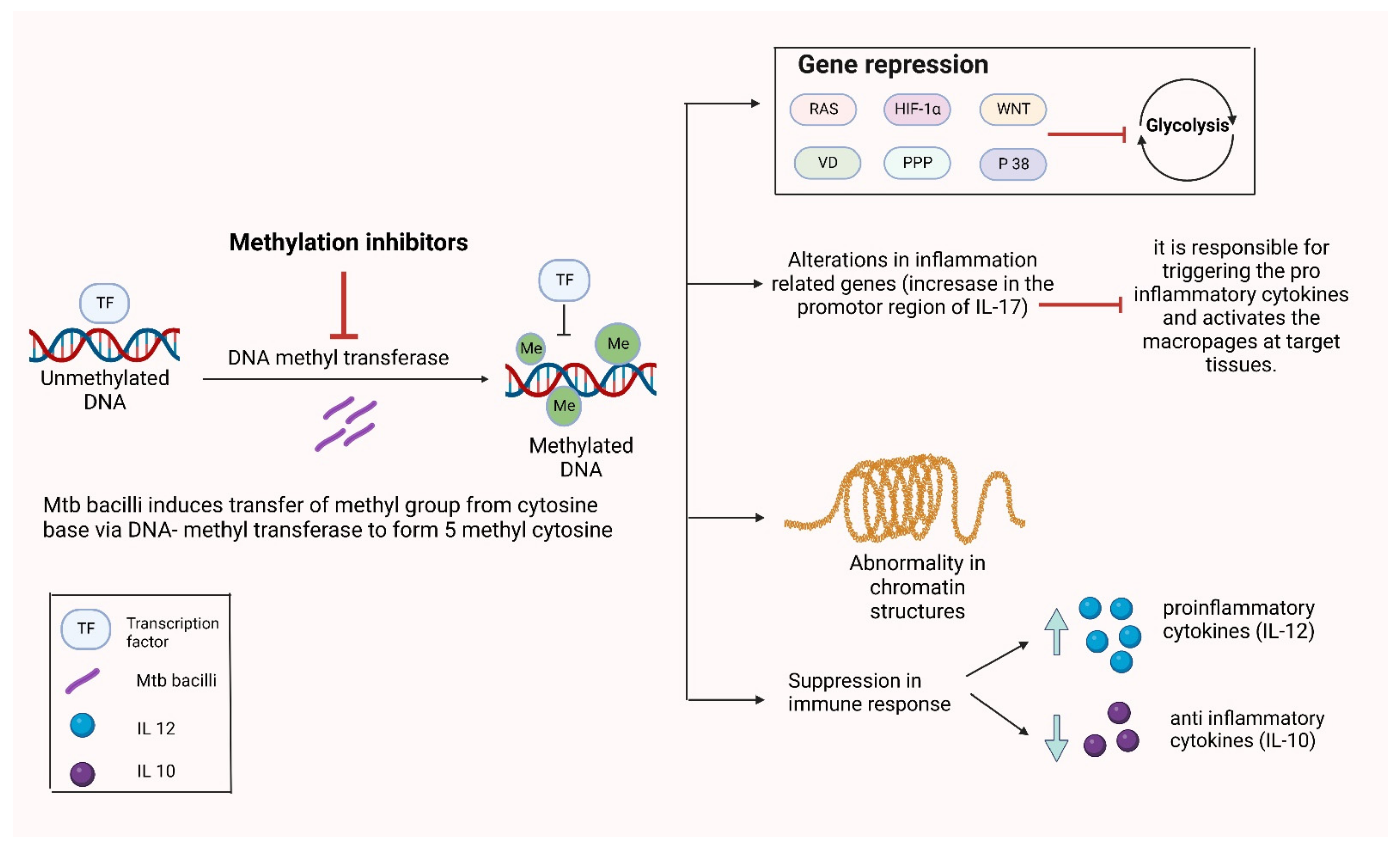
3.3. Alterations in DNA Methylation
4. Therapies Targeting Epigenetic Modifications for Mycobacterium tuberculosis
5. Way Forward
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nadjane Batista Lacerda, S.; de Abreu Temoteo, R.C.; Ribeiro Monteiro de Figueiredo, T.M.; Darliane Tavares de Luna, F.; Alves Nunes de Sousa, M.; de Abreu, L.L.; Luiz Affonso Fonseca, F. Individual and social vulnerabilities upon acquiring tuberculosis: A literature systematic review. Int. Arch. Med. 2014, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO Report on Infectour Diseas. 20 August 1999. Available online: http://www.who.int/infectious-disease-report/ (accessed on 10 October 2022).
- WHO. Global Tuberculosis Report 2013; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Chakraborty, A.K. Epidemiology of tuberculosis: Current status in India. Indian J. Med. Res. 2004, 120, 248–276. [Google Scholar] [PubMed]
- Deutsch-Feldman, M.; Pratt, R.H.; Price, S.F.; Tsang, C.A.; Self, J.L. Tuberculosis—United States, 2020. Morb. Mortal. Wkly. Rep. 2021, 70, 409. [Google Scholar] [CrossRef]
- Sotgiu, G.; Nahid, P.; Loddenkemper, R.; Abubakar, I.; Miravitlles, M.; Migliori, G.B. The ERS-endorsed official ATS/CDC/IDSA clinical practice guidelines on treatment of drug-susceptible tuberculosis. Eur. Respir. J. 2016, 48, 963–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaberg, T.; Forssbohm, M.; Hauer, B.; Kirsten, D.; Kropp, R.; Loddenkemper, R.; Loytved, G.; Magdorf, K.; Rieder, H.L.; Sagebiel, D. Guidelines for Drug Treatment of Tuberculosis in Adults and Childhood. Pneumologie 2001, 55, 494–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, H.L. Probability distribution of drug-resistant mutants in unselected populations of Mycobacterium tuberculosis. Appl. Microbiol. 1970, 20, 810–814. [Google Scholar] [CrossRef]
- Falzon, D.; Jaramillo, E.; Schünemann, H.; Arentz, M.; Bauer, M.; Bayona, J.; Blanc, L.; Caminero, J.; Daley, C.; Duncombe, C. WHO guidelines for the programmatic management of drug-resistant tuberculosis: 2011 update. Eur. Respir. J. 2011, 38, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Diacon, A.H.; Pym, A.; Grobusch, M.; Patientia, R.; Rustomjee, R.; Page-Shipp, L.; Pistorius, C.; Krause, R.; Bogoshi, M.; Churchyard, G.; et al. The Diarylquinoline TMC207 for Multidrug-Resistant Tuberculosis. N. Engl. J. Med. 2009, 360, 2397–2405. [Google Scholar] [CrossRef] [Green Version]
- Frimodt-Moller, J.; Thomas, J.; Parthasarathy, R. Observations on the Protective Effect of Bcg Vaccination in a South Indian Rural Population. Bull. World Health Organ. 1964, 30, 545–574. [Google Scholar]
- Young, D.B. Current tuberculosis vaccine development. Clin. Infect. Dis. 2000, 30 (Suppl. 3), S254–S256. [Google Scholar] [CrossRef]
- Van Hest, N.; Aldridge, R.; De Vries, G.; Sandgren, A.; Hauer, B.; Hayward, A.; de Oñate, W.A.; Haas, W.; Codecasa, L.; Caylà, J.J.E. Tuberculosis control in big cities and urban risk groups in the European Union: A consensus statement. Eurosurveillance 2014, 19, 20728. [Google Scholar] [CrossRef] [PubMed]
- Hwang, L.Y.; Grimes, C.Z.; Beasley, R.P.; Graviss, E.A. Latent tuberculosis infections in hard-to-reach drug using population-detection, prevention and control. Tuberculosis (Edinburgh, Scotland). Tuberculosis 2009, 89 (Suppl. 1), S41–S45. [Google Scholar] [CrossRef]
- Giosuè, S.; Casarini, M.; Alemanno, L.; Galluccio, G.; Mattia, P.; Pedicelli, G.; Rebek, L.; Bisetti, A.; Ameglio, F. Effects of Aerosolized Interferon- α in Patients with Pulmonary Tuberculosis. Am. J. Respir. Crit. Care Med. 1998, 158, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Raad, I.; Hachem, R.; Leeds, N.; Sawaya, R.; Salem, Z.; Atweh, S. Use of adjunctive treatment with interferon-γ in an immunocompromised patient who had refractory multidrug-resistant tuberculosis of the brain. Clin. Infect. Dis. 1996, 22, 572–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arevalo, I.; Ward, B.; Miller, R.; Meng, T.-C.; Najar, E.; Alvarez, E.; Matlashewski, G.; Llanos-Cuentas, A. Successful treatment of drug-resistant cutaneous leishmaniasis in humans by use of imiquimod, an immunomodulator. Clin. Infect. Dis. 2001, 33, 1847–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermudez, L.E.; Petrofsky, M.; Wu, M.; Young, L.S. Clarithromycin Significantly Improves Interleukin-12-Mediated Anti-Mycobacterium aviumActivity and Abolishes Toxicity in Mice. J. Infect. Dis. 1998, 178, 896–899. [Google Scholar] [CrossRef] [Green Version]
- Steward, W.P. Granulocyte and granulocyte-macrophage colony-stimulating factors. Lancet 1993, 342, 153–157. [Google Scholar] [CrossRef]
- Singh, M.M.; Kumar, P.; Malaviya, A.N.; Kumar, R. Levamisole as an adjunct in the treatment of pulmonary tuberculosis. Am. Rev. Respir. Dis. 1981, 123, 277–279. [Google Scholar] [CrossRef]
- Durban Immunotherapy Trial Group. Immunotherapy with Mycobacterium vaccae in patients with newly diagnosed pulmonary tuberculosis: A randomised controlled trial. Lancet 1999, 354, 116–119. [Google Scholar] [CrossRef]
- Migliori, G.; Hopewell, P.; Blasi, F.; Spanevello, A.; Raviglione, M. Improving the TB case management: The International Standards for Tuberculosis Care. Eur. Respir. Soc. 2006, 28, 687–690. [Google Scholar] [CrossRef] [Green Version]
- Parida, A.; Bairy, K.; Chogtu, B.; Magazine, R.; Vidyasagar, S. Comparison of directly observed treatment short course (DOTS) with self-administered therapy in pulmonary tuberculosis in Udupi District of Southern India. J. Clin. Diagn. Res. 2014, 8, HC29–HC31. [Google Scholar] [PubMed]
- Frieden, T.R.; Sbarbaro, J.A. Promoting adherence to treatment for tuberculosis: The importance of direct observation. Bull. World Health Organ. 2007, 85, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Marimani, M.; Ahmad, A.; Duse, A. The role of epigenetics, bacterial and host factors in progression of Mycobacterium tuberculosis infection. Tuberculosis 2018, 113, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Kathirvel, M.; Mahadevan, S. The role of epigenetics in tuberculosis infection. Epigenomics 2016, 8, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Churchyard, G.; Kim, P.; Shah, N.S.; Rustomjee, R.; Gandhi, N.; Mathema, B.; Dowdy, D.; Kasmar, A.; Cardenas, V. What We Know About Tuberculosis Transmission: An Overview. J. Infect. Dis. 2017, 216, S629–S635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, T.R.; Borel, S.; Gutierrez, M.G. The innate immune response in human tuberculosis. Cell. Microbiol. 2015, 17, 1277–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs. pathogen evasion. Cell. Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Sepehri, Z.; Kiani, Z.; Kohan, F.; Ghavami, S. Toll-Like Receptor 4 as an Immune Receptor AgainstMycobacterium tuberculosis: A Systematic Review. Lab. Med. 2019, 50, 117–129. [Google Scholar] [CrossRef]
- Mortaz, E.; Adcock, I.M.; Tabarsi, P.; Masjedi, M.R.; Mansouri, D.; Velayati, A.A.; Casanova, J.-L.; Barnes, P.J. Interaction of Pattern Recognition Receptors with Mycobacterium Tuberculosis. J. Clin. Immunol. 2015, 35, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.P.J.; Meintjes, G.; Wilkinson, K.A.; Graham, C.M.; Marais, S.; Van der Plas, H.; Deffur, A.; Schutz, C.; Bloom, C.; Munagala, I.; et al. HIV–tuberculosis-associated immune reconstitution inflammatory syndrome is characterized by Toll-like receptor and inflammasome signalling. Nat. Commun. 2015, 6, 8451. [Google Scholar] [CrossRef] [Green Version]
- Sheneef, A.; Hussein, M.T.; Mohamed, T.; Mahmoud, A.; Yousef, L.M.; A Alkady, O. Pentraxin 3 Genetic Variants and The Risk of Active Pulmonary Tuberculosis. Egypt. J. Immunol. 2017, 24, 21–27. [Google Scholar]
- Mayer-Barber, K.D.; Barber, D.L. Innate and adaptive cellular immune responses to Mycobacterium tuberculosis infection. Cold Spring Harb. Perspect. Med. 2015, 5, a018424. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Lodi, L.; Galli, L.; Chiappini, E. Immune response to Mycobacterium tuberculosis: A narrative review. Front. Pediatr. 2019, 7, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, S.; Mittal, E.; A Philips, J. Tuberculosis and the art of macrophage manipulation. Pathog. Dis. 2018, 76, fty037. [Google Scholar] [CrossRef] [Green Version]
- Jasenosky, L.D.; Scriba, T.J.; Hanekom, W.A.; Goldfeld, A.E. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol. Rev. 2015, 264, 74–87. [Google Scholar] [CrossRef]
- Hunter, R.L. Tuberculosis as a three-act play: A new paradigm for the pathogenesis of pulmonary tuberculosis. Tuberculosis 2016, 97, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Rehn, A.; Rahman, J.; Andersson, J.; Svensson, M.; Brighenti, S. Pulmonary tuberculosis patients with a vitamin D deficiency demonstrate low local expression of the antimicrobial peptide LL-37 but enhanced FoxP3+ regulatory T cells and IgG-secreting cells. Clin. Immunol. 2015, 156, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Luevano, S.P.; Rodriguez-Carlos, A.; Jacobo-Delgado, Y.; Valdez-Miramontes, C.; Enciso-Moreno, J.A.; Rivas-Santiago, B. Steroid hormone modulates the production of cathelicidin and human β-defensins in lung epithelial cells and macrophages promoting Mycobacterium tuberculosis killing. Tuberculosis 2021, 128, 102080. [Google Scholar] [CrossRef]
- Noble, D. Conrad Waddington and the origin of epigenetics. J. Exp. Biol. 2015, 218, 816–818. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, A.; Guttman, J.A.; Finlay, B.B. Manipulation of host-cell pathways by bacterial pathogens. Nature 2007, 449, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Cossart, P. Post-translational modifications in host cells during bacterial infection. FEBS Lett. 2010, 584, 2748–2758. [Google Scholar] [CrossRef] [PubMed]
- Bobak, C.A.; Abhimanyu; Natarajan, H.; Gandhi, T.; Grimm, S.L.; Nishiguchi, T.; Koster, K.; Longlax, S.C.; Dlamini, Q.; Kahari, J.; et al. Increased DNA methylation, cellular senescence and premature epigenetic aging in guinea pigs and humans with tuberculosis. Aging 2022, 14, 2174–2193. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Curry, H.M.; Zwilling, B.S.; Lafuse, W. Mycobacteria Inhibition of IFN-γ Induced HLA-DR Gene Expression by Up-Regulating Histone Deacetylation at the Promoter Region in Human THP-1 Monocytic Cells. J. Immunol. 2005, 174, 5687–5694. [Google Scholar] [CrossRef] [Green Version]
- Fatima, S.; Kumari, A.; Agarwal, M.; Pahuja, I.; Yadav, V.; Dwivedi, V.P.; Bhaskar, A. Epigenetic code during mycobacterial infections: Therapeutic implications for tuberculosis. FEBS J. 2021, 289, 4172–4191. [Google Scholar] [CrossRef]
- Koo, M.-S.; Subbian, S.; Kaplan, G. Strain specific transcriptional response in Mycobacterium tuberculosis infected macrophages. Cell Commun. Signal. 2012, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Peterson, C.L.; Laniel, M.-A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef] [Green Version]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA Demethylation Dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Vymetalkova, V.; Vodicka, P.; Vodenkova, S.; Alonso, S.; Schneider-Stock, R. DNA methylation and chromatin modifiers in colorectal cancer. Mol. Asp. Med. 2019, 69, 73–92. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaseen, I.; Kaur, P.; Nandicoori, V.; Khosla, S. Mycobacteria modulate host epigenetic machinery by Rv1988 methylation of a non-tail arginine of histone H3. Nat. Commun. 2015, 6, 8922. [Google Scholar] [CrossRef]
- Singh, V.; Prakhar, P.; Rajmani, R.S.; Mahadik, K.; Borbora, S.M.; Balaji, K.N. Histone Methyltransferase SET8 Epigenetically Reprograms Host Immune Responses to Assist Mycobacterial Survival. J. Infect. Dis. 2017, 216, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Houghton, A.M. Matrix metalloproteinases in destructive lung disease. Matrix Biol. 2015, 44, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Loffek, S.; Schilling, O.; Franzke, C.-W. Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moores, R.C.; Brilha, S.; Schutgens, F.; Elkington, P.T.; Friedland, J.S. Epigenetic Regulation of Matrix Metalloproteinase-1 and -3 Expression in Mycobacterium tuberculosis Infection. Front. Immunol. 2017, 8, 602. [Google Scholar] [CrossRef] [Green Version]
- Aung, H.T.; Schroder, K.; Himes, S.R.; Brion, K.; Van Zuylen, W.; Trieu, A.; Suzuki, H.; Hayashizaki, Y.; Hume, D.A.; Sweet, M.J.J.T.F.J. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 2006, 20, 1315–1327. [Google Scholar] [CrossRef]
- Khosla, S.; Sharma, G.; Yaseen, I. Learning epigenetic regulation from mycobacteria. Microb. Cell 2016, 3, 92–94. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Nayak, B.; Meuli, M.; Sander, P.; Mishra, S.; Sonawane, A. Mycobacterium tuberculosis phosphoribosyltransferase promotes bacterial survival in macrophages by inducing histone hypermethylation in autophagy-related genes. Front. Cell. Infect. Microbiol. 2021, 11, 676456. [Google Scholar] [CrossRef]
- Chandran, A.; Antony, C.; Jose, L.; Mundayoor, S.; Natarajan, K.; Kumar, R.A. Mycobacterium tuberculosis Infection Induces HDAC1-Mediated Suppression of IL-12B Gene Expression in Macrophages. Front. Cell. Infect. Microbiol. 2015, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Nath, L.; Kamal, M.A.; Varshney, A.; Jain, A.; Singh, S.; Rao, K.V. Genome-wide Analysis of the Host Intracellular Network that Regulates Survival of Mycobacterium tuberculosis. Cell 2010, 140, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Almeida Da Silva, P.E.; Palomino, J.C. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: Classical and new drugs. J. Antimicrob. Chemother. 2011, 66, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Upadhyay, S.; Srilalitha, M.; Nandicoori, V.K.; Khosla, S. The interaction of mycobacterial protein Rv2966c with host chromatin is mediated through non-CpG methylation and histone H3/H4 binding. Nucleic Acids Res. 2015, 43, 3922–3937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef]
- Sontheimer, E.J.; Carthew, R.W. Silence from within: Endogenous siRNAs and miRNAs. Cell 2005, 122, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Maudet, C.; Mano, M.; Eulalio, A. MicroRNAs in the interaction between host and bacterial pathogens. FEBS Lett. 2014, 588, 4140–4147. [Google Scholar] [CrossRef] [Green Version]
- Bentwich, I.; Avniel, A.; Karov, Y.; Aharonov, R.; Gilad, S.; Barad, O.; Barzilai, A.; Einat, P.; Einav, U.; Meiri, E.; et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005, 37, 766–770. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Hua, Z.; Lv, Q.; Ye, W.; Wong, C.-K.A.; Cai, G.; Gu, D.; Ji, Y.; Zhao, C.; Wang, J.; Yang, B.B.J.P.o. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE 2006, 1, e116. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennini, M.E.; Liu, Y.; Yang, J.; Croniger, C.M.; Boom, W.H.; Harding, C.V. CCAAT/Enhancer-Binding Protein β and δ Binding to CIITA Promoters Is Associated with the Inhibition of CIITA Expression in Response toMycobacterium tuberculosis19-kDa Lipoprotein. J. Immunol. 2007, 179, 6910–6918. [Google Scholar] [CrossRef] [PubMed]
- Sharbati, J.; Lewin, A.; Kutz-Lohroff, B.; Kamal, E.; Einspanier, R.; Sharbati, S. Integrated microRNA-mRNA-analysis of human monocyte derived macrophages upon Mycobacterium avium subsp. hominissuis infection. PLoS ONE 2011, 6, e20258. [Google Scholar] [CrossRef]
- Shi, G.; Mao, G.; Xie, K.; Wu, D.; Wang, W. MiR-1178 regulates mycobacterial survival and inflammatory responses in Mycobacterium tuberculosis-infected macrophages partly via TLR4. J. Cell. Biochem. 2018, 119, 7449–7457. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Q. MicroRNA-708-5p regulates mycobacterial vitality and the secretion of inflammatory factors in Mycobacterium tuberculosis-infected macrophages by targeting TLR4. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8028–8038. [Google Scholar]
- Niu, W.; Sun, B.; Li, M.; Cui, J.; Huang, J.; Zhang, L. TLR-4/microRNA-125a/NF-κB signaling modulates the immune response to Mycobacterium tuberculosis infection. Cell Cycle 2018, 17, 1931–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.; Song, Z.; Wu, Y.; Gao, Y.; Gao, M.; Liu, F.; Wang, F.; Zhang, Y. MicroRNA-27b Modulates Inflammatory Response and Apoptosis duringMycobacterium tuberculosisInfection. J. Immunol. 2018, 200, 3506–3518. [Google Scholar] [CrossRef] [Green Version]
- Wen, Q.; Zhou, C.; Xiong, W.; Su, J.; He, J.; Zhang, S.; Du, X.; Liu, S.; Wang, J.; Ma, L. MiR-381-3p Regulates the Antigen-Presenting Capability of Dendritic Cells and Represses Antituberculosis Cellular Immune Responses by Targeting CD1c. J. Immunol. 2016, 197, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Singh, Y.; Kaul, V.; Mehra, A.; Chatterjee, S.; Tousif, S.; Dwivedi, V.P.; Suar, M.; Van Kaer, L.; Bishai, W.R.; Das, G. Mycobacterium tuberculosis Controls MicroRNA-99b (miR-99b) Expression in Infected Murine Dendritic Cells to Modulate Host Immunity. J. Biol. Chem. 2013, 288, 5056–5061. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Liu, K.; Jia, Q.; Zhang, H.; Bie, Q.; Zhang, B. The Roles of Host Noncoding RNAs in Mycobacterium tuberculosis Infection. Front. Immunol. 2021, 12, 664787. [Google Scholar] [CrossRef]
- Skvortsova, K.; Stirzaker, C.; Taberlay, P. The DNA methylation landscape in cancer. Essays Biochem. 2019, 63, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wu, X.-m.; He, X.; Wang, R.-z.; Yin, X.-y.; Zhou, F.; Ji, M.-h.; Shen, J.-c.J.P.B. Contribution of DNA methyltransferases to spared nerve injury induced depression partially through epigenetically repressing Bdnf in hippocampus: Reversal by ketamine. Pharmacol. Biochem. Behav. 2021, 200, 173079. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, A.R.; Rajapakshe, K.; Nishiguchi, T.; Grimm, S.L.; Mtetwa, G.; Dlamini, Q.; Kahari, J.; Mahapatra, S.; Kay, A.W.; Maphalala, G.; et al. DNA hypermethylation during tuberculosis dampens host immune responsiveness. J. Clin. Investig. 2020, 130, 3113–3123. [Google Scholar] [CrossRef]
- Jose, L.; Ramachandran, R.; Bhagavat, R.; Gomez, R.L.; Chandran, A.; Raghunandanan, S.; Omkumar, R.V.; Chandra, N.; Mundayoor, S.; Kumar, R.A.J.T.F.j. Hypothetical protein Rv3423. 1 of Mycobacterium tuberculosis is a histone acetyltransferase. FEBS J. 2016, 283, 265–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shell, S.S.; Prestwich, E.G.; Baek, S.-H.; Shah, R.R.; Sassetti, C.M.; Dedon, P.C.; Fortune, S.M. DNA Methylation Impacts Gene Expression and Ensures Hypoxic Survival of Mycobacterium tuberculosis. PLoS Pathog. 2013, 9, e1003419. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Sowpati, D.T.; Singh, P.; Khan, M.Z.; Ganji, R.; Upadhyay, S.; Banerjee, S.; Nandicoori, V.K.; Khosla, S.J.S.r. Genome-wide non-CpG methylation of the host genome during M. tuberculosis infection. Sci. Rep. 2016, 6, 25006. [Google Scholar] [CrossRef]
- Lyu, M.; Zhou, J.; Jiao, L.; Wang, Y.; Zhou, Y.; Lai, H.; Xu, W.; Ying, B. Deciphering a TB-related DNA methylation biomarker and constructing a TB diagnostic classifier. Mol. Ther. Nucleic Acids 2022, 27, 37–49. [Google Scholar] [CrossRef]
- Yadav, V.; Dwivedi, V.P.; Bhattacharya, D.; Mittal, A.; Moodley, P.; Das, G.J.J.o.G.; Research, G. Understanding the host epigenetics in Mycobacterium tuberculosis infection. J. Genet. Genome Res. 2015, 2. [Google Scholar] [CrossRef]
- Siddle, K.J.; Deschamps, M.; Tailleux, L.; Nédélec, Y.; Pothlichet, J.; Lugo-Villarino, G.; Libri, V.; Gicquel, B.; Neyrolles, O.; Laval, G.; et al. A genomic portrait of the genetic architecture and regulatory impact of microRNA expression in response to infection. Genome Res. 2014, 24, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Leung, E.T.; Wong, H.; Lui, C.Y.G.; Lee, N.; To, K.-F.; Choy, K.W.; Chan, R.C.; Ip, M. Unraveling methylation changes of host macrophages in Mycobacterium tuberculosis infection. Tuberculosis 2016, 98, 139–148. [Google Scholar] [CrossRef]
- Kumar, P.; Agarwal, R.; Siddiqui, I.; Vora, H.; Das, G.; Sharma, P.J.I. ESAT6 differentially inhibits IFN-γ-inducible class II transactivator isoforms in both a TLR2-dependent and-independent manner. Immunol. Cell Biol. 2012, 90, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Frantz, F.G.; Castro, R.C.; Fontanari, C.; Bollela, V.R.; Zambuzi, F.A. DNA Methylation impairs monocyte function in tuberculosis leading to disease progression. Am. Assoc. Immnol. 2019, 202 (Suppl. 1), 125.10. [Google Scholar]
- Crossman, D.K. Characterization of a Novel Acetyltransferase Found Only in Pathogenic Strains of Mycobacterium Tuberculosis; The University of Alabama at Birmingham: Birmingham, AL, USA, 2007. [Google Scholar]
- Andraos, C.; Koorsen, G.; Knight, J.C.; Bornman, L. Vitamin D receptor gene methylation is associated with ethnicity, tuberculosis, and TaqI polymorphism. Hum. Immunol. 2011, 72, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo-Patino, M.; Heater, S.; Simmons, J.; Peterson, G.; Stein, C.; Mayanja-Kizza, H.; Boom, W.; Hawn, T. Selective HDAC3 Inhibitor Restricts Mycobacterial Growth and Modulates Macrophage Immune Responses. Am. Thorac. Soc. 2019, 199, A4425. [Google Scholar] [CrossRef]
- Moreira, J.D.; Koch, B.E.V.; van Veen, S.; Walburg, K.V.; Vrieling, F.; Guimarães, T.M.P.D.; Meijer, A.H.; Spaink, H.; Ottenhoff, T.H.M.; Haks, M.C.; et al. Functional Inhibition of Host Histone Deacetylases (HDACs) Enhances in vitro and in vivo Anti-mycobacterial Activity in Human Macrophages and in Zebrafish. Front. Immunol. 2020, 11, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, T.; Lugrin, J.; LE Roy, D.; Goy, G.; Mombelli, M.; Koessler, T.; Ding, X.C.; Chanson, A.-L.; Reymond, M.K.; Miconnet, I.; et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011, 117, 1205–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, B.; Fukudome, E.Y.; Kochanek, A.R.; Finkelstein, R.A.; Chong, W.; Jin, G.; Lu, J.; Demoya, M.A.; Velmahos, G.C.; et al. Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery 2010, 148, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.; Coleman, A.; Gogan, K.; Dunne, P.; Basdeo, S.; Keane, J. Vorinostat (SAHA) promotes innate and adaptive immunity to Mycobacterium tuberculosis. Access Microbiol. 2020, 2, 29. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, Z.; Liu, B.; Zhao, T.; Li, Y.; Alam, H.B. Selective histone deacetylase 6 inhibition prolongs survival in a lethal two-hit model. J. Surg. Res. 2015, 197, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Ariffin, J.K.; Das Gupta, K.; Kapetanovic, R.; Iyer, A.; Reid, R.C.; Fairlie, D.; Sweet, M.J. Histone Deacetylase Inhibitors Promote Mitochondrial Reactive Oxygen Species Production and Bacterial Clearance by Human Macrophages. Antimicrob. Agents Chemother. 2016, 60, 1521–1529. [Google Scholar] [CrossRef] [Green Version]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, S.; Iyer, L.R.; Muley, M.; Singh, P.K.; Shastry, A.; Saxena, A.; Kulathingal, J.; Vijaykanth, G.; Raghul, J.; Rajesh, N.; et al. Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. Int. Immunopharmacol. 2013, 16, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Clocchiatti, A.; Di Giorgio, E.; Ingrao, S.; Meyer-Almes, F.; Tripodo, C.; Brancolini, C. Class IIa HDACs repressive activities on MEF2-depedent transcription are associated with poor prognosis of ER + breast tumors. FASEB J. 2013, 27, 942–954. [Google Scholar] [CrossRef] [Green Version]
- Hamers, A.A.; Vos, M.; Rassam, F.; Marinković, G.; Kurakula, K.; van Gorp, P.J.; de Winther, M.P.; Gijbels, M.J.; de Waard, V.; de Vries, C.J. Bone Marrow–Specific Deficiency of Nuclear Receptor Nur77 Enhances Atherosclerosis. Circ. Res. 2012, 110, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.N.; Shaked, I.; Hubbeling, H.G.; Punt, J.A.; Wu, R.; Herrley, E.; Zaugg, C.; Pei, H.; Geissmann, F.; Ley, K.; et al. NR4A1 (Nur77) Deletion Polarizes Macrophages Toward an Inflammatory Phenotype and Increases Atherosclerosis. Circ. Res. 2012, 110, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Etna, M.P.; Giacomini, E.; Severa, M.; Coccia, E.M. Pro- and anti-inflammatory cytokines in tuberculosis: A two-edged sword in TB pathogenesis. Semin. Immunol. 2014, 26, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, S.; Tremblay, A.M.; Xiao, L.; Yang, Q.; Ma, K.; Nie, J.; Mao, Z.; Wu, Z.; Giguère, V.; Yang, X.-J.J.J.o.B.C. Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J. Biol. Chem. 2006, 281, 4423–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Yu, S.; Chai, Q.; Wang, J.; Wu, T.; Liu, R.; Liu, Y.; Liu, C.H.; Pang, Y. HDAC6 contributes to human resistance against Mycobacterium tuberculosis infection via mediating innate immune responses. FASEB J. 2021, 35, e22009. [Google Scholar] [CrossRef]
- Wang, X.; Tang, X.; Zhou, Z.; Huang, Q. Histone deacetylase 6 inhibitor enhances resistance to Mycobacterium tuberculosis infection through innate and adaptive immunity in mice. Pathog. Dis. 2018, 76, fty064. [Google Scholar] [CrossRef] [Green Version]
- Campo, M.; Heater, S.; Peterson, G.J.; Simmons, J.D.; Skerrett, S.J.; Mayanja-Kizza, H.; Stein, C.M.; Boom, W.H.; Hawn, T.R. HDAC3 inhibitor RGFP966 controls bacterial growth and modulates macrophage signaling during Mycobacterium tuberculosis infection. Tuberculosis 2021, 127, 102062. [Google Scholar] [CrossRef]
- Lugrin, J.; Ciarlo, E.; Santos, A.; Grandmaison, G.; Dos Santos, I.; Le Roy, D.; Roger, T. The sirtuin inhibitor cambinol impairs MAPK signaling, inhibits inflammatory and innate immune responses and protects from septic shock. Biochim. Biophys. Acta 2013, 1833, 1498–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swamy, B.N.; Suma, T.; Rao, G.V.; Reddy, G.C. Synthesis of isonicotinoylhydrazones from anacardic acid and their in vitro activity against Mycobacterium smegmatis. Eur. J. Med. Chem. 2007, 42, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhou, M.-M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Smale, S.T.; Tarakhovsky, A.; Natoli, G. Chromatin Contributions to the Regulation of Innate Immunity. Annu. Rev. Immunol. 2014, 32, 489–511. [Google Scholar] [CrossRef]
- Chan, C.H.; Fang, C.; Qiao, Y.; Yarilina, A.; Prinjha, R.K.; Ivashkiv, L.B. BET bromodomain inhibition suppresses transcriptional responses to cytokine-Jak-STAT signaling in a gene-specific manner in human monocytes. Eur. J. Immunol. 2015, 45, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenis, D.S.; Medzikovic, L.; van Loenen, P.B.; van Weeghel, M.; Huveneers, S.; Vos, M.; Gogh, I.J.E.-V.; Bossche, J.V.D.; Speijer, D.; Kim, Y.; et al. Nuclear Receptor Nur77 Limits the Macrophage Inflammatory Response through Transcriptional Reprogramming of Mitochondrial Metabolism. Cell Rep. 2018, 24, 2127–2140.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harth, G.; Zamecnik, P.C.; Tang, J.-Y.; Tabatadze, D.; Horwitz, M.A. Treatment of Mycobacterium tuberculosis with antisense oligonucleotides to glutamine synthetase mRNA inhibits glutamine synthetase activity, formation of the poly-L-glutamate/glutamine cell wall structure, and bacterial replication. Proc. Natl. Acad. Sci. USA 2000, 97, 418–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harth, G.; Zamecnik, P.C.; Tabatadze, D.; Pierson, K.; Horwitz, M.A. Hairpin extensions enhance the efficacy of mycolyl transferase-specific antisense oligonucleotides targeting Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7199–7204. [Google Scholar] [CrossRef] [Green Version]
- Skvortsova, Y.V.; Salina, E.G.; Burakova, E.A.; Bychenko, O.S.; Stetsenko, D.A.; Azhikina, T.L. A New Antisense Phosphoryl Guanidine Oligo-2′-O-Methylribonucleotide Penetrates Into Intracellular Mycobacteria and Suppresses Target Gene Expression. Front. Pharmacol. 2019, 10, 1049. [Google Scholar] [CrossRef]
- Das, A.R.; Dattagupta, N.; Sridhar, C.N.; Wu, W.-K. A novel thiocationic liposomal formulation of antisense oligonucleotides with activity against Mycobacterium tuberculosis. Scand. J. Infect. Dis. 2003, 35, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.-C.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Azacitidine (5-azacytidine, Vidaza™) for Injectable Suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine). In Seminars in Hematology; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Cheng, J.C.; Matsen, C.; Gonzales, F.A.; Ye, W.; Greer, S.; Marquez, V.E.; Jones, P.A.; Selker, E.U. Inhibition of DNA Methylation and Reactivation of Silenced Genes by Zebularine. JNCI J. Natl. Cancer Inst. 2003, 95, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Kearns, N.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9–histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.; Valentini, D.; Zumla, A.; Maeurer, M. Evaluation of the efficacy of valproic acid and suberoylanilide hydroxamic acid (vorinostat) in enhancing the effects of first-line tuberculosis drugs against intracellular Mycobacterium tuberculosis. Int. J. Infect. Dis. 2018, 69, 78–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rekha, R.S.; Mily, A.; Sultana, T.; Haq, A.; Ahmed, S.; Kamal, S.M.M.; Van Schadewijk, A.; Hiemstra, P.S.; Gudmundsson, G.H.; Agerberth, B.; et al. Immune responses in the treatment of drug-sensitive pulmonary tuberculosis with phenylbutyrate and vitamin D3 as host directed therapy. BMC Infect. Dis. 2018, 18, 303. [Google Scholar] [CrossRef]
- Mily, A.; Rekha, R.S.; Kamal, S.M.M.; Arifuzzaman, A.S.M.; Rahim, Z.; Khan, L.; Haq, A.; Zaman, K.; Bergman, P.; Brighenti, S.; et al. Significant Effects of Oral Phenylbutyrate and Vitamin D3 Adjunctive Therapy in Pulmonary Tuberculosis: A Randomized Controlled Trial. PLoS ONE 2015, 10, e0138340. [Google Scholar] [CrossRef]
- Hammitzsch, A.; Tallant, C.; Fedorov, O.; O’Mahony, A.; Brennan, P.E.; Hay, D.A.; Martinez, F.O.; Al-Mossawi, M.H.; de Wit, J.; Vecellio, M.; et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc. Natl. Acad. Sci. USA 2015, 112, 10768–10773. [Google Scholar] [CrossRef] [Green Version]
- Gauba, K.; Gupta, S.; Shekhawat, J.; Sharma, P.; Yadav, D.; Banerjee, M. Immunomodulation by epigenome alterations in Mycobacterium tuberculosis infection. Tuberculosis 2021, 128, 102077. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Liu, C.; Zhang, L.; Lv, D.; Weng, Y.; Cheng, Z.; Chen, X.; Zhan, J.; Zhang, H. Isonicotinylation is a histone mark induced by the anti-tuberculosis first-line drug isoniazid. Nat. Commun. 2021, 12, 5548. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Shariq, M.; Quadir, N.; Singh, J.; Sheikh, J.A.; Hasnain, S.E.; Ehtesham, N.Z. Mycobacterium tuberculosis Protein PE6 (Rv0335c), a Novel TLR4 Agonist, Evokes an Inflammatory Response and Modulates the Cell Death Pathways in Macrophages to Enhance Intracellular Survival. Front. Immunol. 2021, 12, 696491. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein | Epigenetic Target | Effect |
|---|---|---|
| Rv3423.1 | Histone acetyl transferase | It leads to increase in no. of bacteria in host intracellular environment and their survival [86]. |
| Rv2966c | DNA methylation (non CpG context) | It showed the ability to localize the host’s cell nucleus and repression of specific genes [66]. |
| Rv1988 | Histone methyltransferase | It demethylates the amino acid arginine specifically at 42nd position in the histone H3, which has a profound effect on host gene transcription due to its capacity to localize with the chromatin in the host nucleus [54]. |
| Rv3763 | Histone acetyl transferase | It is responsible for histone hypoacetylation at CIITA promoter via suppressing IFN-γ induced genes [66]. |
| Rv3263 | DNA methylation | It is responsible for all detectable methylation modification by a particular Mtb strain, H37Rv [87]. |
| Epigenetic Modifications | Targets Involved | Therapeutic Agents |
|---|---|---|
| Histone modifications | MMP IL-12b, IL-1β CIITA, HLA-DR, CD64 NF-κB JAK-STAT pathways | Nonspecific HDAC inhibitors: SAHA (vorinostat) Trichostatin A (TSA) Phenylbutyrate (PBA) HDAC6 specific: Tubustatin A MC2780 HDAC3 specific: RGFP966 Bromodomain inhibitors HAT inhibitor: Anacardiac acid |
| Alterations in expression of non-coding RNA | DCs TLR4 NF-κB CD1c | Antisense mRNA targeting agents: Phosphorothioate-modified antisense oligodeoxyribonucleotides (PS-ODNs) Phosphoryl guanidine oligo-2′-O-methylribonucleotides (2′-OMe PGOs) Phosphorothioate antisense oligonucleotides (PAOs) |
| Alteration in DNA methylation | IFN-γ Monocyte derived-DCs TLR2 IL-17, IL-12, IL-10 | Methylation inhibitors: 5-Azacytidine Zebularine |
| Target of the Therapy | Therapeutic Agent | Nature of STUDY | Results |
|---|---|---|---|
| HDAC non-specific inhibitors | Vorinostat Valproic acid | In vitro (H37Rv cultures) | Vorinostat and Valproic acid showed 1.5 and 2 log reduction in CFU, respectively. Both improved the efficacy of rifampicin against Mtb [130]. |
| Trichostatin A | In vivo (M. marinum infected zebrafish) | 32% reduction of bacterial burden in pre-treated model [98]. | |
| Phenylbutyrate with Vitamin D | In vivo (Humans) | Decline in concentrations of cytokines such as TNFα, CCL11/5, PDGF-β Increase in LC3 expression [131] | |
| Phenylbutyrate with Vitamin D | In vivo (Humans- RCT) | 28.8% increase in rate of patients’ culture conversion compared to placebo [132]. | |
| HDAC 6 inhibitors | Tubastatin A | In vivo (C57BL/6 mice model infected with Mtb) | 6 log reduction in CFU after 14 days Upregulation of TNFα, IL-12, IFNγ and downregulation of IL-10 [112]. |
| Tubastatin A | In vitro (THP-1 cell line) | Inhibition of TNFα and IL-6 in the cell line [105]. | |
| Bromodomain inhibitor | CBP30 | In vitro (heparinized human blood) | No effect on expression of TNFα [133]. |
| HAT inhibitors | Isonicotinoylhydrazones derived from anacardic acid | In vitro (M. smegmatis mc2155 cells) | MIC of 4 µg/mL Synergistic actions with isoniazid [115]. |
| DNA methylation inhibitors | Azacytidine | In vivo (Humans) | The trial is still ongoing (NCT03941496) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khadela, A.; Chavda, V.P.; Postwala, H.; Shah, Y.; Mistry, P.; Apostolopoulos, V. Epigenetics in Tuberculosis: Immunomodulation of Host Immune Response. Vaccines 2022, 10, 1740. https://doi.org/10.3390/vaccines10101740
Khadela A, Chavda VP, Postwala H, Shah Y, Mistry P, Apostolopoulos V. Epigenetics in Tuberculosis: Immunomodulation of Host Immune Response. Vaccines. 2022; 10(10):1740. https://doi.org/10.3390/vaccines10101740
Chicago/Turabian StyleKhadela, Avinash, Vivek P. Chavda, Humzah Postwala, Yesha Shah, Priya Mistry, and Vasso Apostolopoulos. 2022. "Epigenetics in Tuberculosis: Immunomodulation of Host Immune Response" Vaccines 10, no. 10: 1740. https://doi.org/10.3390/vaccines10101740